

Abstract
The recent discovery of novel high-affinity and selective dopamine D3 receptor (DA D3R) antagonists and partial agonists has provided tools with which to further elucidate the role DA D3R plays in substance abuse. The present study was conducted to evaluate the transport, metabolism, pharmacokinetics, and brain uptake of the DA D3R-selective fluorenyl amides, NGB 2904 [N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)-9H-fluorene-2-carboxamide] fumarate) and JJC 4-077 [N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-3-hydroxybutyl)-9H-fluorene-2-carboxamide hydrochloride], and the 2-pyridylphenyl amides, CJB 090 [N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)-4-(pyridine-2-yl)benzamide hydrochloride] and PG 01037 [N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-trans-but-2-enyl)-4-(pyridine-2-yl)benzamide hydrochloride], all of which have been studied in animal models of psychostimulant abuse. Additional screening with a panel of human and rat Supersomes was performed for NGB 2904 and PG 01037. Drug-stimulated ATPase activation assays and bidirectional transport and efflux assays were used to test for substrate specificity of NGB 2904 and PG 01037 for human and rat efflux transporters. All compounds exhibited moderate elimination half-lives, ranging from 1.49 to 3.27 h, and large volumes of distribution (5.95–14.19 l/kg). The brain-to-plasma ratios ranged from 2.93 to 11.81 and were higher than those previously reported for cocaine. Brain exposure levels of NGB 2904 and PG 01037 were significantly reduced after intraperitoneal administration compared with intravenous administration. The metabolism of these compounds was mediated primarily by CYP3A subfamilies. PG 01037 was a P-glycoprotein-transported substrate. Higher doses of these compounds are often required for in vivo action, suggesting decreased bioavailability via extravascular administration that may be attributed to high drug efflux and hepatic metabolism. These studies provide important preclinical information for optimization of next-generation D3R selective agents for the treatment of drug addiction.
Cocaine and methamphetamine are potent central nervous system stimulants known for their abuse potential and addictive liability. Cocaine's effects are shorter than that of methamphetamine (half-life 0.5 h for cocaine versus 11 h for methamphetamine), making its high abuse liability and chronic use a significant health concern. Despite numerous controlled studies of more than 60 medications, there are currently no conclusive data to support the efficacy of any particular pharmacological agent to treat cocaine abuse (Ross and Peselow, 2009).
For decades, it has been known that the reinforcing effects produced by cocaine and other psychostimulants are associated with activation of dopamine (DA) receptors but elucidating the role of the individual DA receptor subtypes D1–D5 has been hampered by the lack of subtype-selective ligands. The discrete location of the dopamine D3 receptors (DA D3Rs) in rodent and human brain, coupled with evidence that their expression is modified after long-term exposure to drugs of abuse, suggest a role for this dopamine receptor subtype in drug addiction (for reviews see Heidbreder et al., 2005; Newman et al., 2005). According to recent reports from imaging studies in humans and nonhuman primates, DA D3Rs may play an important role in the abuse-related effects of cocaine (Volkow et al., 2004; Schwarz et al., 2007). Support for pursuing the DA D3Rs as novel targets for drug abuse pharmacotherapies comes from the hypothesis that selective blockade of DA D3Rs may antagonize drug reward and/or reinforcement while obviating the possibility of extrapyramidal side effects typically associated with the blockade of DA D2Rs (Sokoloff et al., 1990). Studies have also linked the D3 receptors to l-DOPA-induced dyskinesia in Parkinson's disease (Kumar et al., 2009; Visanji et al., 2009) and behavioral aberrations associated with schizophrenia (Richtand, 2006), suggesting additional utility of D3 receptor ligands.
Significant research efforts have been directed toward the development of selective DA D3R agents (Micheli and Heidbreder, 2008), yielding novel antagonists and partial agonists with high affinity and selectivity for D3R. Important structure–activity relationships have been derived from the 4-phenylpiperazine class of DA D3R ligands, based on prototypic BP897 [N-[4-[4-(2-methoxyphenyl)piperazin-1-yl]butyl]naphthalene-2-carboxamide] (Pilla et al., 1999) and NGB 2904 [N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)-9H-fluorene-2-carboxamide fumarate] (Yuan et al., 1998), and have been reviewed (Boeckler and Gmeiner, 2006). Both of these compounds, and an NGB 2904 analog, CJB 090 [N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)-4-(pyridine-2-yl)benzamide hydrochloride], have demonstrated efficacy in animal models of drug abuse (Gilbert et al., 2005; Xi et al., 2006; Martelle et al., 2007; Spiller et al., 2008). For example, NGB 2904 attenuated cue-induced reinstatement of cocaine self-administration (Gilbert et al., 2005) and inhibited the enhancement of cocaine-enhanced (Xi et al., 2006) or methamphetamine-enhanced brain stimulation reward in rats (Spiller et al., 2008). Furthermore, CJB 090 (Newman et al., 2003) attenuated the discriminative stimulus effects of cocaine and decreased cocaine-maintained responding in rhesus monkeys without cocaine-like effects (Martelle et al., 2007). Another compound, PG 01037 [N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-trans-but-2-enyl)-4-(pyridine-2-yl)benzamide hydrochloride], has been shown to effectively reduce methamphetamine's rewarding efficacy for drug-seeking behaviors in rats (Higley et al., 2010). NGB 2904, PG 01037, and CJB 090 were effective in all of these animal models at doses that did not induce adverse motor side effects associated with nonselective D2-like antagonists (Achat-Mendes et al., 2009; Higley et al., 2010).
Nevertheless, high doses of these ligands were required in some cases to elicit in vivo action, despite pharmacological magnetic resonance imaging studies showing that significantly lower doses localize in DA D3R-rich regions of the brain (Grundt et al., 2007). It is not known whether the need for higher doses is caused by low blood-brain barrier (BBB) permeability, high peripheral metabolism, or large distribution of these agents into other tissues. Thus, in vivo pharmacokinetic and brain uptake studies were conducted in rats on NGB 2904 and three analogs [JJC 4-077 (N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-3-hydroxybutyl)-9H-fluorene-2-carboxamide hydrochloride), CJB 090, and PG 01037]. In addition, in vitro transport and metabolism studies were undertaken by using the DA D3R antagonists NGB 2904 and PG 01037 as representatives of the fluorenyl amide and 2-pyridylphenyl amide groups, respectively, in a screen of a series of human and rat cytochrome P450 enzymes to understand mechanisms for elimination of these agents. Interactions with the efflux transporters were also assessed to determine the potential impact of drug efflux on their permeability at the BBB.
D3R-selective molecules have typically suffered from suboptimal physical properties such as poor water solubility, undesirable pharmacokinetics, predicted metabolism, or side effects unrelated to D3R binding that have prevented their in vivo investigation or marred interpretation of results. The goal of the current study was to identify the structural motifs responsible for undesirable bioavailability properties, thus guiding future drug design.
Materials and Methods
Materials
Four compounds were analyzed in this study: NGB 2904, JJC 4-077, CJB 090, and PG 01037. PG 01030 [N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-trans-but-2-enyl)-benzothiopen-2-carboxamide hydrochloride] was used as an internal standard. Figure 1 depicts the structures of the five compounds. 2-Hydropropyl-β-cyclodextrin (HPBCD) and polyethyleneglycol 400 (PEG 400) were purchased from Sigma-Aldrich (St. Louis, MO). Saline (0.9% sodium chloride) was obtained from Baxter (Deerfield, IL). Acetonitrile was purchased from American Bioanalytical (Natick, MA). All chemicals and solvents were of high-performance liquid chromatography (HPLC) grade. Water was obtained from a Barnstead water purification system (Thermo Fisher Scientific, Waltham, MA). Madin-Darby canine kidney (MDCK)-MDR1 cells were a gift from Dr. Peter Swaan (University of Maryland, Baltimore, MD). Cell culture supplies [Dulbecco's modified Eagle's medium, phosphate-buffered saline with Ca2+ and Mg2+ (PBS), 100× l-glutamine, nonessential amino acids, fetal bovine serum, 0.25% trypsin-1 mM EDTA, and penicillin G-streptomycin sulfate antibiotic mixture] were purchased from Invitrogen (Carlsbad, CA). [14C]Mannitol (46 mCi/mmol), sodium phosphate dibasic, and verapamil HCl were purchased from Sigma-Aldrich. [3H]saquinavir (3500 mCi/mmol) was purchased from Moravek Biochemicals (Brea, CA). Transwell clusters were purchased from Corning Life Sciences (Acton, MA). Human and rat P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), and multidrug resistance protein 2 (MRP2) Spodotera frugiperda (Sf9) membranes and microsomes from baculovirus-infected insect cells expressing human CYP1A2, 2A6, 2B6, 2C8, 2C9*1(Arg144), 2C19, 2D6*1, 2E1, and 3A4 and rat CYP1A2, 2B1, 2C11, 2D1, 2E1, and 3A1 (Supersomes), and insect cell control and rat P450 reductase insect cell control Supersomes were purchased from BD Gentest (Woburn, MA). The P450s were coexpressed with their corresponding human or rat cytochrome P450 reductase. In addition, human CYP2A6, 2B6, 2C8, 2C9*1, 2C19, 2E1, and 3A4 and rat CYP2B1, 2C11, 2D1, 2E1, and 3A1 were coexpressed with human cytochrome b5. Pooled human liver microsomes, pooled male Sprague-Dawley rat liver microsomes, and NADPH-regenerating systems were also purchased from BD Gentest.
Fig. 1.

Structures of the fluorenyl amides NGB 2904 (A) and JJC 4-077 (B) and the 2-pyridylphenyl amides CJB 090 (C), PG 01037 (D), and PG 01030 (E).
Animal Pharmacokinetic Studies
Animals.
Male Sprague-Dawley rats (250–275 g) were purchased from Harlan (Indianapolis, IN). The rats were housed in the animal facility of the University of Maryland School of Pharmacy and maintained on a 12-h light/dark cycle at a temperature of 72 ± 2°F. Food (Purina 5001 Rodent Chow; Purina, St. Louis, MO) and water were available ad libitum. The animal study protocol was approved by the Institutional Animal Care and Use Committee of the School of Pharmacy, University of Maryland. Animal facilities are accredited by the American Association of the Accreditation of Laboratory Animal Care, and all experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Pharmacokinetics and Brain Distribution Studies of Fluorenyl Amides and 2-Pyridylphenylamides.
The compounds were dissolved in HPBCD solution [10% HPBCD/ ethanol (9:1)] for NGB 2904 and JJC 4-077 or PEG 400 solution [30% PEG 400/ethanol (9:1)] for CJB 090 and PG 01037, and a dose of 10 mg/kg was administered intraveneously via tail vein or intraperitoneally (NGB 2904 and PG 01037) to the rats at a volume of 2 ml/kg. Those doses were based on a compilation of doses and routes of administration used by numerous investigators studying these compounds in vivo. Cohorts of three animals were sacrificed by carbon dioxide asphyxiation at predose and 5, 10, 30, 60, 120, 240, and 360 min postdose. Blood was collected by heart puncture using heparinized syringes and centrifuged for 10 min at 10,000 rpm (Denville Scientific, Metuchen, NJ), and the plasma was stored at −80°C until analysis. Brain tissue was immediately excised, blotted dry, weighed, and stored at −80°C until analysis.
Quantification of Fluorenyl Amides and 2-Pyridylphenyl Amides.
A validated HPLC method with a UV detector was used to quantify the compounds in plasma and brain samples. Preparation of plasma and brain samples involved liquid–liquid extraction methods with organic solvents, ethyl acetate/hexane (3:1, v/v) for NGB 2904 and JJC 4-077 and ethyl acetate/hexane (1:3, v/v) for CJB 090 and PG 01037. Brain samples were homogenized in phosphate-buffered saline using a PowerGen125 from Fisher Scientific (Pittsburgh, PA). A 100-μl aliquot of sample was spiked with internal standard (10 μg/ml of PG 01037 in methanol for NGB 2904 and JJC 4-077; 10 μg/ml of PG 01030 in methanol for CJB 090 and NGB 2904), vortexed slightly, and extracted with 0.9 ml of organic solvents described above. The upper organic layer of 0.8 ml was aliquoted into clean tubes and evaporated to dryness under nitrogen stream at 37°C. The residue was reconstituted with 100 μl of mobile phase, and a 50-μl aliquot was injected into the HPLC system. The HPLC system consisted of a 1525 binary pump and a 717 autosampler from Waters (Milford, MA), and the separation was performed on a Symmetry C-18 analytical column (4.6 × 150 mm, 5 μm) from Waters attached to a C-18 guard column (4.6 × 30 mm, 5 μm) from Phenomenex (Torrance, CA). The mobile phase was an isocratic condition of acetonitrile and 10 mM ammonium acetate (pH 4.7) [52:48 (v/v) for CJB 090, 46:54 (v/v) for PG 01037, and 68:32 (v/v) for JJC 4-077 and NGB 2904] at a flow rate of 1.0 ml/min. UV detection wavelengths were 283 nm for CJB 090 and PG 01037, 305 nm for JJC 4-077, and 310 nm for NGB 2904. The lower limit of quantification for all compounds was 0.05 μg/ml, with a linearity range of 0.05 to 10 μg/ml. Accuracy and precision of all compounds were determined by replicate injection of quality-control samples. Both precision and accuracy of all compounds were of satisfactory results below 15.5% of CV.
Characterization of Human and Rat Cytochrome P450 Enzymes Involved in the Metabolism of NGB 2904 and PG 01037
NGB 2904 and PG 01037 were screened for the enzymes involved in their metabolism. Compounds were incubated with human CYP1A2, 2A6, 2B6, 2C8, 2C9*1(Arg144), 2C19, 2D6*1, 2E1, and 3A4 and rat CYP1A2, 2B1, 2C11, 2D1, 2E1, and 3A1 Supersomes for 60 min. For each enzyme tested, the reaction mixture consisted of 50 pmol/ml P450, NADPH-regenerating system (1.3 mM NADP+, 3.3 mM glucose 6-phosphate, 0.4 U/ml glucose-6-phosphate dehydrogenase, and 3.3 mM magnesium chloride), and 10 μM NGB 2904 or PG 01037 in 100 mM potassium phosphate buffer, pH 7.4 (final volume 500 μl). The reactions were initiated by adding ice-cold Supersomes to the prewarmed mixture of buffer, substrate, and cofactors. After a 60-min incubation period at 37°C, the reactions were stopped by the addition of 250 μl of acetonitrile and centrifuged at 10,000g for 5 min. Two hundred microliters of supernatant was injected onto the HPLC for determination of unchanged compound concentrations. Similar incubations with insect cell control and rat P450 reductase insect cell control Supersomes were performed to control for the native activities and non-P450-specific effects. Metabolism incubations were performed in triplicate.
Determination of the Time Course of NGB 2904 and PG 01037 Metabolism
The time course of metabolism of NGB 2904 and PG 01037 (5 μM final concentration; n = 3) by pooled human liver microsomes and pooled male rat liver microsomes was determined. The microsomes were used at a concentration of 0.8 mg/ml. The cofactor and buffer concentrations were similar to that described above with a final reaction volume of 1500 μl. The reactions were initiated by adding the drug to the prewarmed reaction mixture. After 0, 5, 10, 20, 30, 40, and 60 min of incubation at 37°C, 200 μl of the reaction mixture was sampled, immediately vortexed with 100 μl of acetonitrile to terminate the reaction, and centrifuged at 10,000g for 5 min. Aliquots of the supernatant were then collected for HPLC analysis.
DA D3R Compound-Stimulated ATPase Activity
Drug-stimulated transporter activity was estimated for the D3R antagonists NGB 2904 and PG 01037 by measuring inorganic phosphate released from ATP according to the manufacturer's protocol (BD Gentest). DA D3R compounds were tested at concentrations of 5 to 100 μM. Based on previously published reports, this concentration range provides adequate ATPase activation for a majority of compounds (Litman et al., 1997; Polli et al., 2001). Membranes (20 or 25 μg/well) were prepared in Tris-4-morpholineethanesulfonic acid buffer, pH 6.8 [50 mM Tris-4-morpholineethanesulfonic acid (pH 6.8), 50 mM KCl, 5 mM sodium azide, 2 mM EGTA, and 2 mM dl-dithiothreitol] and incubated at 37°C for 5 min with test compounds or positive controls (20 μM Verapamil; 50 μM 2-amino-1-methyl-6-phenylimidazo(4.5-b)pyridine; 1 mM Probenecid) in the presence or absence of 200 or 400 μM sodium orthovanadate in triplicate wells on a 96-well plate. The reaction was initiated by the addition of 12 or 15 mM Mg-ATP (20 μl). Incubation time depended on both the species and transporter being analyzed and was carried out according to the manufacturer's recommendations (BD Gentest). The reaction was then terminated by the addition of 10% SDS containing antifoam A. The inorganic phosphate released was detected by incubation at 37°C for 20 min with 200 μl of detection reagent [1:4 (v/v) mixture of 35 mM ammonium molybdate in 15 mM zinc acetate (pH 5.0) and 10% ascorbic acid]. Phosphate standards were prepared in each plate. The absorbance was measured at 800 nm by using a Spectramax Gemini UV-visible spectrophotometer (Molecular Devices, Sunnyvale, CA). The drug-stimulated ATPase activity (pmol/min/mg protein) was determined as the difference between the amounts of inorganic phosphate released from ATP in the absence and presence of vanadate. The drug-stimulated ATPase activity was reported as fold-stimulation relative to the basal ATPase activity in the absence of drug [dimethyl sulfoxide (DMSO) control].
Cell Culture
MDCK-MDR1 cells were grown at 37°C, 95% relative humidity, and 5% CO2 atmosphere on 12-well Costar inserts (Transwell; 0.4-μm pore polyester membrane, 1 cm2 in diameter). MDCK-MDR1 cells were seeded at a density of 425,000 cells/cm2 and cultured for 4 days in 1× Dulbecco's modified Eagle's medium, containing 10% fetal bovine serum, 2% glutamine, 1% nonessential amino acids, and 1% penicillin-streptomycin, with the medium changed daily. The formation of restrictive monolayers was monitored by microscopic examination and measurement of transepithelial electrical resistance by using a Millicell-ERS meter (Millipore Corporation, Billerica, MA).
Characterization of MDCK-MDR1 Cell Monolayers
Expression of P-gp in MDCK-MDR1 Cells.
P-gp expression was monitored by Western blot analysis. MDCK-MDR1 total cell lysates were prepared by using radioimmunoprecipitation assay buffer (Sigma-Aldrich) according to the manufacturer's protocol. Protein concentration was determined with a bicinchoninic acid protein assay kit (Thermo Fisher Scientific). Cells lysates were resolved on 10% SDS-polyacrylamide precast gels and transferred to a polyvinylidene fluoride membrane (Bio-Rad Laboratories, Hercules, CA). The membrane was blocked in 5% milk and then incubated with C219 anti-P-gp antibody (EMD Chemicals, San Diego, CA) diluted 50-fold in buffer overnight at 4°C. The blot was washed, incubated with horseradish peroxidase-conjugated secondary antibody (GE Healthcare, Little Chalfont, Buckinghamshire, UK), and then washed again. Bands were visualized by using an ECL Plus detection system (GE Healthcare). Equivalence of protein loading was confirmed by secondary immunoblotting with an anti-β-actin antibody (Sigma-Aldrich).
Evaluation of Monolayer Integrity and Function of P-gp in MDCK-MDR1 Cells.
Before conducting transport studies, monolayer integrity was determined by measuring the permeability of [14C]mannitol. P-gp expression was functionally tested by conducting bidirectional transport studies with the P-gp substrate [3H]saquinavir and determining its efflux ratio across MDCK-MDR1 monolayers.
PG 01037 Bidirectional Transport and Inhibition Studies
These studies were conducted to confirm the results of the ATPase assay and verify that PG 01037 is a P-gp-transported substrate. Transport experiments were performed in both the apical-to-basolateral (A−B) and the basolateral-to-apical (B-A) directions across MDCK-MDR1 monolayers in the presence and absence of the P-gp inhibitor verapamil. At the time of the experiment, the culture medium was removed from both the apical and basolateral chambers and the monolayers were washed twice with PBS. The monolayers (n = 3/group) were incubated with either 200 μM verapamil in PBS or PBS alone for 30 min. After the preincubation period, mixtures of 0.1 mM PG 01037 with either 200 μM verapamil in PBS or PBS alone were added to the donor compartments. The receiver compartments solution consisted of either 200 μM verapamil in PBS (transport in presence of verapamil) or PBS (transport in absence of verapamil). For the A−B study, the inserts were moved to new Transwells containing 1.5 ml of the corresponding receiver compartment solution at 30, 60, 90, and 120 min. For the B-A study, samples were drawn from the apical chamber at the same time points and replaced with equivalent volumes of fresh receiver compartment solution. Transport experiments were performed at 37°C with continuous agitation on a plate shaker (50 cycles/min). Samples were stored at −80°C until the time of analysis.
Data Analysis
Pharmacokinetic Data Analysis.
The destructive sampling data obtained from the pharmacokinetic studies were analyzed by the naive averaging method. For a given compound, the plasma concentrations from three animals at each time point were averaged. Compartmental modeling was used to estimate various pharmacokinetic parameters by using WinNonlin software (version 4.1; Pharsight, Mountain View, CA). Several compartmental models were evaluated to determine the best fit model. A variety of weighting schemes were also analyzed including equal weight, 1/y, 1/ŷ, 1/y2, and 1/ŷ2, where y is the observed drug concentration, and ŷ is the model-predicted drug concentration. Goodness of fit was based on visual inspection, weighted residual sum of squares, random distribution of residuals, precision of parameter estimates, Akaike's information criteria, and Schwarz criteria. Brain uptake of compounds was represented as a brain-to-plasma (B/P) concentration ratio in accordance with the equation of B/P = Cbrain/Cplasma, where Cbrain and Cplasma are the concentration in brain and plasma at a specific time point, respectively. Bailer's method was used to calculate the variance associated with both the plasma area under the curve (AUC) and brain AUC (Bailer, 1988). The S.E. associated with the secondary pharmacokinetic parameter estimates is a measure of the accuracy of the model predictions. Statistical comparisons among plasma and brain AUC values were determined by using Student's t test at p < 0.05. The cLogP values of the compounds were predicted by using the ACD/ChemSketch software program (version 11.0; Advanced Chemistry Development, Inc., Toronto, Canada).
Metabolism Data Analysis.
The human and rat P450 isoforms involved in the metabolism of NGB 2904 and PG 01037 were identified by analyzing the differences in mean substrate concentrations remaining after 60-min incubations. Statistical significance was determined by one-way analysis of variance followed by Dunnett's multiple comparisons of P450 incubation versus control incubations. The percentage of the mean control concentration remaining after 60-min incubation was calculated according to the following equation:
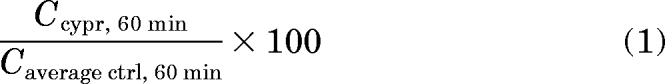 |
where Ccypr, 60 min is the substrate concentration from the rth replicate after 60 min of incubation with a particular P450 Supersome, and Caverage ctrl, 60 min is the average (n = 3) substrate concentration after 60-min incubation with insect cell control Supersomes (for human P450s). The percentages are represented as mean and standard deviation from triplicate reactions.
The intrinsic clearance values were calculated based on the substrate disappearance rate as described previously (Naritomi et al., 2003). The concentration of NGB 2904 and PG 01037 (4 μM) was significantly lower than the Km value estimated in the pilot study (data not shown). Assuming first-order disappearance of substrate, the disappearance rate constant (Ke) was calculated from the slope of log Ct versus time profile based on the following equation:
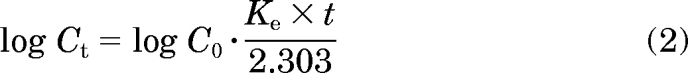 |
where Ct is the concentration of the substrate at the different time points, and C0 is the substrate concentration at time 0. The initial metabolic rate (V0) (picomoles per minute per picomole of P450 or milligrams of microsomal protein) was calculated from the following equation:
 |
where PMS is P450 concentration (picomoles per milliliter) or microsomal protein concentration (milligrams per milliliter). V0 can also be described by using Michaelis-Menten equation as follows:
 |
Assuming that C0 ≪ Km, eq. 4 can be written as follows:
 |
Accordingly, the intrinsic clearance was calculated based on the formula:
 |
The intrinsic clearance values were calculated separately from each of the replicates and compared statistically by using one-way analysis of variance followed by Newman-Keuls post test. CLint values are presented as mean ± S.D. from the three replicates performed for each reaction.
ATPase Activation Assay Data Analysis.
Only those concentrations of compounds that demonstrate a >2 fold stimulation were evaluated for being significantly different from the no compound control, as they are representative of a transporter substrate (Polli et al., 2001). Statistical significance was determined by Student's t test (p < 0.05).
Transport Data Analysis.
The apparent permeability coefficients were determined at sink conditions by using the following equation:
 |
where dQ/dt is equal to the linear appearance rate of compound in the receiver compartment, A is the cross-sectional area of the insert filters, and C0 is the donor concentration at time 0. All values are represented as mean ± standard deviation from three Transwell inserts. Efflux ratios across the monolayers were calculated by using the equation:
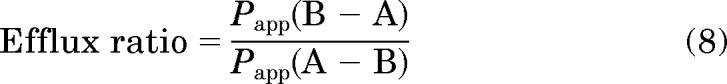 |
where Papp (B−A) is the permeability from the B−A direction, and Papp (A−B) is the permeability from the A−B direction. The statistical significance of the effect of verapamil on the permeability of PG 01037 was determined with two-tailed Student's t test at p = 0.05.
Results
Physicochemical Properties of Fluorenyl Amides and 2-Pyridylphenyl Amides.
The predicted cLogP, molecular mass, number of hydrogen bond donors, number of hydrogen bond acceptors, and number of freely rotating bonds of NGB 2904, JJC 4-077, CJB 090, and PG 01037 were calculated by using ACD/ChemSketch (Table 1). The cLogP of PG 01037 (5.32) was the lowest among the four compounds.
TABLE 1.
Predicted physicochemical properties and in vitro function data on dopamine D3 receptor of NGB 2904, JJC 4-077, CJB 090, and PG 01037
| Parameter | NGB 2904 | JJC 4-077 | CJB 090 | PG 01037 |
|---|---|---|---|---|
| IC50 (nM)a | 14.4 ± 0.5 | 42.0 ± 1.3 (26)b | 6.3 ± 1.7 (30)b | 3.0 ± 0.7 |
| D2 Kin (nM)a | 112 ± 22 | 168 ± 29 | 24.8 ± 8.6 | 93 ± 12 |
| D3 Kin (nM)a | 2.0 ± 0.4 | 1.5 ± 0.1 | 0.4 ± 0.02 | 0.7 ± 0.1 |
| Molecular mass (Da) | 619.32 | 583.38 | 556.26 | 572.35 |
| cLogP | 7.00 | 6.55 | 5.36 | 5.32 |
From Grundt et al., (2007).
Partial agonist activity: EC50 (% stimulation).
Pharmacokinetics of Fluorenyl Amides after Intravenous Administration in Rats.
The observed and the predicted plasma concentrations of NGB 2904 and JJC 4-077 after single intravenous dose administration of 10 mg/kg in male Sprague-Dawley rats was derived based on the best fit achieved with WinNonlin (Fig. 2A). The pharmacokinetics of both compounds were best described with a two-compartment model. Secondary pharmacokinetic parameters of NGB 2904 and JJC 4-077 are shown in Table 2. Plasma concentrations of both compounds showed relatively rapid decline in the first hour after dosing and then moderate decline thereafter. Plasma exposure (AUCplasma) of JJC 4-077 (2.13 ± 0.09 h·μg/ml) was significantly less than that of NGB 2904 (3.45 ± 0.21 h·μg/ml) (p < 0.05). The elimination half-life, systemic clearance, and volume of distribution of JJC 4-077 (3.27 ± 0.73 h, 3.44 ± 0.49 l/h/kg, and 14.19 ± 1.59 l/kg, respectively) were higher than those of NGB 2904 (2.60 ± 0.52 h, 2.46 ± 0.19 l/h/kg, and 7.85 ± 1.14 l/kg, respectively).
Fig. 2.

Plasma and brain pharmacokinetic profiles of D3R analogs in male Sprague-Dawley rats (n = 3/time point). A, observed (mean ± S.D.) and predicted plasma concentration versus time profiles of the fluorenyl amides based on the naive averaging analysis conducted with WinNonlin. B, observed brain concentrations (mean ± S.D.) versus time profiles of the fluorenyl amides. C, observed (mean ± S.D.) and predicted plasma concentration versus time profiles of the 2-pyridylphenyl amides. D, observed brain concentrations (mean ± S.D.) versus time profiles of the 2-pyridylphenyl amides.
TABLE 2.
Pharmacokinetic parameters of NBG 2904, JJC 4-077, CJB 090 and PG 01037 after intravenous and intraperitoneal administration of a 10.0 mg/kg dose
The S.E. of the parameter estimate is a measure of the accuracy of the predictions. Brain t1/2 was calculated by using noncompartmental analysis.
| Parameter | JJC 4-077 (Intravenous) | NGB 2904 (Intravenous) | NGB 2904 (Intraperitoneal) | CJB 090 (Intravenous) | PG 01037 (Intravenous) | PG 01037 (Intraperitoneal) |
|---|---|---|---|---|---|---|
| Plasma t1/2 (h) | 3.27 ± 0.73 | 2.60 ± 0.52 | 2.58 ± 1.15 | 1.49 ± 0.13 | 2.37 ± 0.15 | 1.83 ± 0.20 |
| AUCplasma0→last (h·μg/ml) | 2.13 ± 0.09 | 3.45 ± 0.21 | 0.66 ± 0.07 | 3.26 ± 0.25 | 2.80 ± 0.25 | 2.36 ± 0.14 |
| AUCbrain0→last (h·μg/ml) | 18.62 ± 0.23 | 40.78 ± 1.52 | 4.97 ± 1.40 | 23.47 ± 2.38 | 10.97 ± 1.21 | 6.91 ± 0.44 |
| CL (l/h/kg) | 3.44 ± 0.49 | 2.46 ± 0.19 | 13.84 ± 3.51 | 2.91 ± 0.24 | 2.99 ± 0.09 | 3.93 ± 0.33 |
| Vss (l/kg) | 14.19 ± 1.59 | 7.85 ± 1.14 | NE | 5.95 ± 0.70 | 9.83 ± 0.47 | NE |
| AUCbrain/AUCplasma | 8.73 | 11.81 | 7.55 | 7.19 | 3.92 | 2.93 |
| Brain t1/2 (h) | 2.50 | 2.48 | NE | 1.35 | 2.63 | NE |
NE, this parameter was not estimated with the pharmacokinetic model.
Brain Uptake of Fluorenyl Amides after Intravenous Administration in Rats.
Brain concentrations of NGB 2904 and JJC 4-077 were significantly higher than plasma concentrations at all sampling time points (p < 0.05) (Fig. 2B). Brain concentrations of NGB 2904 and JJC 4-077 remained elevated up to 2 h postdosing and then declined with a moderate elimination phase, which paralleled that of their plasma concentration. The brain exposure (AUCbrain) of NGB 2904 and JJC 4-077 was significantly different from their plasma exposure (AUCplasma) (p < 0.05). Moreover, brain exposure (AUCbrain) of NGB 2904 (40.78 ± 1.52 h·μg/ml) was significantly higher than that of JJC 4-077 (18.62 ± 0.23 h·μg/ml) (p < 0.05). The overall brain-to-plasma exposure ratios (AUCbrain/AUCplasma) were 11.81 and 8.73 for NGB 2904 and JJC 4-077, respectively. The brain uptake for NGB 2904 ranged from 3.43 to 19.36 and from 3.39 to 14.58 for JJC 4-077 (Fig. 3A).
Fig. 3.

B/P ratio versus time profiles of the fluorenyl amides NGB 2904 and JJC 4-077 (n = 3) (A) and 2-pyridylphenyl amides CJB 090 and PG 01037 (n = 3) (B).
Pharmacokinetics of 2-Pyridylphenyl Amides after Intravenous Administration in Rats.
The observed and predicted plasma concentration versus time profiles for CJB 090 and PG 01037 are indicated in Fig. 2A. The plasma concentrations of CJB 090 rapidly declined after dosing, exhibiting a monoexponential decay best described by a one-compartment open model. PG 01037 plasma concentrations followed biexponential decay and were best described by a two-compartment open model. The secondary pharmacokinetic parameters are listed in Table 2. The elimination half-life of CJB 090 was shorter than that of PG 01037. The plasma AUC of CJB 090 was significantly higher than plasma AUC of PG 01037 (p < 0.05). The systemic clearance of both CJB 090 and PG 01037 was similar. The volume of distribution at steady state of CJB 090 was less than that of PG 01037.
Brain Uptake of 2-Pyridylphenyl Amides after Intravenous Administration in Rats.
Brain concentrations of CJB 090 were significantly higher than plasma concentrations at all sampling times, whereas for PG 01037 significant differences (p < 0.05) were observed only at the later time points (120, 240, and 360 min). Whereas the brain exposure (AUCbrain) of CJB 090 (23.47 ± 2.38 h·μg/ml) was significantly higher than that of PG 01037 (10.97 ± 1.21 h·μg/ml) (p < 0.05), the brain exposures (AUCbrain) of CJB 090 and PG 01037 were significantly different from their plasma exposure (AUCplasma) (p < 0.05). Overall AUCbrain/AUCplasma ratios were 7.19 and 3.92 for CJB 090 and PG 01037, respectively. The brain uptake for CJB 090 ranged from 4.81 to 11.20 and from 3.36 to 5.44 for PG 01037 (Fig. 3B).
Pharmacokinetics and Brain Uptake of NGB 2904 and PG 01037 after Intraperitoneal Administration in Rats.
NGB 2904 and PG 01037 were selected for further evaluation of their pharmacokinetics and brain distribution by using the intraperitoneal route of administration because they both represent a DA D3R antagonist from each of the studied classes (i.e., fluorenyl amides and 2-pyridylphenyl amides) without any partial agonist activity (Table 1) and have strikingly different brain distribution profiles after intravenous administration (Fig. 2 and Table 2). The pharmacokinetics of NGB 2904 and PG 01037 after intraperitoneal administration were best described with a one-compartment model (Fig. 2A). The pharmacokinetic parameters for intraperitoneal administration are listed with the intravenous results in Table 2. AUCbrain and AUCplasma of NGB 2904 and PG 01037 were significantly reduced compared with the same dose administered intravenously (p < 0.05). Compared with results from intravenous doses, AUCbrain and AUCplasma ratios were reduced 36 and 25.4% for NGB 2904 and PG 01037, respectively. Comparing the individual intravenous and intraperitoneal B/P ratios at discrete sampling times, significant differences were observed only at 5 and 120 min for PG 01037 and 5, 30, and 120 min for NGB 2904 (Fig. 3). Despite route-dependent differences in brain uptake, brain levels were significantly higher than plasma levels after intraperitoneal administration in accordance with intravenous administration. The clearance values were also increased when the compounds were administered intraperitoneally (Table 2).
Characterization of Human and Rat Cytochrome P450 Enzymes Involved in the Metabolism of NGB 2904 and PG 01037.
The objective of these studies was to screen for the enzymes involved in the metabolism of NGB 2904 and PG 01037, which were selected as representative compounds for each class. Because of their structural similarities, no class-specific differences are expected in the enzymes involved in the metabolism of these compounds. Among the P450 isoforms tested, rat CYP3A1, CYP2B1, CYP2D1, CYP2C11, and CYP1A2 and human CYP2A6, CYP3A4, CYP2B6, and CYP2E1 were involved in NGB 2904 metabolism (Fig. 4, A and B). For PG 01037, rat CYP3A1, CYP2C11, and CYP1A2 and human CYP3A4 were significantly involved in its metabolism after 60-min incubation compared with the control (p < 0.05) (Fig. 4, C and D). In accordance with the aforementioned findings, rat CYP3A1 and CYP2C11 and human CYP3A4 appeared to be the most significant metabolizing enzymes for these compounds.
Fig. 4.

Metabolism of NGB 2904 (A and B) and PG 01037 (C and D) by the human recombinant P450s (A and C, respectively) and the rat recombinant P450s (B and D, respectively). Compounds (10 μM) were incubated with Supersomes (50 pmol/ml) and cofactors for 60 min followed by termination of reactions and determination of unchanged substrate. Data are presented as mean percentage of the average control ± S.D. from triplicate reactions. *, p < 0.05 based on Dunnett's test.
Intrinsic Clearance of NGB 2904 and PG 01037 in Pooled Human and Rat Liver Microsomes.
Under similar experimental conditions, NGB 2904 and PG 01037 followed first-order reaction kinetics with the concentration of the substrate declining monoexponentially with time. The calculated intrinsic clearance values of both compounds are reported in Table 3. There was no significant difference (p > 0.05) between the intrinsic clearance values of PG 01037 in rat and human liver microsomes. However, the intrinsic clearance of NGB 2904 in rat liver microsomes was nearly 2-fold higher than in human liver microsomes and 2-fold higher than the intrinsic clearance of PG 01037 in both human and rat liver microsomes (Table 3).
TABLE 3.
Intrinsic clearance values for 5 μM NGB 2904 and PG 01037 in human and male Sprague-Dawley rat pooled liver microsomes (0.8 mg/ml)
Intrinsic clearance is reported as microliters per minute per milligram of protein (n = 3/reaction).
| Microsomal Preparation | Intrinsic Clearance (5 μM) |
|
|---|---|---|
| NGB 2904 | PG 01037 | |
| Pooled human liver microsomes | 29.95 ± 3.83 | 25.62 ± 1.52 |
| Pooled male rat liver microsomes | 55.85 ± 3.05a,b | 26.20 ± 5.67 |
P < 0.05 based on Newman-Keuls test, intrinsic clearance of NGB 2904 in human liver microsomes compared with rat liver microsomes.
P < 0.05 based on Newman-Keuls test, intrinsic clearance of NGB 2904 in rat liver microsomes compared with intrinsic clearance of PG 01037 in human and rat liver microsomes.
Effect of DA D3R Analogs on Efflux Transporter ATPase Activity.
Increasing concentrations of NGB 2904 and PG 01037 were examined to determine their effects on P-gp, BCRP, and MRP2 ATPase activity. Each compound, together with a known excess of ATP, was incubated with recombinant human or rat transporter membranes. The compound was classified as a stimulator or inhibitor if the fold stimulation was more or less than 2-fold compared with the DMSO control (Polli et al., 2001). The positive control compound in each transporter assay significantly (p < 0.05) stimulated inorganic phosphate release with a fold stimulation more than two. Treatment with increasing concentrations of PG 01037 but not NGB 2904 produced a dose-dependent fold stimulation of human and rat P-gp (Fig. 5, A and B, respectively). BCRP and MRP2 ATPase activity were not significantly altered (<2-fold stimulation) by increasing concentrations of NGB 2904 or PG 01037 (Fig. 5, C–F).
Fig. 5.

Stimulation of human P-gp (A), rat P-gp (B), human BCRP (C), rat BCRP (D), human MRP2 (E), and rat MRP2 (F) ATPase activity by increasing concentrations of NGB 2904 and PG 01037. Stimulation of ATPase activity was estimated by measuring inorganic phosphate released from ATP in the presence or absence of their respective controls or D3R analogs (NGB 2904 and PG 01037; 5–100 μM). The drug-stimulated ATPase activity was reported as fold-stimulation relative to the basal ATPase activity in the absence of drug (DMSO control). Data are represented as mean fold stimulation of the average control from triplicate reactions mean ± S.D. *, p < 0.05.
PG 01037 Bidirectional Transport and Inhibition Studies in MDCK-MDR1 Cell Monolayers.
A high level of P-gp expression was noted in the MDCK-MDR1 cells from each passage used for transport studies (passages 18–24) (Fig. 6A). Mannitol permeability (Papp) ranged from 4.74 × 10−6 to 7.01 × 10−6 cm/s across MDCK-MDR1 cell monolayers, and transepithelial electrical resistance values were >400 Ω cm2. The saquinavir efflux ratio was 2.8 ± 0.06 and was significantly decreased (p < 0.05) to 1.6 ± 0.11 in the presence of verapamil. This validates the existence of functional P-gp in these cell monolayers.
Fig. 6.

Effect of the competitive inhibitor verapamil (200 μM) on the permeability and P-gp-mediated efflux of PG 01037 across MDCK-MDR1 monolayers. A, P-gp expression levels over the period in which transport studies were conducted (passages 18–24). B, efflux ratios (mean ± S.D., n = 3) across MDCK-MDR1 monolayers in the absence (−) and presence (+) of verapamil. C, apical-to-basolateral permeability in the absence (−) and presence (+) of verapamil across MDCK-MDR1 monolayers (data represented as mean ± S.D., n = 3). *, p < 0.05.
PG 01037 showed polarized transport with a higher B−A permeability (19.04 ± 0.09 × 10−6 cm/s) than A−B permeability (4.41 ± 0.19 × 10−6 cm/s). The efflux ratio for PG 01037 was 4.3 ± 0.24 (Fig. 6B). Verapamil was used to verify the involvement of P-gp in mediating the observed efflux of PG 01037. Verapamil caused a significant (p < 0.05) inhibition of P-gp-mediated efflux of PG 01037 (efflux ratio decreased from 4.3 to 0.9) and increased the A−B permeability of PG 01037 in the MDCK-MDR1 cells (Fig. 6, B and C). Previous results indicate that 200 μM verapamil does not affect the viability of MDCK-MDR1 cells (Othman et al., 2007).
Discussion
The clinical development of selective DA D3R agents involves optimizing their physical and “drug-like” properties to increase bioavailability without diminishing pharmacological efficacy. In addition, an optimal drug must display an appropriate absorption, distribution, metabolism, excretion, and toxicity profile and achieve adequate concentrations at its target site. The compounds evaluated in this study were developed to achieve Lipinski's criterion for absorption (Lipinski et al., 2001) while adhering to accepted guidelines for central nervous system penetration (van de Waterbeemd et al., 1998). Structure–activity relationship studies have shown that optimal pharmacological selectivity and high D3R affinity come from compounds that have high molecular weights (>400) and often cLogP values (>5), predisposing them to have poor bioavailability or unacceptable metabolic profiles. Thus, it is important to identify structural motifs that might be modified to improve the bioavailability of future candidates targeting the DA D3R for therapeutic use.
All four compounds entered the brain quickly and demonstrated high brain penetration when administered intravenously. Among the physiochemical parameters, cLogP directly correlated with AUCbrain/AUCplasma (Tables 1 and 2), suggesting lipophilicity as a major factor for determining BBB penetration of the structural analogs. In fact, both the fluorenyl amides (NGB 2904 and JJC 4-077) and 2-pyridylphenyl amides (CJB 090 and PG 01037) had brain concentrations that were significantly higher than the corresponding plasma concentrations, with statistically significant higher AUCbrain than AUCplasma values. The fluorenyl amides displayed relatively higher B/P ratios than those of the 2-pyridylphenyl amides, likely attributed to their higher lipophilicity. Despite possible variation associated with differences in species, dose, and route of administration, the high brain penetration supports the behavioral data using these compounds in models of yawning, brain stimulation reward, and drug-seeking behavior (Collins et al., 2005, 2009; Gilbert et al., 2005; Martelle et al., 2007; Spiller et al., 2008; Achat-Mendes et al., 2009; Higley et al., 2010) in the sense that high brain uptake and D3R binding of these compounds are critical to elicit behavioral responses.
The DA D3R analogs displayed plasma half-lives (t1/2) in the range of 1.71 to 3.27 h, which are substantially longer than cocaine (t1/2 = 0.5 h) (Raje et al., 2003), and their elimination from the brain (1.35–2.63 h) is much slower than that of cocaine (0.6 h) (Raje et al., 2003) (Table 2), which are required characteristics for the inhibition of cocaine's downstream effects. NGB 2904 and JJC 4-077 are structurally identical except for the presence of a hydroxyl group (3-OH) in the linking chain (Fig. 1). The faster systemic clearance of JJC 4-077 compared with NGB 2094 (Table 2) could be the result of enhanced biotransformation of this OH group by phase II enzymes. In contrast, substitution of a fluorenyl amide (NGB 2904 and JJC 4-077) with a 2-pyridylphenyl amide (CJB 090 and PG 01037) did not affect systemic clearance (Table 2).
NGB 2904 has served as a prototypical D3R antagonist in numerous behavioral studies, especially animal models of addiction (Xi and Gardner, 2007). Nevertheless, its poor water solubility and high lipophilicity likely preclude this compound from being a viable candidate for clinical development. NGB 2904 has, however, served as an important research tool and a template for the generation of structurally similar analogs with improved physiochemical properties (Grundt et al., 2007; Newman et al., 2009). For example, substitution of the fluorenyl moiety (NGB 2904 and JJC 4-077) with a 2-pyridylphenyl group (CJB 090 and PG 01037) decreased lipophilicity and increased DA D3R affinity (Table 1). One of these resulting compounds, PG 01037, is a water-soluble and highly potent DA D3R antagonist. Subsequent testing has shown that PG 01037 rapidly enters the brain and localizes in D3 but not D2 receptor-rich regions in rat brain (Grundt et al., 2007), a finding supported by relatively low Ki values of PG 01037 for D3 receptors compared with D2 receptors (Table 1).
NGB 2904 and PG 01037 have diverse pharmacokinetic properties (Table 2) but behave similarly in their ability to attenuate drug-induced behaviors in vivo (Spiller et al., 2008; Higley et al., 2010). Many behavioral evaluations have been conducted by using the intraperitoneal route of administration and observed compound-specific behavioral effects from a wide range of doses (i.e., 0.30–56.0 mg/kg) (Spiller et al., 2008; Baladi et al., 2010). Thus, there is the possibility that these analogs could differ in their rate and/or extent of absorption from the peritoneal cavity. Although brain levels remained substantially higher than plasma levels, there were significant (p < 0.05) decreases in plasma and brain AUCs of both NGB 2904 and PG 01037 after intraperitoneal administration compared with intravenous administration (Table 2 and Fig. 2). Their clearance values were also increased after intraperitoneal administration (Table 2). Together, these results support the initial supposition of compromised bioavailability and could explain the need for relatively high extravascular drug administration for in vivo action. The limited bioavailability of both compounds after intraperitoneal administration may be caused by the influence of dispositional factors such as metabolizing enzymes and drug transporters.
Drug efflux transporters can have significant effects on in vivo disposition and pharmacokinetics of drugs. PG 01037 demonstrated dose-dependent stimulation of both human and rat P-gp ATPase activity, evidenced by an increase in fold-stimulation values (Fig. 5, A and B). NGB 2904 did not stimulate P-gp ATPase activity, suggesting that replacement of the fluorenyl moiety of NGB 2904 with the 2-pyridylphenyl group of PG 01037 enhanced brain efflux by increasing P-gp interactions. To confirm the P-gp-mediated efflux of PG 01037, in vitro transport studies were conducted in the presence and absence of verapamil. Verapamil enhanced A−B permeability and significantly decreased the efflux ratio of PG 01037, indicating that P-gp mediated PG 01037 efflux. P-gp efflux at the BBB could, in part, account for the decreased B/P ratio of PG 01037 compared with NGB 2904 in rats (Table 2). On the other hand, the relatively high lipophilicity (cLogP > 5) of PG 01037 (Table 1) may explain why brain uptake of PG 01037 remained high. MRP2 and BCRP are also expressed at the BBB (Yousif et al., 2007). However, neither NGB 2904 nor PG 01037 modulated their ATPase activity (Fig. 5, C–F).
In general, compounds administered intraperitoneally are absorbed primarily through the portal circulation and, therefore, must pass through the liver before reaching systemic circulation and target organs (Lukas et al., 1971). The significant decreases in AUCplasma and AUCbrain values of NGB 2904 and PG 01037 after intraperitoneal administration in comparison with intravenous administration indicates that these compounds may be metabolized by the liver before reaching the brain. The route of administration affected the brain penetration of NGB 2904 to a greater extent than it did for PG 01037, suggesting that NGB 2904 is more susceptible to metabolism, possibly through its fluorenyl amide linking group (Fig. 3). The results of the in vitro metabolism study show that human and rat CYP3A subfamilies are involved primarily in the metabolism of both NGB 2904 and PG 01037 (Fig. 4). Thus, the liver may be the main site for metabolism of the D3R analogs when they are administered intraperitoneally. Our data further indicate that the intrinsic clearance of NGB 2904 is faster in pooled rat liver microsomes than in pooled human liver microsomes (Table 1), which could be attributed to its substrate specificity for many of the evaluated CYP450 enzymes (Fig. 4, A and B). The low metabolic stability of NGB 2904 in rats correlates with a rate of systemic clearance that is substantially greater when administered intraperitoneally. The overall contribution of drug metabolism to the systemic clearance of NGB 2904 needs to be determined as does its metabolism by phase II enzymes.
In conclusion, the evolution of D3R agents, beginning with NGB 2904, has led to the development of highly potent D3R compounds. CJB 090 is a D3R partial agonist with a similar D3R selectivity profile to NGB 2904, but somewhat less lipophilic. By replacing the saturated butyl linking chain of CJB 090, the resulting PG 01037 is significantly more D3R-selective and a full antagonist. This compound has served as an important tool for in vivo studies, but high doses required for behavioral activity support further optimization. JJC 4-077, like CJB 090, is a partial agonist, but it is the most D3R-selective ligand in this set. Although the 3-OH group in the linking chain resulted in high D3R selectivity, the studies herein suggest that it may be a metabolic liability, suggesting that additional modification at this position might be preferred. In this study, PG 01037 demonstrated the most favorable overall pharmacokinetic profile, BBB permeation, and physicochemical characteristics, including improved aqueous solubility. Nevertheless, further chemical modification may improve these properties. The 4-phenylpiperazines appear to be a promising class of compounds for further development; however, PG 01037 is characterized by significant P-gp efflux and hepatic metabolism, which is predictive of low bioavailability in humans and provides a plausible explanation for the requirement of high doses for activity in animals. Overall, these studies present critical preclinical information for the design and development of the next generation of D3R-selective agents for the treatment of drug addiction.
Acknowledgments
We thank Dr. Peter Swaan for providing the MDCK-MDR1 cell line.
This work was supported by the Intramural Research Program of the National Institutes of Health National Institute of Drug Abuse [Grant R01-DA1671503].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.109.165084.
- DA D3R
- dopamine D3 receptor
- DA
- dopamine
- DAT
- dopamine transporter
- BBB
- blood-brain barrier
- B/P
- brain to plasma
- CJB 090
- N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)-4-(pyridine-2-yl)benzamide hydrochloride
- NGB 2904
- N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)-9H-fluorene-2-carboxamide fumarate
- JJC 4-077
- N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-3-hydroxybutyl)-9H-fluorene-2-carboxamide hydrochloride
- PG 01037
- N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-trans-but-2-enyl)-4-(pyridine-2-yl)benzamide hydrochloride
- BP897
- N-[4-[4-(2-methoxyphenyl)piperazin-1-yl]butyl]naphthalene-2-carboxamide
- PG 01030
- N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-trans-but-2-enyl)-benzothiopen-2-carboxamide hydrochloride
- HPLC
- high-performance liquid chromatography
- DMSO
- dimethyl sulfoxide
- MDCK
- Madin-Darby canine kidney
- P-gp
- P-glycoprotein
- MRP2
- multidrug resistance protein 2
- BCRP
- breast cancer resistance protein
- HPBCD
- 2-hydropropyl-β-cyclodextrin
- PEG 400
- polyethyleneglycol 400
- PBS
- phosphate-buffered saline with Ca2+ and Mg2+
- A−B
- apical to basolateral
- B−A
- basolateral to apical
- AUC
- area under the curve.
References
- Achat-Mendes et al., 2009.Achat-Mendes C, Platt DM, Newman AH, Spealman RD. (2009) The dopamine D3 receptor partial agonist CJB 090 inhibits the discriminative stimulus but not the reinforcing or priming effects of cocaine in squirrel monkeys. Psychopharmacology (Berl) 206:73–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailer, 1988.Bailer AJ. (1988) Testing for the equality of area under the curves when using destructive measurement techniques. J Pharmacokinet Biopharm 16:303–309 [DOI] [PubMed] [Google Scholar]
- Baladi et al., 2010.Baladi MG, Newman AH, France CP. (2010) Dopamine D3 receptors mediate the discriminative stimulus effects of quinpirole in free-feeding rats. J Pharmacol Exp Ther 332:308–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckler and Gmeiner, 2006.Boeckler F, Gmeiner P. (2006) The structural evolution of dopamine D3 receptor ligands: structure–activity relationships and selected neuropharmacological aspects. Pharmacol Ther 112:281–333 [DOI] [PubMed] [Google Scholar]
- Collins et al., 2009.Collins GT, Truccone A, Haji-Abdi F, Newman AH, Grundt P, Rice KC, Husbands SM, Greedy BM, Enguehard-Gueiffier C, Gueiffier A, et al. (2009) Proerectile effects of dopamine D2-like agonists are mediated by the D3 receptor in rats and mice. J Pharmacol Exp Ther 329:210–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins et al., 2005.Collins GT, Witkin JM, Newman AH, Svensson KA, Grundt P, Cao J, Woods JH. (2005) Dopamine agonist-induced yawning in rats: a dopamine D3 receptor-mediated behavior. J Pharmacol Exp Ther 314:310–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert et al., 2005.Gilbert JG, Newman AH, Gardner EL, Ashby CR, Jr, Heidbreder CA, Pak AC, Peng XQ, Xi ZX. (2005) Acute administration of SB-277011A, NGB 2904, or BP 897 inhibits cocaine cue-induced reinstatement of drug-seeking behavior in rats: role of dopamine D3 receptors. Synapse 57:17–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundt et al., 2007.Grundt P, Prevatt KM, Cao J, Taylor M, Floresca CZ, Choi JK, Jenkins BG, Luedtke RR, Newman AH. (2007) Heterocyclic analogues of N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)arylcarboxamides with functionalized linking chains as novel dopamine D3 receptor ligands: potential substance abuse therapeutic agents. J Med Chem 50:4135–4146 [DOI] [PubMed] [Google Scholar]
- Heidbreder et al., 2005.Heidbreder CA, Gardner EL, Xi ZX, Thanos PK, Mugnaini M, Hagan JJ, Ashby CR., Jr (2005) The role of central dopamine D3 receptors in drug addiction: a review of pharmacological evidence. Brain Res Brain Res Rev 49:77–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley et al., 2010.Higley AE, Spiller K, Grundt P, Newman AH, Kiefer SW, Xi ZZ, Gardner EL. (2010) PG01037, a novel dopamine D3 receptor antagonist, inhibits the effects of methamphetamine in rats. J Psychopharmacol, doi:10.1177/0269881109358201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar et al., 2009.Kumar R, Riddle L, Griffin SA, Grundt P, Newman AH, Luedtke RR. (2009) Evaluation of the D3 dopamine receptor selective antagonist PG01037 on l-DOPA-dependent abnormal involuntary movements in rats. Neuropharmacology 56:944–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski et al., 2001.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26 [DOI] [PubMed] [Google Scholar]
- Litman et al., 1997.Litman T, Nielsen D, Skovsgaard T, Zeuthen T, Stein WD. (1997) ATPase activity of P-glycoprotein related to emergence of drug resistance in Ehrlich ascites tumor cell lines. Biochim Biophys Acta 1361:147–158 [DOI] [PubMed] [Google Scholar]
- Lukas et al., 1971.Lukas G, Brindle SD, Greengard P. (1971) The route of absorption of intraperitoneally administered compounds. J Pharmacol Exp Ther 178:562–564 [PubMed] [Google Scholar]
- Martelle et al., 2007.Martelle JL, Claytor R, Ross JT, Reboussin BA, Newman AH, Nader MA. (2007) Effects of two novel D3-selective compounds, NGB 2904 [N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)-9H-fluorene-2-carboxami de] and CJB 090 [N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)-4-(pyridin-2-yl)benzami de], on the reinforcing and discriminative stimulus effects of cocaine in rhesus monkeys. J Pharmacol Exp Ther 321:573–582 [DOI] [PubMed] [Google Scholar]
- Micheli and Heidbreder, 2008.Micheli F, Heidbreder C. (2008) Selective D3 receptor antagonists. A decade of progress: 1997–2007. Expert Opin Ther Patents 18:821–840 [DOI] [PubMed] [Google Scholar]
- Naritomi et al., 2003.Naritomi Y, Terashita S, Kagayama A, Sugiyama Y. (2003) Utility of hepatocytes in predicting drug metabolism: comparison of hepatic intrinsic clearance in rats and humans in vivo and in vitro. Drug Metab Dispos 31:580–588 [DOI] [PubMed] [Google Scholar]
- Newman et al., 2003.Newman AH, Cao J, Bennett CJ, Robarge MJ, Freeman RA, Luedtke RR. (2003) N-(4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butyl, butenyl, and butynyl)arylcarboxamides as novel dopamine D(3) receptor antagonists. Bioorg Med Chem Lett 13:2179–2183 [DOI] [PubMed] [Google Scholar]
- Newman et al., 2009.Newman AH, Grundt P, Cyriac G, Deschamps JR, Taylor M, Kumar R, Ho D, Luedtke RR. (2009) N-(4-(4-(2,3-dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J Med Chem 52:2559–2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman et al., 2005.Newman AH, Grundt P, Nader MA. (2005) Dopamine D3 receptor partial agonists and antagonists as potential drug abuse therapeutic agents. J Med Chem 48:3663–3679 [DOI] [PubMed] [Google Scholar]
- Othman et al., 2007.Othman AA, Syed SA, Newman AH, Eddington ND. (2007) Transport, metabolism, and in vivo population pharmacokinetics of the chloro benztropine analogs, a class of compounds extensively evaluated in animal models of drug abuse. J Pharmacol Exp Ther 320:344–353 [DOI] [PubMed] [Google Scholar]
- Pilla et al., 1999.Pilla M, Perachon S, Sautel F, Garrido F, Mann A, Wermuth CG, Schwartz JC, Everitt BJ, Sokoloff P. (1999) Selective inhibition of cocaine-seeking behavior by a partial dopamine D3 receptor agonist. Nature 400:371–375 [DOI] [PubMed] [Google Scholar]
- Polli et al., 2001.Polli JW, Wring SA, Humphreys JE, Huang L, Morgan JB, Webster LO, Serabjit-Singh CS. (2001) Rational use of in vitro P-glycoprotein assays in drug discovery. J Pharmacol Exp Ther 299:620–628 [PubMed] [Google Scholar]
- Raje et al., 2003.Raje S, Cao J, Newman AH, Gao H, Eddington ND. (2003) Evaluation of the blood-brain barrier transport, population pharmacokinetics, and brain distribution of benztropine analogs and cocaine using in vitro and in vivo techniques. J Pharmacol Exp Ther 307:801–808 [DOI] [PubMed] [Google Scholar]
- Richtand, 2006.Richtand NM. (2006) Behavioral sensitization, alternative splicing, and D3 dopamine receptor-mediated inhibitory function. Neuropsychopharmacology 31:2368–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross and Peselow, 2009.Ross S, Peselow E. (2009) Pharmacotherapy of addictive disorders. Clin Neuropharmacol 32:277–289 [DOI] [PubMed] [Google Scholar]
- Schwarz et al., 2007.Schwarz AJ, Gozzi A, Reese T, Heidbreder CA, Bifone A. (2007) Pharmacological modulation of functional connectivity: the correlation structure underlying the phMRI response to d-amphetamine modified by selective dopamine D3 receptor antagonist SB277011A. Magn Reson Imaging 25:811–820 [DOI] [PubMed] [Google Scholar]
- Sokoloff et al., 1990.Sokoloff P, Giros B, Martres MP, Bouthenet ML, Schwartz JC. (1990) Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 347:146–151 [DOI] [PubMed] [Google Scholar]
- Spiller et al., 2008.Spiller K, Xi ZX, Peng XQ, Newman AH, Ashby CR, Jr, Heidbreder C, Gaal J, Gardner EL. (2008) The selective dopamine D3 receptor antagonists SB-277011A and NGB 2904 and the putative partial D3 receptor agonist BP-897 attenuate methamphetamine-enhanced brain stimulation reward in rats. Psychopharmacology (Berl) 196:533–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Waterbeemd et al., 1998.van de Waterbeemd H, Camenisch G, Folkers G, Chretien JR, Raevsky OA. (1998) Estimation of blood-brain barrier crossing of drugs using molecular size and shape, and H-bonding descriptors. J Drug Target 6:151–165 [DOI] [PubMed] [Google Scholar]
- Visanji et al., 2009.Visanji NP, Fox SH, Johnston T, Reyes G, Millan MJ, Brotchie JM. (2009) Dopamine D3 receptor stimulation underlies the development of l-DOPA-induced dyskinesia in animal models of Parkinson's disease. Neurobiol Dis 35:184–192 [DOI] [PubMed] [Google Scholar]
- Volkow et al., 2004.Volkow ND, Fowler JS, Wang GJ, Swanson JM. (2004) Dopamine in drug abuse and addiction: results from imaging studies and treatment implications. Mol Psychiatry 9:557–569 [DOI] [PubMed] [Google Scholar]
- Xi and Gardner, 2007.Xi ZX, Gardner EL. (2007) Pharmacological actions of NGB 2904, a selective dopamine D3 receptor antagonist, in animal models of drug addiction. CNS Drug Rev 13:240–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi et al., 2006.Xi ZX, Newman AH, Gilbert JG, Pak AC, Peng XQ, Ashby CR, Jr, Gitajn L, Gardner EL. (2006) The novel dopamine D3 receptor antagonist NGB 2904 inhibits cocaine's rewarding effects and cocaine-induced reinstatement of drug-seeking behavior in rats. Neuropsychopharmacology 31:1393–1405 [DOI] [PubMed] [Google Scholar]
- Yousif et al., 2007.Yousif S, Marie-Claire C, Roux F, Scherrmann JM, Declèves X. (2007) Expression of drug transporters at the blood-brain barrier using an optimized isolated rat brain microvessel strategy. Brain Res 1134:1–11 [DOI] [PubMed] [Google Scholar]
- Yuan et al., 1998.Yuan J, Chen X, Brodbeck R, Primus R, Braun J, Wasley JW, Thurkauf A. (1998) NGB 2904 and NGB 2849: two highly selective dopamine D3 receptor antagonists. Bioorg Med Chem Lett 8:2715–2718 [DOI] [PubMed] [Google Scholar]