

Abstract
The EP1 prostanoid receptor is one of four subtypes whose cognate physiological ligand is prostaglandin-E2 (PGE2). It is in the family of G-protein-coupled receptors and is known to activate Ca2+ signaling, although relatively little is known about other aspects of E-type prostanoid receptor (EP) 1 receptor signaling. In human embryonic kidney (HEK) cells expressing human EP1 receptors, we now show that PGE2 stimulation of the EP1 receptor up-regulates the expression of hypoxia-inducible factor-1α (HIF-1α), which can be completely blocked by pertussis toxin, indicating coupling to Gi/o. This up-regulation of HIF-1α occurs under normoxic conditions and could be inhibited with wortmannin, Akt inhibitor, and rapamycin, consistent with the activation of a phosphoinositide-3 kinase/Akt/mammalian target of rapamycin (mTOR) signaling pathway, respectively. In contrast to the hypoxia-induced up-regulation of HIF-1α, which involves decreased protein degradation, the up-regulation of HIF-1α by the EP1 receptor was associated with the phosphorylation of ribosomal protein S6 (rpS6), suggesting activation of the ribosomal S6 kinases and increased translation. Stimulation of endogenous EP1 receptors in human HepG2 hepatocellular carcinoma cells recapitulated the normoxic up-regulation of HIF-1α observed in HEK cells, was sensitive to pertussis toxin, and involved the activation of mTOR signaling and phosphorylation of rpS6. In addition, treatment of HepG2 cells with sulprostone, an EP1-selective agonist, up-regulated the mRNA expression of vascular endothelial growth factor-C, a HIF-regulated gene. HIF-1α is known to promote tumor growth and metastasis and is often up-regulated in cancer. Our findings provide a potential mechanism by which increased PGE2 biosynthesis could up-regulate the expression of HIF-1α and promote tumorigenesis.
E-type prostanoid receptors (EP) are the receptors that mediate the actions of prostaglandin E2 (PGE2) and are members of the superfamily of G-protein coupled receptors. There are four primary subtypes of EP receptors, named EP1, EP2, EP3, and EP4. The EP1, EP2, and EP3 receptors were initially classified on the basis of their pharmacology and upon differences in their functional effects on various types of smooth muscle, as well as their activation of second-messenger signaling pathways (Coleman et al., 1994). Thus, PGE2 stimulation of EP1 receptors produced contractile responses that could be selectively blocked with 8-chloro-dibenz[b,f][1,4]oxazepine-10(11H)-carboxy-(2-acetyl)hydrazide (SC-19220) and were involved in the mobilization of intracellular Ca2+. PGE2 stimulation of EP2 receptors produced smooth muscle relaxation that was associated with activation of adenylyl cyclase, whereas, stimulation of EP3 receptors produced smooth muscle contraction that was associated with inhibition of adenylyl cyclase. EP2 and EP3 receptors could also be differentiated pharmacologically, EP2 receptors being selectively activated by butaprost and EP3 (and EP1) receptors being selectively activated by sulprostone. EP4 receptors were first recognized as EP2-like receptors that were insensitive to butaprost; thus, stimulation with PGE2 caused smooth muscle relaxation that could not be mimicked with butaprost or sulprostone (Lawrence and Jones, 1992). However, a full appreciation of their relationship to the EP2 receptors required the molecular cloning of their genes, which established that EP4 receptors were coupled to both the activation of adenylyl cyclase and the stimulation of phosphatidylinositol 3-kinase (PI3K) signaling (Regan, 2003).
Although it is well accepted that the PGE2 stimulation of the EP1 receptor is linked to smooth muscle contraction and increased intracellular Ca2+, it is unclear whether this involves exclusive coupling to Gq/11 and typical activation of phospholipase C (PLC) and phosphatidylinositol hydrolysis. For example, in Chinese hamster ovary cells stably transfected with the mouse EP1 receptor, sulprostone induced intracellular Ca2+ mobilization by two pathways (Katoh et al., 1995). One pathway involved transient Ca2+ release from internal stores that could be blocked with the PLC inhibitor 1-[6-[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino] hexyl]-1H-pyrrole-2,5-dione (U73122) and a second involved extracellular Ca2+ influx that was U73122-insensitive. Coexpression of the mouse EP1 receptor with a receptor-activated Ca2+ channel (TPR5) in Xenopus laevis oocytes produced similar findings (Tabata et al., 2002). Thus, PGE2 stimulated an initial endogenous transient inward current that followed by a large inward Ca2+-dependent current, but only the former could be inhibited with antisense Gαq/11 mRNA.
Further work on the signaling properties of the EP1 prostanoid receptor is limited. There is a report showing that endogenous EP1 receptors in human trophoblasts activate intracellular Ca2+ signaling by a mechanism involving the activation of PLC (Nicola et al., 2005). Thus, the EP1 receptor-mediated release of intracellular Ca2+ was found to be primarily dependent on internal Ca2+ stores and not on extracellular Ca2+. This is in contrast to earlier findings with recombinant EP1 receptors (Watabe et al., 1993) and with endogenous EP1 receptors in guinea pig trachea (Creese and Denborough, 1981), in which Ca2+ responses were almost entirely dependent upon extracellular Ca2+. Recent evidence has suggested that stimulation of endogenous EP1 receptors can inhibit an Akt kinase signaling pathway that potentially contributes to apoptotic neuronal cell death after oxygen/glucose deprivation (Zhou et al., 2008). The molecular mechanism of this signaling is unclear, although it seems to involve an EP1 receptor-mediated decrease in the phosphorylation of phosphatase and tensin homolog deleted on chromosome 10 (PTEN), a lipid phosphatase.
To further examine the signaling properties of the human EP1 prostanoid receptor, we conducted an exploratory gene array screen to identify possible target genes regulated by this receptor (X. B. Chen and J. W. Regan, unpublished observations). Two genes related to angiogenesis were found to be up-regulated after PGE2 treatment. Hypoxia-inducible factor-1α (HIF-1α) is a master transcription factor that controls the expression of many genes involved in the hypoxic response, including several that regulate angiogenesis (Semenza, 2001). The cellular expression of HIF-1α is typically controlled by a post-translational mechanism, and its mRNA expression was unchanged in our microarray screen. We hypothesized, however, that post-transcriptional up-regulation of HIF-1α by the EP1 receptor might be responsible for the up-regulation of the angiogenic genes observed in our microarray screen. As described in the following report, we confirmed this hypothesis and found that the up-regulation of HIF-1α by the human EP1 receptor involved unexpected coupling to a pertussis toxin sensitive G-protein and activation of a PI3K/Akt/mammalian target of rapamycin (mTOR) signaling pathway.
Materials and Methods
Materials.
TRIzol Reagent, Dulbecco's modified Eagle's medium, Opti-MEM, hygromycin B, G418 (Geneticin), gentamicin, pcDNA3, pCEP4, and human embryonic kidney (HEK) 293-Epstein Barr nuclear antigen (EBNA) cells were from Invitrogen (Carlsbad, CA). iScript cDNA kit was from Bio-Rad Laboratories (Hercules, CA). Antibodies against HIF-1α were from BD Sciences (San Jose, CA). Anti-mouse IgG conjugated with horseradish peroxidase and anti-ubiquitin and anti-vinculin antibodies were from Sigma-Aldrich (St. Louis, MO). Polyvinylidene difluoride membranes were from Bio-Rad Laboratories. Cell lysis buffer and antibodies against phospho-ribosomal protein S6 (Ser235/Ser236; product #2211) were from Cell Signaling Technology (Waltham, MA). PGE2 and sulprostone were from Cayman Chemical Company (Ann Arbor, MI). [3H]PGE2 and [myo-2-3H(N)]inositol were from PerkinElmer Life and Analytical Sciences (Waltham, MA). Pertussis toxin, wortmannin, rapamycin, Akt inhibitor, bisindolylmaleimide I, and BAPTA-AM were from Calbiochem (San Diego, CA). FuGENE 6 was from Roche (San Francisco, CA). The Dual Luciferase Reporter Assay System and the Renilla reniformis luciferase reporter pRL-CMV were from Promega (Madison, WI). TaqMan Gene Expression Assays were from Applied Biosystems (Foster City, CA) and corresponded to the following gene symbols: glyceraldehyde 3-phosphate dehydrogenase (GAPDH), HIF-1α, early growth response factor-1 (EGR-1), phosphoglycerate kinase 1 (PKG1), connective tissue growth factor (CTGF), glucose transporter 1 (GLUT1), vascular endothelial growth factor (VEGF)-A, VEGF-C, and erythropoietin (EPO).
Cell Culture.
A cell line stably expressing the recombinant human EP1 prostanoid receptor was generated using HEK cells expressing the EBNA. In brief, the polymerase chain reaction (PCR) was used to amplify a product from human kidney cDNA containing the coding sequence (nucleotides 1–1209) of the human EP1 receptor (Funk et al., 1993), which was then cloned into the EcoRV site of the expression vector, pcDNA3, to generate hEP1/pcDNA3. hEP1/pcDNA3 was digested with HindIII and XhoI, and the small fragment containing the EP1 coding sequence was cloned into the corresponding sites of the vector pCEP4 to yield hEP1/pCEP4. HEK-EBNA cells were transfected with hEP1/pCEP4 using calcium phosphate precipitation and were selected by resistance to hygromycin B. Clones were obtained by limiting dilution. Clonal cell lines stably expressing the EP1 receptor (HEK-hEP1) were identified by PGE2 stimulation of inositol phosphates (IP) formation and by the radioligand binding of [3H]PGE2. A control cell line expressing the “empty” pCEP4 vector (HEK-pCEP4) was prepared in a similar manner. Cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 250 μg/ml G418, 100 μg/ml gentamicin, and 200 μg/ml hygromycin B. Human hepatocellular carcinoma HepG2 cells were obtained from the American Type Culture Collection (Manassas, VA) and cultured in modified Eagle's medium containing 10% fetal bovine serum. Cells were cultured in a humidified incubator at 37°C in 5% CO2/95% air.
Inositol Phosphate Assay.
Cells were split into six-well plates and grown to ∼90% confluence. Starting ∼18 h before the assay, the cells were incubated with Opti-MEM containing 3.25 μCi/ml [myo-2-3H(N)]inositol. The cells were pretreated with 10 mM LiCl, followed by 1 h of incubation with varying concentrations of PGE2. The treated cells were harvested and inositol phosphate levels were determined by anion exchange chromatography as described previously (Fujino et al., 2000).
Western Blotting.
Cells were split into six-well plates at a density of 106 cells/well and incubated overnight. They were then treated with 1 μM PGE2 for indicated times, and immunoblotting was performed essentially as described previously (Fujino et al., 2003). In short, cell lysates were prepared and measured for protein content using the Bradford assay. Approximately 100 μg of protein was electrophoresed on 10% SDS-polyacrylamide gel electrophoresis gels and transferred to polyvinylidene difluoride membranes. Membranes were incubated overnight at 4°C with primary antibodies and then for 1 h at room temperature with the secondary antibodies. Anti-phospho-ribosomal S6 kinase-1 and anti-HIF-1α antibodies were used at a dilution of 1:1000 and 1:200, respectively, in 3% nonfat milk. Horseradish peroxidase-conjugated secondary antibodies were used at a dilution of 1:10,000 in 3% nonfat milk. After incubation with secondary antibodies, the membranes were washed and immunoreactivity was detected by enhanced chemiluminescence. To ensure equal loading of proteins, the membranes were stripped and re-probed with anti-vinculin antibodies. For immunoprecipitation experiments, lysates were incubated with anti-HIF-1α antibodies and protein-A beads overnight at 4°C. The beads were washed, resuspended in SDS-polyacrylamide gel electrophoresis sample buffer, then electrophoresed and immunoblotted as above with either anti-ubiquitin or anti-HIF-1α antibodies.
HRE Reporter Gene Assay.
Cells were split into six-well plates and grown to ∼75% confluence; ∼24 h later, the cells were cotransfected with 2 μg/well of pGL3/HRE-Luc27 firefly luciferase reporter plasmid under the control of a HIF response element (Welsh et al., 2002) and 10 ng/well of the R. reniformis luciferase reporter pRL-CMV using 5 μl of FuGENE 6. Cultures were then treated with either vehicle (0.1% dimethyl sulfoxide in phosphate-buffered saline solution) or 1 μM PGE2 for 18 h. Cells were harvested and 2 μl of the lysates were taken to measure luciferase activity using the Dual Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. The data were normalized by calculating ratios of firefly luciferase scores to the corresponding R. reniformis luciferase values.
Quantitative Real-Time PCR.
Total RNAs were isolated using either TRIzol Reagent or Absolutely RNA Miniprep kits according to the manufacturer's instructions. The RNA integrity was verified by 1% agarose gel electrophoresis, and the quantity was determined by spectrophotometry. cDNAs were prepared from ∼500 ng of total RNA using the iScript cDNA synthesis kit. mRNA expression was determined using the TaqMan Gene Expression Assay primers listed under Materials. PCR reactions were subjected to 40 cycles of 95°C for 15 s and 60°C for 45 s in an ABI 7500 real-time PCR system. Threshold cycle values (Ct) were determined automatically by the system software and relative mRNA expression was analyzed by the comparative ΔΔCt method. Data were normalized to the mRNA expression of GAPDH.
Results
[3H]PGE2 Binding and Stimulation of IP Formation by the Recombinant Human EP1 Receptor Expressed in HEK Cells.
Although it is generally assumed that EP1 receptors are coupled to Gq/11, previous studies have failed to show a robust stimulation of intracellular IP formation. For example, PGE2 stimulation of the mouse EP1 receptor expressed in Chinese hamster ovary cells increased IP formation only ∼1.2-fold (Watabe et al., 1993), whereas agonist stimulation of the ovine FP receptor, another Gq/11 coupled prostanoid receptor, increased IP formation nearly 10-fold (Fujino et al., 2000). Some authors have even indicated that EP1 receptors do not stimulate IP formation and that the coupling of these receptors to intracellular Ca2+ mobilization may involve an unknown G protein (Hata and Breyer, 2004; Sugimoto and Narumiya, 2007). To further characterize the signaling properties of these receptors, we prepared HEK cells stably expressing human EP1 prostanoid receptors and initially examined the radioligand binding of [3H]PGE2- and PGE2-stimulated IP formation in these cells. Figure 1A shows the results of PGE2 competition curves for the whole-cell specific binding of [3H]PGE2 to HEK cells stably expressing either the empty vector control plasmid (HEK-pCEP4) or plasmid encoding the human EP1 receptor (HEK-hEP1). In cells stably transfected with the empty vector, there was no significant displacement of [3H]PGE2 by PGE2, whereas in the EP1-expressing cells, PGE2 competed for the binding of [3H]PGE2 in a simple monophasic manner with an IC50 of 3.6 ± 0.2 nM.
Fig. 1.
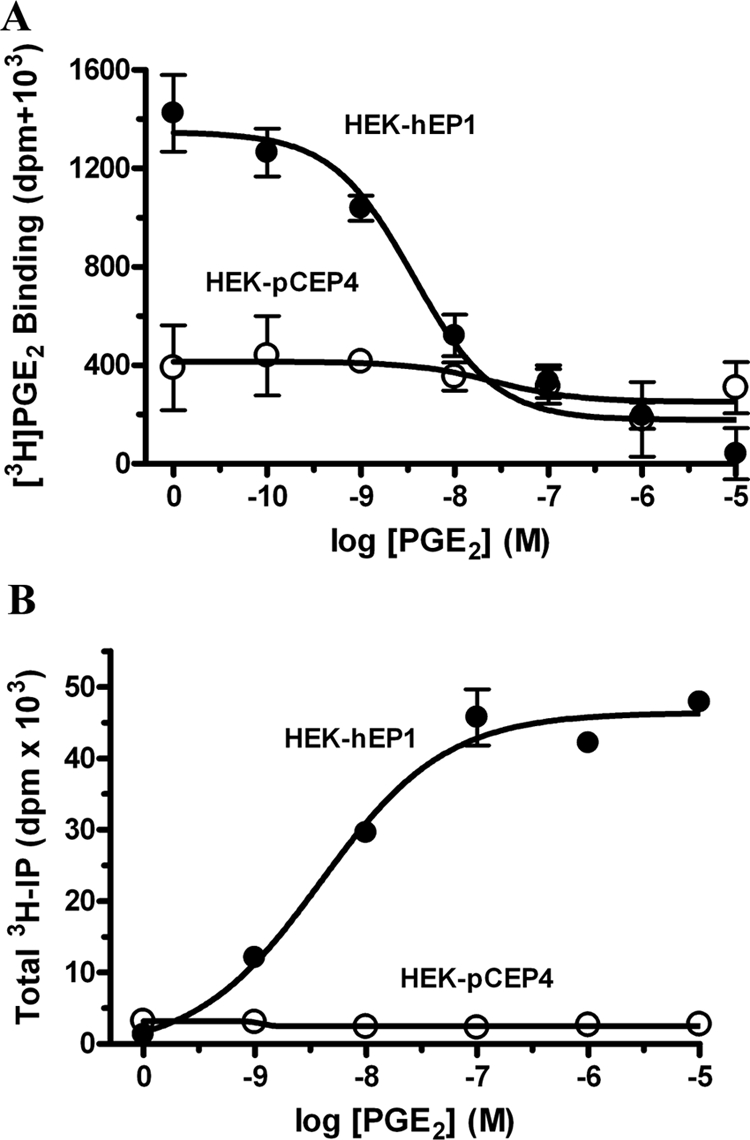
Specific whole-cell binding of [3H]PGE2 (A) and PGE2 stimulation of total inositol phosphates formation (B) in HEK cells stably expressing either empty vector (HEK-pCEP4) or vector encoding the human EP1 prostanoid receptor (HEK-hEP1). A, PGE2 competition curves for the specific binding of [3H]PGE2 were done essentially as described previously (Regan et al., 1994) by incubating whole cells for 1 h at room temperature in a final concentration of 2.5 nM [3H]PGE2 in the presence of various concentrations of nonradioactive PGE2. Data are the means ± S.E.M. of triplicate measurements from a representative experiment that was repeated three times. B, total IPs were determined as described under Materials and Methods on HEK-pCEP4 and HEK-hEP1 cells that were incubated for 1 h at 37°C with the indicated concentrations of PGE2. Data are the means ± S.E.M. of duplicate measurements from a representative experiment that was repeated three times.
Figure 1B shows the results for the stimulation of total intracellular IP formation by PGE2 in control HEK-pCEP4 cells and in HEK-hEP1 cells. In contrast to the control cells in which no response was observed, PGE2 treatment of HEK-hEP1 cells produced a robust dose-dependent stimulation of IP formation with an EC50 of 4.8 ± 0.2 nM, consistent with the coupling of these receptors to Gq/11.
Normoxic Up-Regulation of HIF-1α Expression by PGE2 in HEK Cells Expressing the Human EP1 Receptor.
A preliminary DNA microarray study was conducted to profile the expression of genes that were potentially regulated by PGE2 interaction with the human EP1 prostanoid receptor (X. B. Chen and J. W. Regan, unpublished observations). In brief, HEK-hEP1 cells were treated with 1 μM PGE2 for various periods of time, and changes in gene expression relative to vehicle treated cells were examined using Affymetrix human genome arrays. Among the genes that were found to be most strongly up-regulated were two related to the regulation of angiogenesis. Although the gene expression (mRNA) of HIF-1α was unchanged by treatment with PGE2, we hypothesized that changes in the protein expression of HIF-1α might be responsible for global changes in angiogenic gene expression. Therefore, we used Western blot analysis to examine potential PGE2-stimulated up-regulation of HIF-1α expression in HEK-hEP1 cells maintained under the routine normoxic conditions of cell culture (95% air/5% CO2). Figure 2A shows an immunoblot for the expression of HIF-1α and vinculin in HEK-hEP1 cells after treatment with 1 μM PGE2 for various periods of time between 0 (untreated) and 12 h. A low level of HIF-1α expression was present at 0 h and was unchanged after 1 h treatment with PGE2; after 3, 6, and 12 h of treatment, however, a marked up-regulation of HIF-1α was observed. On the other hand, the expression of vinculin remained essentially constant at all time points.
Fig. 2.

Time courses for the PGE2-stimulated up-regulation of HIF-1α protein (A), mRNA expression (C), and stimulation of HIF-1 reporter gene activity (B) in HEK cells stably expressing the human EP1 receptor. A, HEK-hEP1 cells were incubated with 1 μM PGE2 at 37°C for the indicated times and were subjected to immunoblot analysis using antibodies against human HIF-1α or vinculin as described under Materials and Methods. A representative immunoblot is shown from one of three independent experiments. B, HIF-responsive luciferase reporter gene activity in control HEK-pCEP4 cells or HEK-hEP1 cells after treatment with either vehicle (veh) or 1 μM PGE2. Cells were transiently transfected with a reporter plasmid under the control of an HRE, and luciferase activity was determined as described under Materials and Methods. Data are the means ± S.E.M. of quadruplicate measurements from a representative experiment that was repeated three times. ∗∗∗, p < 0.001 compared with corresponding vehicle treatment; two-way ANOVA, followed by Bonferroni post test. C, HEK-hEP1 cells were incubated with 1 μM PGE2 at 37°C for the indicated times and then RNA was isolated and used for quantitative real-time PCR with primers specific for either HIF-1α or GAPDH as described under Materials and Methods. Data were analyzed by the comparative ΔΔCt method relative to the expression of GAPDH. Data are the means ± S.E.M. (n = 6) of the pooled data from two independent experiments, each done in triplicate.
A luciferase reporter gene construct under the control of a HIF response element (HRE) was used to examine whether the up-regulation of HIF-1α after PGE2 stimulation of the EP1 receptor could potentially stimulate transcriptional activity of HIF-1 target genes. Figure 2B shows that in HEK cells expressing the human EP1 receptor, PGE2 stimulated HRE-mediated luciferase activity nearly 3-fold but had no significant effect in control cells expressing the empty pCEP4 vector. These results indicate that the up-regulation of HIF-1α by agonist stimulation of the EP1 receptor results in functional interactions with HIF-1β and activation of the HRE.
Up-Regulation of HIF-1α Expression Mediated by the EP1 Receptor Does Not Involve an Increase in Gene Transcription.
Quantitative real-time PCR was used to examine the expression of HIF-1α mRNA in HEK-hEP1 cells treated with 1 μM PGE2, and the results are shown in Fig. 2C. In contrast to the up-regulation of HIF-1α protein expression observed in Fig. 2A, there were no changes in the expression of HIF-1α mRNA at 1 and 3 h after treatment with PGE2. These results indicate that PGE2-stimulated up-regulation of HIF-1α expression mediated by the EP1 receptor does not involve changes in gene transcription, which is consistent with known mechanisms for the up-regulation of HIF-1α in response to hypoxia.
Up-Regulation of HIF-1α Expression Mediated by the EP1 Receptor Appears to Be Translational and Does Not Involve a Decrease in 26s Proteasome Activity or Decreased Ubiquitination of HIF-1α.
It is well established that under normoxic conditions, HIF-1α is constitutively expressed and subsequently degraded by the activity of the 26S proteasome so that accumulation does not occur and the levels of HIF-1α protein expression remain very low (Semenza, 2001). Recognition of HIF-1α by the 26S proteasome requires the ubiquitination of HIF-1α by the von Hippel-Lindau protein/ubiquitin E3 ligase complex, which in turn depends upon the hydroxylation of regulatory proline residues in HIF-1α by oxygen-sensitive prolyl hydroxylases (Schofield and Ratcliffe, 2004). Thus, the up-regulation of HIF-1α expression mediated by the EP1 receptor under normoxic conditions could potentially involve decreased protein degradation, either by a global decrease in 26S proteosome activity or by a specific decrease in the hydroxylation and/or ubiquitination of HIF-1α.
Therefore, we used immunoprecipitation and immunoblot analysis with antibodies against HIF-1α and ubiquitin to examine the ratio of ubiquitinated HIF-1α to total HIF-1α after treatment of HEK-hEP1 cells with either vehicle or 1 μM PGE2 for 6 h. For these experiments, the cells were pretreated for 4 h with 25 μM N-benzoyloxycarbonyl (Z)-Leu-Leu-leucinal (MG132) to inhibit the constitutive degradation of HIF-1α by the 26S proteosome. Figure 3A shows a representative immunoblot and a bar graph of the pooled data from three experiments after immunoprecipitation with antibodies against HIF-1α and then immunoblotting with antibodies against either ubiquitin or HIF-1α. As shown in the bar graph, treatment with PGE2 resulted in similar, ∼1.5-fold increases in the expression of ubiquitinated HIF-1α and total HIF-1α; thus, the ratio of ubiquitinated HIF-1α to total HIF-1α did not change after treatment with PGE2. These findings indicate that the EP1 receptor induced up-regulation of HIF-1α is not a consequence of decreased ubiquitin mediated degradation resulting from either decreased prolyl hydroxylation of HIF-1α or decreased activity of the E3 ubiquitin ligase.
Fig. 3.

Immunoblot analysis showing the PGE2-stimulated up-regulation of ubiquitinated HIF-1α or total HIF-1α (A) and time course of the up-regulation of HIF-1α (B) in HEK cells stably expressing the human EP1 receptor under control conditions or after pretreatment with the proteosome inhibitor MG132. A, HEK-hEP1 cells were pretreated with MG132 for 4 h and then treated with either vehicle (veh) or 1 μM PGE2 for 6 h at 37°C. Lysates were prepared and HIF-1α was immunoprecipitated (IP) with antibodies against HIF-1α and then immunoblotted (IB) with antibodies against either ubiquitin or HIF-1α as described under Materials and Methods. Shown is a representative immunoblot and a bar graph of the pooled data from three independent experiments analyzed by densitometry. Data are the means ± S.E.M. ∗∗∗, p < 0.001 compared with corresponding vehicle; one-way ANOVA, followed by Bonferroni post test. B, HEK-hEP1 cells were pretreated with either vehicle (control) or MG132 for 4 h and were then treated with 1 μM PGE2 for indicated times at 37°C. Lysates were prepared and subjected to immunoblot analysis with antibodies against either HIF-1α or vinculin as described under Materials and Methods. C, HEK-hEP1 cells were pretreated with MG132 for 4 h and then treated with either vehicle (veh) or 1 μM PGE2 for 6 h at 37°C. Lysates were prepared and subjected to immunoblot analysis with antibodies against either HIF-1α or vinculin as above and were then analyzed by densitometry with the expression of HIF-1α normalized to the expression of vinculin. Shown is a bar graph of the pooled data from three independent experiments. Data are the means ± S.E.M. ∗∗, p < 0.01 compared with vehicle; one-way unpaired t test.
The 26S proteasome inhibitor, MG132, was also used to examine whether the up-regulation of HIF-1α could be attributed solely to a generalized decrease in the activity of 26S proteasome. Figure 3B shows an immunoblot of the time courses for the PGE2-stimulated up-regulation of HIF-1α expression in HEK-hEP1 cells under control conditions or after pretreatment with MG132. It is immediately apparent that pretreatment with MG132 caused a dramatic up-regulation in the expression of HIF-1α at all time points, including the zero time point. This result confirms that under normoxic conditions, HIF-1α expression in HEK cells is largely under the control of a classic mechanism involving constitutive proteasome-mediated degradation. However, it is also evident that even after inhibiting the 26S proteasome, the expression of HIF-1α was up-regulated after 3 and 6 h of treatment with 1 μM PGE2. Three additional experiments were conducted to examine the effect of MG132 on the expression of HIF-1α after the treatment of HEK-hEP1 cells with either vehicle or 1 μM PGE2 for 6 h. For these experiments, the expression of HIF-1α was analyzed by immunoblot analysis and then quantified by densitometry. As shown in Fig. 3C, treatment with PGE2 resulted in a statistically significant 1.6-fold increase in the expression of HIF-1α after the inhibition of the 26S proteosome with MG132. Although decreased degradation of HIF-1α cannot be completely excluded, these findings strongly suggest that an additional mechanism, such as increased translation, is responsible for the up-regulation of HIF-1α by the EP1 receptor.
Up-Regulation of HIF-1α Expression Mediated by the EP1 Receptor Involves the Activation of PI3K, Akt, and mTOR Signaling.
An important mechanism of translational control involves the activity of the ribosomal S6 kinases, which in turn are regulated by the activity of the PI3K, Akt, and the mTOR signaling pathways (Dufner and Thomas, 1999). We therefore decided to examine the potential involvement of PI3K, Akt, and mTOR signaling in the EP1 receptor mediated up-regulation of HIF-1α expression through the use of wortmannin, Akt inhibitor, and rapamycin, which are selective inhibitors of PI3K, Akt, and mTOR, respectively. Figure 4A shows that pretreatment of HEK-hEP1 cells with any one of these inhibitors completely blocked the up-regulation of HIF-1α protein expression mediated by PGE2 and suggest that the EP1 receptor-mediated up-regulation of HIF-1α occurs as a result of increased translation that is driven by the activation of PI3K, Akt, and mTOR signaling.
Fig. 4.

Immunoblots showing the PGE2-stimulated up-regulation of HIF-1α expression in HEK cells stably expressing the human EP1 receptor under control conditions or after pretreatment with either the PI3K inhibitor wortmannin, the Akt inhibitor, or the mTOR inhibitor rapamycin (A); after pretreatment with either the protein kinase C inhibitor BIM or the Ca2+ chelator BAPTA-AM (B); or after pretreatment with the Gi/o inhibitor pertussis toxin (C). A, HEK-hEP1 cells were pretreated with vehicle (control), 10 μM wortmannin (wort), 10 μM Akt inhibitor (Akt I), or 4 μM rapamycin (rapa) for 30 min at 37°C and were then treated with vehicle (veh) or 1 μM PGE2 for 6 h at 37°C. B, HEK-hEP1 cells were pretreated with vehicle (control), 10 μM BIM, or 10 μM BAPTA-AM (BAPTA) for 30 min at 37°C and were then treated with vehicle (veh) or 1 μM PGE2 for 6 h at 37°C. C, HEK-hEP1 cells were pretreated with vehicle (control) or 5 nM pertussis toxin (PTX) overnight and were then treated with vehicle (veh) or 1 μM PGE2 at 37°C for 6 h. Cell lysates were prepared and subjected to immunoblot analysis with antibodies against HIF-1α or vinculin as described under Materials and Methods. Data are representative of at least three independent experiments for each antibody and condition.
Pretreatment of HEK-hEP1 Cells with Pertussis Toxin Blocks the PGE2 Stimulated Up-Regulation of HIF-1α Expression by the EP1 Receptor Indicating Coupling to Gi/o.
Given the coupling of the EP1 receptor to Gq/11 and its ability to stimulate intracellular Ca2+ mobilization, we examined the effects of the intracellular Ca2+ chelator BAPTA/AM and the protein kinase C (PKC) inhibitor bisindolylmaleimide I (BIM) on the PGE2-stimulated up-regulation of HIF-1α by the human EP1 receptor. As shown in Fig. 4B, pretreatment of HEK-hEP1 cells with either BIM or BAPTA/AM unexpectedly failed to significantly inhibit the PGE2-stimulated up-regulation of the expression of HIF-1α, suggesting that coupling to Gq/11 and activation of Ca2+/PKC signaling were not involved in mediating this response. We have recently found that the human EP4 prostanoid receptor can activate PI3K signaling by coupling to a pertussis toxin-sensitive G-protein (Fujino and Regan, 2006). In addition, a number of GPCRs that were traditionally considered to couple exclusively to Gq/11 have been found to couple to Gi/o and activate PI3K signaling (e.g., Voss et al., 2007). We therefore decided to use the Gi/o inhibitor pertussis toxin to examine the potential involvement of Gi/o in up-regulation of HIF-1α expression mediated by the human EP1 receptor. Figure 4C shows that pretreatment of HEK-hEP1 cells with pertussis toxin completely blocked the PGE2-stimulated up-regulation of HIF-1α compared with the vehicle-treated control cells. These data indicate that EP1 prostanoid receptor mediated up-regulation of HIF-1α requires coupling to Gi/o.
PGE2 Stimulation of the EP1 Receptor Induces Phosphorylation of Ribosomal Protein S6 and Requires Gi/o-Mediated Activation of PI3K Signaling.
Ribosomal protein S6 (rpS6) is one of the primary targets of the ribosomal S6 kinases and is a key regulator of translation and cell growth (Ruvinsky and Meyuhas, 2006). The activity of the ribosomal S6 kinases is in turn regulated both directly and indirectly by the activation of PI3K, Akt, and/or mTOR signaling. Two key sites of rpS6 phosphorylation that reflect activation of the ribosomal S6 kinases are Ser235/Ser236. Therefore, we examined the phosphorylation of rpS6 on Ser235/Ser236 after the treatment of HEK-hEP1 cells for various periods of time with 1 μM PGE2. As shown by the immunoblot in Fig. 5A, PGE2 treatment of HEK-hEP1 cells increased the phosphorylation of rpS6 over basal levels after just 1 h and was maintained for up to 24 h. The evidence that the PGE2-induced phosphorylation of rpS6 precedes the up-regulation of HIF-1α supports a putative role of activation of the ribosomal S6 kinases in the translation control of HIF-1α expression after activation the human EP1 receptor.
Fig. 5.

Immunoblots showing the PGE2-stimulated phosphorylation of rpS6 (phospho-rpS6) in HEK cells stably expressing the human EP1 receptor after a time course of PGE2 treatment (A); after pretreatment of cells with the Gi/o inhibitor pertussis toxin (B); or after pretreatment of cells with the PI3K inhibitor wortmannin (C). A, HEK-hEP1 cells were treated with 1 μM PGE2 for indicated times at 37°C, and lysates were prepared and subjected to immunoblot analysis with antibodies against phospho-rpS6 (Ser235/Ser236) or with vinculin as described under Materials and Methods. B, HEK-hEP1 cells were pretreated with either vehicle (control) or 5 nM pertussis toxin (PTX) overnight and were then treated with either vehicle (veh) or 1 μM PGE2 at 37°C for 6 h. Lysates were prepared and subjected to immunoblot analysis as above. C, HEK-hEP1 cells were pretreated with either vehicle (control) or 10 μM wortmannin (wort) for 30 min and then treated with 1 μM PGE2 at 37°C. Lysates were prepared and subjected to immunoblot analysis as above. Data are representative of at least three independent experiments for each antibody and condition.
Next, HEK-hEP1 cells were pretreated with either pertussis toxin or wortmannin to examine the potential involvement of coupling to Gi/o and PI3K signaling in the PGE2-induced phosphorylation of rpS6. Figure 5, B and C, show that pretreatment of cells with either pertussis toxin or wortmannin respectively, decreased the PGE2-stimulated phosphorylation of rpS6 compared with the vehicle-treated control cells and suggests that coupling to Gi/o and activation of PI3K signaling by PGE2 stimulation of the EP1 receptor results in the activation of ribosomal S6 kinase activity.
EP1 and EP2, but Not EP3 and EP4, Prostanoid Receptors Are Expressed in the HepG2 Hepatocellular Carcinoma Cell Line.
HepG2 cells were used to examine the potential of natively expressed endogenous EP1 receptors to couple to Gi/o and up-regulate the expression of HIF-1α. HepG2 cells were chosen because it has been previously shown that prostaglandin synthesis contributes to the growth of these cells by a mechanism involving the activation of Akt, although the specific prostanoid receptors contributing to this response are unknown (Leng et al., 2003). In addition, it is well known that the expression of HIF-1α is important in cancer growth and metastasis (Semenza, 2001), and we were interested in the potential normoxic up-regulation of HIF-1α by PGE2 in cancer cells. PCR with primers specific for the EP1, EP2, EP3, and EP4 receptors was used to examine the expression of the EP prostanoid receptor subtypes in RNA prepared from HepG2 cells and from SH-SY5Y cells, a neuroblastoma cell line known to express endogenous EP1 receptors (Hoshino et al., 2007). As shown in Fig. 6, HepG2 cells expressed the EP1 and EP2 subtypes, but not the EP3 and EP4 subtypes. SH-SY5Y cells, on the other hand, expressed all four EP receptor subtypes.
Fig. 6.

Photographs of ethidium-stained 2% agarose gels showing the products obtained after reverse transcription and PCR with primers specific for the EP1 (A), EP2 (B), EP3 (C), and EP4 (D) prostanoid receptor subtypes and template RNA isolated from HEK cells stably expressing either the EP1, EP2, EP3, or EP4 receptor subtype and from human hepatocellular carcinoma cells (HepG2) and SH-SY5Y human neuroblastoma cells. Reverse transcription and PCR were performed as described previously (Fujino et al., 2007) with an initial incubation at 94°C for 5 min, followed by 35 cycles of 94°C for 20 s, 60°C for 30 s, and 72°C for 60 s. The EP receptor primers were exactly according to Shoji et al. (2004). Molecular size standards are in the far left lanes, and nontemplate control (NTC) reactions are in the far right lanes. Gels shown are from a representative of three independent experiments.
Sulprostone Stimulates the Up-Regulation of HIF-1α Protein Expression, but not mRNA, in HepG2 Cells.
HepG2 cells were used to examine the potential up-regulation of HIF-1α protein and mRNA expression by sulprostone, a selective agonist of the EP1 and EP3 receptors. Figure 7A, top, shows an immunoblot of the expression of HIF-1α and vinculin after treatment of HepG2 cells with 1 μM sulprostone for various periods of time. In untreated cells (0 h) and after 1 h of treatment with sulprostone, there was a low basal expression of HIF-1α, which increased progressively after 3, 6, 12, and 24 h of treatment. Treatment of HepG2 cells with sulprostone did not affect the expression of vinculin. Figure 7A, bottom, shows the results of quantitative real-time PCR for the expression of HIF-1α mRNA. As can be seen, there were no significant changes in the expression of HIF-1α mRNA after the treatment of HepG2 cells with sulprostone. These findings mirror the results obtained in HEK-hEP1 cells and show that sulprostone does not up-regulate the expression of HIF-1α in HepG2 cells by increased transcription. In separate studies, we have found that the human EP1 receptor also mediates the up-regulation of EGR-1 in HEK-hEP1 cells by increased transcription (J.W.R. and R.J., unpublished observations). As a positive control for the measurement of transcriptional changes, we also examined the up-regulation of EGR-1 protein and mRNA expression by sulprostone in HepG2 cells. The immunoblot in Fig. 7B, top, shows that the expression of EGR-1 in HepG2 cells was increased after 1, 2, and 3 h of treatment with 1 μM sulprostone. In contrast to the results obtained for HIF-1α, however, Fig. 7B, bottom, shows that EGR-1 mRNA expression increased ∼3.5-fold after 1 h of treatment with sulprostone and then returned to baseline by 3 h.
Fig. 7.

Time courses for the up-regulation of protein and mRNA expression for HIF-1α (A) and early growth response factor-1 (B) after the treatment of HepG2 cells with sulprostone, an EP1/EP3-selective agonist. Cells were treated with 1 μM sulprostone at 37°C for the indicated times and then subjected to immunoblot analysis with antibodies against either HIF-1α, EGR-1, or vinculin or to quantitative real-time PCR analysis with primers specific for either HIF-1α, EGR-1 or GAPDH as described under Materials and Methods. Shown are representative immunoblots from one of at least three independent experiments for each antibody and condition. PCR data were analyzed by the comparative ΔΔCt method relative to the expression of GAPDH at each time point and were then normalized to expression at time 0 for each gene. Bar graphs represent the mean ± S.E.M. (n = 6) of the pooled data from two independent experiments each performed in triplicate; ∗∗∗, p < 0.001 compared with time 0; one-way ANOVA, followed by Bonferroni post test.
Pretreatment of HepG2 cells with Pertussis Toxin or Rapamycin Blocks the Sulprostone Stimulated Up-Regulation of HIF-1α Expression by the EP1 Receptor.
HepG2 cells were pretreated with pertussis toxin and then stimulated with sulprostone to determine whether the up-regulation of HIF-1α observed with PGE2 treatment involved coupling of an EP1 receptor to Gi/o. As shown by the immunoblot in Fig. 8A, treatment of HepG2 cells with sulprostone under control conditions resulted in a marked increase in the expression of HIF-1α with no observable change in the expression of vinculin. Furthermore, pretreatment of HepG2 cells with pertussis toxin completely blocked the sulprostone-stimulated increase in HIF-1α expression indicating coupling of the endogenous EP1 receptor to Gi/o.
Fig. 8.

Immunoblots showing the up-regulation of HIF-1α expression (A and B) or phosphorylation of ribosomal protein S6 (C) by sulprostone in HepG2 cells either alone (C) or after pretreatment of cells with either the Gi/o inhibitor pertussis toxin (A) or the mTOR inhibitor rapamycin (B). A, HepG2 cells were pretreated with either vehicle (control) or 5 nM pertussis toxin (PTX) overnight at 37°C and then treated with either vehicle (veh) or 1 μM sulprostone (SP) for 6 h at 37°C. Lysates were prepared and subjected to immunoblot analysis with antibodies against HIF-1α and vinculin as described under Materials and Methods. B, HepG2 cells were pretreated with either vehicle (control) or 4 μM rapamycin (rapa) for 30 min and were then treated with either vehicle (veh) or 1 μM sulprostone (SP) for 6 h at 37°C. Lysates were prepared and subjected to immunoblot analysis as above. C, HepG2 cells were treated with 1 μM sulprostone (SP) for indicated times at 37°C, and lysates were prepared and subjected to immunoblot analysis either with antibodies against phosphorylated rpS6 (phospho-rpS6) or vinculin as described under Materials and Methods. Data are representative of at least three independent experiments for each antibody and condition.
Cells were pretreated with rapamycin and then stimulated with sulprostone to examine the potential involvement of mTOR signaling in the up-regulation of HIF-1α expression mediated by the endogenous EP1 receptors in HepG2 cells. The immunoblot in Fig. 8B shows that pretreatment of HepG2 cells with rapamycin completely blocked the sulprostone-stimulated increase in the expression of HIF-1α, suggesting that the mechanism involved in the up-regulation of HIF-1α expression by the endogenous EP1 receptor in HepG2 cells is similar to that characterized for the recombinant EP1 receptor expressed in HEK cells and involves the activation of mTOR signaling. Therefore, we examined a time course for the phosphorylation of ribosomal protein S6 (rpS6) after the treatment of HepG2 cells with sulprostone. As shown in Fig. 8C, treatment of HepG2 cells with 1 μM sulprostone resulted in an initial phosphorylation of rpS6 after 3 h of treatment that increased after 6 h and was maintained up to 12 h. This sulprostone-mediated phosphorylation of rpS6 at Ser235/Ser236 is consistent with EP1 receptor stimulation of mTOR signaling and activation of the ribosomal S6 kinases.
Treatment of HepG2 Cells With Sulprostone Results In the Up-Regulation of VEGF-C mRNA Expression.
Numerous studies have documented the HIF-1α-dependent up-regulation of dozens of genes in a wide variety of cell types after hypoxia (Semenza, 2001). These genes include regulators of angiogenesis, such as VEGF-A and VEGF-C; regulators of glycolytic metabolism, such as PKG1 and GLUT1; and other growth factors and hormones, such as CTGF and EPO. Using quantitative real-time PCR, we examined the mRNA expression of these six genes after the treatment of HepG2 cells with sulprostone to determine whether activation of endogenous EP1 receptors under normoxic conditions could regulate the expression of any known HIF-1 regulated target genes. As shown in Fig. 9, treatment of HepG2 cells with 1 μM sulprostone resulted in a clear time-dependent increase in the mRNA expression of VEGF-C, with essentially no effect on the expression of the other genes. Figure 10 shows the relative transcript levels of these six genes at time 0, before treatment of the cells with sulprostone. The mRNA expression of PKG1, CTGF, and GLUT1 were high, approximately half the transcript level of GAPDH, which may have precluded further up-regulation of these genes by a receptor-dependent mechanism. The expression of VEGF-A was approximately 5% of that of GAPDH, whereas the transcript levels of VEGF-C and EPO were more than a thousand times lower than the mRNA expression of GAPDH.
Fig. 9.

Quantitative real-time PCR analysis for the expression of the indicated human genes after treatment of HepG2 cells with 1 μM sulprostone for 1, 3, or 6 h. Data for each time point were analyzed by the comparative ΔΔCt method relative to the expression of GAPDH and were then normalized to the 0 time point for each gene. Shown are the means ± S.E.M. (n = 4) of the pooled data from two independent experiments, each performed in duplicate. ∗∗∗, p < 0.001; ∗, p < 0.05 compared with time 0; one-way ANOVA, followed by Bonferroni post test.
Fig. 10.

Quantitative real-time PCR analysis for the relative expression of the indicated human genes in HepG2 cells at time 0 before treatment with sulprostone. The data obtained at time 0 in the experiments depicted in Fig. 9 were re-analyzed by the comparative ΔΔCt method relative to the expression of GAPDH and were then normalized to the expression of GAPDH. Values for VEGF-C and EPO were 0.0000025 ± 0.0000002 and 0.0002 ± 0.00003, respectively. Shown are the means ± S.E.M. (n = 4); ∗∗∗, p < 0.001 compared with GAPDH; one-way ANOVA, followed by followed by Bonferroni post test.
Discussion
The EP1 prostanoid receptor is one of the four primary receptor subtypes for PGE2, the others being the EP2, EP3, and EP4 receptor subtypes. In terms of its amino acid sequence identity with the other prostanoid receptors, EP1 is actually more closely related to the prostaglandin F and thromboxane A2 receptors, followed by the EP3, EP2, and EP4 receptors (Regan et al., 1994). Of the four EP receptors, the EP1 also has the lowest binding affinity for PGE2 and, like, the prostaglandin F and thromboxane A2 receptors, the EP1 is generally regarded to couple to Gq/11 as opposed to the EP2 and EP4 receptors, which couple primarily to Gs, and the EP3 receptor, which couples primarily to Gi/o. As noted previously, however, the coupling of the EP1 receptor to Gq/11 has occasionally been questioned because of its generally poor ability to stimulate the formation of intracellular IPs. In the present study, however, we have shown that PGE2 clearly stimulated intracellular IP accumulation in HEK cells expressing the human EP1 receptor, consistent with coupling of this receptor to Gq/11. We also show that the human EP1 receptor can couple to Gi/o to up-regulate the expression of HIF-1α through the activation of PI3K, Akt, and mTOR signaling. This up-regulation of HIF-1α by the EP1 receptor occurs under conditions of normoxia and involves activation of the ribosomal S6 kinases and increased translation.
The expression of HIF-1α is traditionally regarded as being regulated by increased protein stability, resulting from decreased protein degradation in response to hypoxia (Semenza, 2001). Thus, HIF-1α is constitutively expressed in most cells, but under normoxic conditions it undergoes continuous ubiquitin-mediated degradation after hydroxylation of regulatory proline residues. The prolyl hydrolases that carry out the hydroxylation of HIF-1α use molecular oxygen and are also sensitive to the concentration of O2 in the cell (Schofield and Ratcliffe, 2004). Decreases in the partial pressure of O2 during hypoxia decrease the activity of these enzymes and result in the stabilization and up-regulation of HIF-1α expression. Although less fully appreciated, HIF-1α up-regulation is known to occur under normoxic conditions in response to receptor-mediated activation by a variety of cytokines and hormones (Zhou and Brüne, 2006). The signaling pathways involved in this receptor mediated up-regulation of HIF-1α under normoxic conditions typically involve the activation of PI3K and Akt signaling and/or MAP kinase signaling. However, in contrast to the decreased degradation of HIF-1α that occurs during hypoxia, the cytokine receptor-mediated up-regulation of HIF-1α that occurs during normoxia is typically the result of increased translation and/or transcription. Although PGE2 has been previously reported to up-regulate the expression of HIF-1α under normoxic conditions in lung and gastric carcinoma cells, the specific mechanism of this activation was not investigated and was considered to be a downstream consequence of the up-regulation of cyclooxygenase-2 (COX-2) expression (Jung et al., 2003; Huang et al., 2005).
Like PGE2, many of the cytokines that can up-regulate the expression of HIF-1α are also involved in inflammatory responses. Well characterized examples include IL-1β and TNF-α, which have been shown to up-regulate HIF-1α under normoxic conditions. For example, in human synovial fibroblasts, IL-1β and TNF-α increased HIF-1α mRNA and functional expression of HIF-1 (Thornton et al., 2000). HIF-1α translocates to the nucleus and combines with HIF-1β to form HIF-1, the functionally active transcription factor. HIF-1 is a master transcription factor that has been shown to affect the expression of gene families, including those involved in angiogenesis, erythropoiesis, energy metabolism, and epithelial-mesenchymal transition (Semenza, 2001). HIF-1 is being increasingly recognized as a critical factor in the inflammatory response. This was clearly demonstrated by a targeted deletion of HIF-1α in the myeloid cells of mice in which the inflammatory response was greatly decreased (Cramer et al., 2003). In addition, the expression of a variety of hypoxia-responsive genes was significantly diminished under both normoxic and hypoxic conditions, indicating the critical role of HIF-1α in both normoxic and hypoxic gene regulation.
Another key player in the inflammatory response is COX-2, which is strongly up-regulated in the earliest phases of inflammation and remains elevated throughout the inflammatory response (Smith et al., 2000). The expression of microsomal prostaglandin-E synthase-1 is typically simultaneously up-regulated, leading to marked increase in the tissue concentration of PGE2. A similar up-regulation of COX-2 has also been well documented in a wide variety of tumors, lending strong support to the idea that chronic inflammation is a key event in tumorigenesis (Mantovani et al., 2008). Hepatocellular carcinoma (HCC) is one example of a cancer in which there is good evidence for a role of COX-2 and prostaglandin signaling in the development of hepatic inflammation and malignant transformation (Breinig et al., 2007). Thus, COX-2 is overexpressed in HCC, and its expression progressively increases as the liver goes from a stage of chronic hepatitis to cirrhosis, to a premalignant condition, and finally to cancer. In addition, the concentration of PGE2 is increased in HCC, and exogenous PGE2 has been shown to drive hepatic cancer cell growth and invasiveness.
Although it is generally assumed that PGE2 contributes to tumorigenesis through a combination of increased cell survival, increased cell proliferation/motility, induction of angiogenesis, and suppression of immune surveillance, very little is known about the specific EP receptors and molecular mechanisms mediating these effects. In addition little is known about specific mechanisms that could potentially link the role of PGE2 in the inflammatory response to its role in carcinogenesis. In this regard, our present findings provide important new information regarding the putative role of the EP1 receptor in inflammation and tumorigenesis. Thus, we have shown both in HEK cells expressing recombinant EP1 receptors and in HepG2 cells expressing endogenous EP1 receptors that PGE2 can rapidly induce the up-regulation of HIF-1α under normoxic cell culture conditions. This means at the very earliest stages of inflammation, after the initial up-regulation of COX-2 and PGE2 synthesis, there is the potential for increased expression of HIF-1α and the activation of HIF-1 responsive genes. These genes even include COX-2 itself, which has previously been shown to be up-regulated in HCC and associated with tumor angiogenesis (Cheng et al., 2004).
A highly significant correlation has recently been found between the expression of HIF-1α and lymphatic metastasis and VEGF-C expression in human esophageal cancer (Katsuta et al., 2005). Likewise, in lymph node-positive invasive breast cancer, a significant correlation has been found between the expression of HIF-1α and VEGF-C and between the expression of HIF-1α and peritumoral lymphangiogenesis (Schoppmann et al., 2006). Although hypoxia and overexpression of HIF-1α have been found to up-regulate the gene expression of VEGF-C (Manalo et al., 2005), the exact mechanism of this up-regulation is unclear because the VEGF-C promoter does not contain a known consensus binding site for HIF-1. It is intriguing, therefore, that we have found that EP1 receptor activation in HepG2 cells can up-regulate the protein expression of HIF-1α and the mRNA expression of VEGF-C and that the up-regulation of HIF-1α seems to precede the up-regulation of VEGF-C. In this regard, a previous study has found that overexpression of COX-2 up-regulates the expression of VEGF-C in human lung adenocarcinoma cells by a mechanism involving the activation of EP1 receptors (Su et al., 2004). These findings suggest the possibility that up-regulation of VEGF-C in cancer involves either a permission interaction between the up-regulation of HIF-1α and EP1 receptor activation or a direct mechanistic relationship, possibly involving an EP1 receptor mediated up-regulation of HIF-1α, followed by a HIF-1α-mediated up-regulation of VEGF-C.
We have shown for the first time that the EP1 receptor-mediated up-regulation of HIF-1α occurs by a mechanism involving coupling of the EP1 receptor to Gi/o and activation of a PI3K/Akt/mTOR signaling pathway. It is well established that the PI3K/Akt/mTOR pathway is dysregulated in many types of cancer (Shaw and Cantley, 2006), and our findings provide a further mechanism by which the EP1 receptor could influence known oncogenic signaling pathways in addition to its up-regulation of HIF-1α itself. Thus, inappropriate activation of PI3K/Akt/mTOR signaling could drive cellular growth through increased translation and promote cell survival by inhibiting apoptosis. Our findings with the HepG2 cells have specific implications for a role of EP1 receptors in HCC. Thus, previous studies have shown that HIF-1α is overexpressed in patients with HCC (Huang et al., 2005) and that stimulation of EP1 receptors in human HCC cells can enhance tumor cell invasion (Han et al., 2006); however, the potential mechanistic relationship between these observations is unknown. Our results suggest that the up-regulation of COX-2, which is known to occur in HCC, could lead to the up-regulation of HIF-1α and VEGF-C through PGE2 stimulation of the EP1 receptor and thereby drive tumor angiogenesis and metastasis. The EP1 prostanoid receptor clearly merits interest as a potential therapeutic target for the treatment of inflammation and cancer.
Acknowledgments
We thank Dr. Garth Powis (University of Texas M. D. Anderson Cancer Center) for generously providing the pGL3/HRE-Luc reporter plasmid and the Arizona Cancer Center Genomics Facility Core, Dr. George Watts specifically, for help with quantitative PCR and the interpretation of microarray data.
This work was supported by the National Institutes of Health National Eye Institute [Grant EY11291] (to J.W.R.); by Allergan Inc.; and by the University of Arizona College of Pharmacy. Portions of this work were also made possible by grants to the Arizona Cancer Center Genomics Facility Core from the National Institutes of Health National Institute of Environmental Health Sciences [Grant 5P30-ES06694-14900209].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.063933.
- EP
- E-type prostanoid receptor
- PGE2
- prostaglandin E2
- SC-19220
- 8-chloro-dibenz[b,f][1,4]oxazepine-10(11H)-carboxy-(2-acetyl)hydrazide
- PI3K
- phosphatidylinositol 3-kinase
- PLC
- phospholipase C
- U73122
- 1-[6-[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl] amino]hexyl]-1H-pyrrole-2,5-dione
- HIF-1α
- hypoxia-inducible factor-1α
- HEK
- human embryonic kidney
- CMV
- cytomegalovirus
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- PKG1
- phosphoglycerate kinase 1
- CTGF
- connective tissue growth factor
- GLUT1
- facilitated glucose transporter member 1
- VEGF-A
- vascular endothelial growth factor A
- VEGF-C
- lymphatic vascular endothelial growth factor C
- EPO
- erythropoietin.
- EBNA
- Epstein Barr nuclear antigen
- PCR
- polymerase chain reaction
- IP
- inositol phosphate
- HEK-hEP1
- HEK cells expressing the human EP1 receptor
- HRE
- HIF response element
- MG132
- N-benzoyloxycarbonyl (Z)-Leu-Leu-leucinal
- AKT
- protein kinase B
- mTOR
- mammalian target of rapamycin
- BAPTA
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- AM
- acetoxymethyl ester
- PKC
- protein kinase C
- BIM
- bisindolylmaleimide I
- rpS6
- ribosomal protein S6
- EGR-1
- early growth response factor-1
- COX-2
- cyclooxygenase-2
- HCC
- hepatocellular carcinoma
- ANOVA
- analysis of variance.
References
- Breinig M, Schirmacher P, Kern MA. (2007) Cyclooxygenase-2 (COX-2)—a therapeutic target in liver cancer? Curr Pharm Des 13:3305–3315 [DOI] [PubMed] [Google Scholar]
- Cheng AS, Chan HL, To KF, Leung WK, Chan KK, Liew CT, Sung JJ. (2004) Cyclooxygenase-2 pathway correlates with vascular endothelial growth factor expression and tumor angiogenesis in hepatitis B virus-associated hepatocellular carcinoma. Int J Oncol 24:853–860 [PubMed] [Google Scholar]
- Coleman RA, Smith WL, Narumiya S. (1994) International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev 46:205–229 [PubMed] [Google Scholar]
- Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, et al. (2003) HIF-1α is essential for myeloid cell-mediated inflammation. Cell 112:645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creese BR, Denborough MA. (1981) The effects of imidazole on contractility and cyclic AMP levels of guinea-pig tracheal smooth muscle. Clin Exp Pharmacol Physiol 9:145–155 [DOI] [PubMed] [Google Scholar]
- Dufner A, Thomas G. (1999) Ribosomal S6 kinase signaling and the control of translation. Exp Cell Res 253:100–109 [DOI] [PubMed] [Google Scholar]
- Fujino H, Chen XB, Regan JW, Murayama T. (2007) Indomethacin decreases EP2 prostanoid receptor expression in colon cancer cells. Biochem Biophys Res Commun 359:568–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino H, Regan JW. (2006) EP4 prostanoid receptor coupling to a pertussis toxin-sensitive inhibitory G-protein. Mol Pharmacol 69:13–18 [DOI] [PubMed] [Google Scholar]
- Fujino H, Srinivasan D, Pierce KL, Regan JW. (2000) Differential regulation of prostaglandin F2α receptor isoforms by protein kinase C. Mol Pharmacol 57:353–358 [PubMed] [Google Scholar]
- Fujino H, Xu W, Regan JW. (2003) Prostaglandin E2 induced functional expression of early growth response factor-1 by EP4, but not EP2, prostanoid receptors via the phosphatidylinositol 3-kinase and extracellular signal-regulated kinases. J Biol Chem 278:12151–12156 [DOI] [PubMed] [Google Scholar]
- Funk CD, Furci L, FitzGerald GA, Grygorczyk R, Rochette C, Bayne MA, Abramovitz M, Adam M, Metters KM. (1993) Cloning and expression of a cDNA for the human prostaglandin E receptor EP1 subtypes. J Biol Chem 268:26767–26772 [PubMed] [Google Scholar]
- Han C, Michalopoulos GK, Wu T. (2006) Prostaglandin E2 receptor EP1 transactivates EGFR/MET receptor tyrosine kinases and enhances invasiveness in human hepatocellular carcinoma cells. J Cell Physiol 207:261–270 [DOI] [PubMed] [Google Scholar]
- Hata AN, Breyer RM. (2004) Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther 103:147–166 [DOI] [PubMed] [Google Scholar]
- Hoshino T, Nakaya T, Homan T, Tanaka K, Sugimoto Y, Araki W, Narita M, Narumiya S, Suzuki T, Mizushima T. (2007) Involvement of prostaglandin E2 in production of amyloid-beta peptides both in vitro and in vivo. J Biol Chem 282:32676–32688 [DOI] [PubMed] [Google Scholar]
- Huang GW, Yang LY, Lu WQ. (2005) Expression of hypoxia-inducible factor 1alpha and vascular endothelial growth factor in hepatocellular carcinoma: impact on neovascularization and survival. World J Gastroenterol 11:1705–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SP, Wu MS, Shun CT, Wang HP, Hsieh CY, Kuo ML, Lin JT. (2005) Cyclooxygenase-2 increases hypoxia-inducible factor-1 and vascular endothelial growth factor to promote angiogenesis in gastric carcinoma. J Biomed Sci 12:229–241 [DOI] [PubMed] [Google Scholar]
- Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L. (2003) IL-1β-mediated up-regulation of HIF-1α via an NFκB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J 17:2115–2117 [DOI] [PubMed] [Google Scholar]
- Katoh H, Watabe A, Sugimoto Y, Ichikawa A, Negishi M. (1995) Characterization of the signal transduction of prostaglandin E receptor EP1 subtype in cDNA-transfected Chinese hamster ovary cells. Biochim Biophys Acta 1244:41–48 [DOI] [PubMed] [Google Scholar]
- Katsuta M, Miyashita M, Makino H, Nomura T, Shinji S, Yamashita K, Tajiri T, Kudo M, Ishiwata T, Naito Z. (2005) Correlation of hypoxia inducible factor-1alpha with lymphatic metastasis via vascular endothelial growth factor-C in human esophageal cancer. Exp Mol Pathol 78:123–130 [DOI] [PubMed] [Google Scholar]
- Lawrence RA, Jones RL. (1992) Investigation of the prostaglandin E (EP-) receptor subtype mediating relaxation of the rabbit jugular vein. Br J Pharmacol 105:817–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng J, Han C, Demetris AJ, Michalopoulos GK, Wu T. (2003) Cyclooxygenase-2 promotes hepatocellular carcinoma cell growth through Akt activation: evidence for Akt inhibition in celecoxib-induced apoptosis. Hepatology 38:756–768 [DOI] [PubMed] [Google Scholar]
- Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. (2005) Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105:659–669 [DOI] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F. (2008) Cancer-related inflammation. Nature 454:436–444 [DOI] [PubMed] [Google Scholar]
- Nicola C, Timoshenko AV, Dixon SJ, Lala PK, Chakraborty C, Lala PK, Chakraborty C. (2005) EP1 receptor-mediated migration of the first trimester human extravillous trophoblast: the role of intracellular calcium and calpain. J Clin Endocrinol Metab 90:4736–4746 [DOI] [PubMed] [Google Scholar]
- Regan JW. (2003) EP2 and EP4 prostanoid receptor signaling. Life Sci 74:143–153 [DOI] [PubMed] [Google Scholar]
- Regan JW, Bailey TJ, Donello JE, Pierce KL, Pepperl DJ, Zhang D, Kedzie KM, Fairbairn CE, Bogardus AM, Woodward DF. (1994) Molecular cloning and expression of human EP3 receptors: evidence of three variants with differing carboxyl termini. Br J Pharmacol 112:377–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvinsky I, Meyuhas O. (2006) Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci 31:342–348 [DOI] [PubMed] [Google Scholar]
- Schofield CJ, Ratcliffe PJ. (2004) Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 5:343–354 [DOI] [PubMed] [Google Scholar]
- Schoppmann SF, Fenzl A, Schindl M, Bachleitner-Hofmann T, Nagy K, Gnant M, Horvat R, Jakesz R, Birner P. (2006) Hypoxia inducible factor-1alpha correlates with VEGF-C expression and lymphangiogenesis in breast cancer. Breast Cancer Res Treat 99:135–141 [DOI] [PubMed] [Google Scholar]
- Semenza GL. (2001) Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med 7:345–350 [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Cantley LC. (2006) Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441:424–430 [DOI] [PubMed] [Google Scholar]
- Smith WL, DeWitt DL, Garavito RM. (2000) Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem 69:145–182 [DOI] [PubMed] [Google Scholar]
- Shoji Y, Takahashi M, Kitamura T, Watanabe K, Kawamori T, Maruyama T, Sugimoto Y, Negishi M, Narumiya S, Sugimura T, et al. (2004) Downregulation of prostaglandin E receptor subtype EP3 during colon cancer development. Gut 53:1151–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su JL, Shih JY, Yen ML, Jeng YM, Chang CC, Hsieh CY, Wei LH, Yang PC, Kuo ML. (2004) Cyclooxygenase-2 induces EP1- and HER-2/Neu-dependent vascular endothelial growth factor-C up-regulation: a novel mechanism of lymphangiogenesis in lung adenocarcinoma. Cancer Res 64:554–564 [DOI] [PubMed] [Google Scholar]
- Sugimoto Y, Narumiya S. (2007) Prostaglandin E receptors. J Biol Chem 282:11613–11617 [DOI] [PubMed] [Google Scholar]
- Tabata H, Tanaka S, Sugimoto Y, Kanki H, Kaneko S, Ichikawa A. (2002) Possible coupling of prostaglandin E receptor EP(1) to TRP5 expressed in Xenopus laevis oocytes. Biochem Biophys Res Commun 298:398–402 [DOI] [PubMed] [Google Scholar]
- Thornton RD, Lane P, Borghaei RC, Pease EA, Caro J, Mochan E. (2000) Interleukin 1 induces hypoxia-inducible factor 1 in human gingival and synovial fibroblasts. Biochem J 350:307–312 [PMC free article] [PubMed] [Google Scholar]
- Voss B, McLaughlin JN, Holinstat M, Zent R, Hamm HE. (2007) PAR1, but not PAR4, activates human platelets through a Gi/o/phosphoinositide-3 kinase signaling axis. Mol Pharmacol 71:1399–1406 [DOI] [PubMed] [Google Scholar]
- Watabe A, Sugimoto Y, Honda A, Irie A, Namba T, Negishi M, Ito S, Narumiya S, Ichikawa A. (1993) Cloning and expression of cDNA for a mouse EP1 subtype of prostaglandin E receptor. J Biol Chem 268:20175–20178 [PubMed] [Google Scholar]
- Welsh SJ, Bellamy WT, Briehl MM, Powis G. (2002) The redox protein thioredoxin-1 (Trx-1) increases hypoxia inducible factor 1α protein expression: Trx-1 overexpression results in increased vascular endothelial growth factor production and enhanced tumor angiogenesis. Cancer Res 62:5089–5095 [PubMed] [Google Scholar]
- Zhou J, Brüne B. (2006) Cytokines and hormones in the regulation of hypoxia inducible factor-1alpha (HIF-1alpha). Cardiovasc Hematol Agents Med Chem 4:189–197 [DOI] [PubMed] [Google Scholar]
- Zhou P, Qian L, Chou T, Iadecola C. (2008) Neuroprotection by PGE2 receptor EP1 inhibition involves the PTEN/AKT pathway. Neurobiol Dis 29:543–551 [DOI] [PMC free article] [PubMed] [Google Scholar]