

Abstract
γ-Hydroxybutyric acid (GHB) is an endogenous neurotransmitter that is abused because of its sedative/hypnotic and euphoric effects. The objectives of this study were to evaluate the concentration-effect relationships of GHB in plasma, cerebrospinal fluid (CSF), brain (whole and discrete brain regions), and brain frontal cortex extracellular fluid. This information is crucial for future studies to evaluate effects of therapeutic interventions on the toxicodynamics of GHB. GHB (200–1000 mg/kg) was administered intravenously to rats, and plasma and frontal cortex microdialysate samples were collected for up to 6 h after the dose, or plasma, CSF, and brain (whole, frontal cortex, striatum, and hippocampus) concentrations were determined at the offset of its sedative/hypnotic effect [return to righting reflex (RRR)]. GHB-induced changes in the brain neurotransmitters γ-aminobutyric acid (GABA) and glutamate were also determined. GHB, GABA, and glutamate concentrations were measured by liquid chromatography/tandem mass spectrometry. GHB-induced sleep time significantly increased in a dose-dependent manner (20-fold increase from 200 to 1000 mg/kg). GHB concentrations in plasma (300–400 μg/ml), whole brain (70 μg/g), discrete brain regions (80–100 μg/g), and brain microdialysate (29–39 μg/ml) correlated with RRR. In contrast, CSF GHB and GABA and glutamate concentrations in discrete brain regions exhibited no relationship with RRR. Our results suggest that GHB-induced sedative/hypnotic effects are mediated directly by GHB and that at high GHB doses, GABA formation from GHB may not contribute to the observed sedative/hypnotic effect. These results support the use of a clinical GHB detoxification strategy aimed at decreasing plasma and brain GHB concentrations after GHB overdoses.
γ-Hydroxybutyrate (GHB) is an endogenous short-chain fatty acid formed from γ-aminobutyric acid (GABA) (Gold and Roth, 1977) and found in the mammalian brain, heart, liver, intestine, and kidney (Maitre, 1997; Tedeschi et al., 2003). Clinically, GHB is marketed in the United States as Xyrem (Jazz Pharmaceuticals, Palo Alto, CA) to treat narcolepsy with cataplexy (Mamelak et al., 1986) and in Europe for the treatment of alcohol withdrawal (Gallimberti et al., 2000). However, the illicit use and abuse of GHB are likely the result of its sedative/hypnotic and euphoric effects. GHB is referred to as a “club drug” based on its widespread use at nightclubs and raves (Carter et al., 2009). Abuse of GHB has led to an increase in overdoses and toxicological effects characterized by dizziness, respiratory depression, vomiting, and unconsciousness, as well as coma and death (Okun et al., 2001; Carter et al., 2009). Current therapeutic strategies for GHB overdose are limited to supportive care, with antidotes such as physostigmine and naloxone having little to no effect (Mason and Kerns, 2002).
GHB pharmacokinetics are nonlinear in rats (Lettieri and Fung, 1979) and humans (Ferrara et al., 1992; Palatini et al., 1993) with decreased total clearance at higher doses. The observed nonlinearity results from capacity-limited metabolism (Lettieri and Fung, 1979; Ferrara et al., 1992; Palatini et al., 1993), transporter-mediated and saturable absorption (Arena and Fung, 1980), and renal reabsorption (Morris et al., 2005). At high doses, the decreased clearance of GHB results in sustained high plasma GHB concentrations and a corresponding increase in sedative/hypnotic effect as measured by loss of righting reflex (LRR) and return to righting reflex (RRR) (Raybon and Boje, 2007; Wang et al., 2008b). Reductions in GHB sedative/hypnotic effect have been observed after the coadministration of l-lactate (Wang et al., 2008a) or luteolin (Wang et al., 2008b). These compounds increase the renal clearance of GHB by inhibiting active renal reabsorption, mediated by monocarboxylate transporters (MCTs), resulting in a concomitant decrease in plasma concentrations.
Whereas the physiological effects of GHB on sleep and sleep disorders (Carai et al., 2001; Mamelak, 2009) are postulated to occur via the GHB receptor, the pharmacological and toxicological effects of GHB are thought to occur through interactions with the GABAB receptor (Maitre, 1997). The observed behavioral effects associated with GHB exposure, including discriminative stimulus (Carter et al., 2006; Koek et al., 2006; Koek and France, 2008), hypolocomotion (Kaupmann et al., 2003), and LRR (Carai et al., 2001), are consistent with the involvement of the GABAB receptor. These behavioral effects are observed with the prototypical GABAB receptor agonist baclofen (Carter et al., 2003, 2006; Smith et al., 2006). However, the mechanism of action contributing to the observed toxicological effects of GHB is controversial and appears to vary based on the dose administered and the behavioral or physiological endpoint that is measured (Carter et al., 2009; Mamelak, 2009). Three potential mechanisms of action have been suggested based on the role of the GABAB receptor: 1) direct interaction with the GABAB receptor (Mathivet et al., 1997); 2) indirect interaction through the metabolic conversion of GHB to GABA (Hechler et al., 1997); or 3) GHB-mediated stimulation of GABA release (Hechler et al., 1997). Additional mechanisms of action for GHB have been suggested, including the involvement of GABAC receptors and GABAB-mediated increase in synthesis of GABAA-modulating neuroactive steroids (Carter et al., 2009; Mamelak, 2009).
Few studies have examined the relationship between GHB toxicokinetics (TK) and its sedative/hypnotic effect (Lettieri and Fung, 1979; Wang et al., 2008a,b). Previous investigations have focused on plasma concentration-effect relationships and have not examined the relationships with respect to cerebrospinal fluid (CSF) and discrete brain region concentrations. The objectives of the present study were 1) to examine exposure-response relationships for GHB-induced sedative/hypnotic effect; 2) to evaluate TK/toxicodynamic (TD) relationships at the offset of the GHB-induced sedative/hypnotic effect in plasma, CSF, and discrete brain regions; and 3) to evaluate the relationship between changes in GABA and glutamate and GHB-induced sedative/hypnotic effects in discrete brain regions.
Materials and Methods
Chemicals and Reagents
GHB (sodium salt) and formic acid were purchased from Sigma-Aldrich (St. Louis, MO). Deuterated GHB (GHB-d6) was obtained from Cerilliant Corporation (Round Rock, TX). Artificial CSF (aCSF) was obtained from Harvard Apparatus Inc. (Holliston, MA). Ketamine, xylazine, heparin, and carprofen were purchased from Henry Schein (Melville, NY). High-performance liquid chromatography (HPLC)-grade acetonitrile, methanol, water, and acetic acid were purchased from Honeywell Burdick & Jackson (Muskegon, MI).
Animals and Surgery
Male Sprague-Dawley rats (280–320 g) were used in all the studies. Rats were randomly assigned to dose groups and were housed individually after surgical procedures. Rats were used in a single study and were administered GHB on a single occasion. Rats were housed under controlled lighting (12-h light/dark cycle) and temperature (20 ± 2°C) conditions with food and water provided ad libitum. All the animal experiments were approved by the Institutional Animal Care and Use Committee, University at Buffalo, State University of New York. All the rats had cannulas implanted as described previously (Morris et al., 2005). In brief, right jugular vein cannulas were implanted under anesthesia after an intraperitoneal injection of 90 mg/kg ketamine and 9 mg/kg xylazine. Animals were allowed 3 days for surgical recovery before the start of experiments. Jugular vein catheters were flushed daily with heparinized saline (40 IU of heparin/ml of saline) to maintain patency.
Microdialysis Study.
Rats were implanted with a jugular vein cannula and were then mounted in a stereotaxic frame (Harvard Apparatus Inc.). Microdialysis guide cannulas (for CMA11 probes; CMA Microdialysis, North Chelmsford, MA) containing dummy probes were implanted in the frontal cortex (anteroposterior +3.2 mm and mediolateral ±2.5 mm from bregma, and dorsoventral −0.5 mm from dura) (Paxinos and Watson, 1986) with the incisor bar set 0.5 mm below the interaural line. The side of the brain in which the cannula was placed was alternated across animals. The cannula was fixed in place using jeweler's screws (Plastics One, Roanoke, VA) and dental resin. Rats were administered carprofen 5 mg/kg twice daily for 2 days after surgery and allowed at least 6 days for surgical recovery before microdialysis probe implantation.
Dosing and Sleep Time Determination
GHB was dissolved in sterile water (200 mg/ml) and filtered with a 0.2-μm filter to ensure sterility. GHB (200, 400, 600, 800, or 1000 mg/kg) was administered by intravenous bolus injection via the jugular vein cannulation to rats. GHB doses were selected based on previous studies to ensure a range of sedative/hypnotic effects. The onset of sedative/hypnotic effect (time of LRR) and offset of sedative/hypnotic effect (RRR) were recorded in all the animals. LRR and RRR are defined as the time at which the animal lost or regained the ability to right itself when placed on its back. Animals were placed on their backs immediately after dose administration and every 30 s thereafter until LRR. The sedative/hypnotic duration of effect (sleep time) of GHB was determined as the difference between RRR and LRR.
Experimental Design
Evaluation of GHB TK/TD.
GHB (200, 400, 600, or 1000 mg/kg) was administered to male Sprague-Dawley rats (n = 7–10 per dose; total = 34). Blood samples (200 μl) for analysis of GHB were withdrawn from the jugular vein cannula at different time points (5, 10, 20, 30, 60, 90, 120, 180, 240, 300, and 360 min) and transferred to heparinized 0.6-ml microcentrifuge tubes. Plasma was separated by centrifugation at 1000g for 10 min at 4°C and stored at −80°C.
Evaluation of GHB TK/TD at Offset of Effect.
Male Sprague-Dawley rats (n = 7–8 per dose; total = 22) were administered GHB (400, 600, or 800 mg/kg i.v.). This study was designed to detect a change of 25% with 15% variability (90% power). At RRR, ketamine/xylazine (90:9 mg/kg) was administered, and blood, CSF, and brain (whole brain, frontal cortex, striatum, and hippocampus) samples were collected. Plasma was separated by centrifugation at 1000g for 10 min at 4°C. Whole brain and brain regions were immediately snap-frozen in liquid nitrogen. All the samples were stored at −80°C until analysis.
Evaluation of GHB TK Using in Vivo Microdialysis.
Microdialysis probes (CMA11; CMA Microdialysis) were prepared according to the manufacturer's instructions and implanted 24 h before the experiment to allow the blood-brain barrier to reform after implantation (de Lange et al., 2000). All the experiments were conducted in metabolic cages in awake and freely moving rats with ad libitum access to water. Microdialysis probes were perfused with blank aCSF at a rate of 2 μl/min and allowed to stabilize for 2 h. Microdialysate fractions were collected every 20 min. After stabilization, the probe was perfused with 1 μg/ml GHB with aCSF for 80 min to calculate in vivo probe recovery of GHB for each rat using the established retrodialysis method (Bouw and Hammarlund-Udenaes, 1998). The retrodialysis was followed by a washout period where blank aCSF was perfused for 80 min to remove any residual GHB. Rats were then administered GHB 400, 600, or 800 mg/kg i.v. (n = 3 per dose; total = 9), dialysate samples collected for 6 h after dose (20-min intervals), and times of LRR and RRR were recorded for each rat. On completion of the time course, rats were sacrificed, and the probe tracks were stained with dye. Brains were collected to confirm probe location. Dialysate samples were stored at −80°C until analysis.
Effects of GHB on Neurotransmitter Levels in Discrete Brain Regions.
Male Sprague-Dawley rats were administered GHB (400, 600, or 800 mg/kg i.v.). Under anesthesia, rats were exsanguinated, followed by collection of the frontal cortex, striatum, and hippocampus at 0, 10, 20, 30, 60, 90, 120, 150, 180, 210, 240, 270, 300, and 330 min after dose (n = 3 per time point; n = 30 per dose; total = 90). Tissue samples were snap-frozen in liquid nitrogen and stored at −80°C until analysis.
Plasma, CSF, and Brain Sample Preparation
GHB was extracted from plasma using an anion exchange solid-phase extraction procedure described previously (Fung et al., 2004; Raybon and Boje, 2007) with slight modifications. In brief, 5 μl of GHB-d6 (1 mg/ml) and 5 μl of double-distilled water were added to 50 μl of plasma. Standards and quality controls were prepared by the addition of 5 μl of GHB-d6 (1 mg/ml) and 5 μl of GHB stock solution to 50 μl of blank plasma. Plasma proteins were precipitated by the addition of 0.4 ml of acetonitrile followed by centrifugation at 10,000g for 20 min. Supernatant (0.2 ml) was aspirated and then diluted with 0.8 ml of double-distilled water. Bond Elute SAX cartridges (100 mg of resin, 1-ml volume; Varian, Inc., Palo Alto, CA) were preconditioned and washed, and samples and standards were eluted as described previously (Raybon and Boje, 2007). The eluent was evaporated under a gentle stream of N2 gas, reconstituted in 1.25 ml of 0.1% formic acid in double-distilled water and 5% acetonitrile, and stored at −80°C until liquid chromatography/tandem mass spectrometry (LC/MS/MS) analysis.
CSF and microdialysate samples were diluted 1:40 and 1:10, respectively, with aCSF to bring GHB concentrations within the range of the standard curve. GHB-d6 (5 μl of 5 μg/ml) was added to 35 μl of diluted CSF or dialysate sample. Standards and quality controls were prepared by the addition of 5 μl of GHB-d6 (5 μg/ml) to 35 μl of GHB stock solution in aCSF. Samples and standards were stored at −80°C until LC/MS/MS analysis.
Whole brain and brain subregion samples were homogenized in 4 ml/g tissue double-distilled water. Ten microliters of GHB-d6 (200 μg/ml) and 10 μl of GABA-d6 (50 μg/ml) were added to 100 μl of sample homogenate. GHB standards and quality controls were prepared by the addition of 10 μl of GHB-d6 (200 μg/ml) and 10 μl of GHB stock solution to 100 μl of blank brain homogenate. The neurotransmitter standards and quality controls (GABA and glutamate combined) were prepared in double-distilled water to which 10 μl of GABA-d6 (50 μg/ml) and 10 μl of double-distilled water were added. Double-distilled water (880 μl) was added to samples and standards, followed by 1 ml of acetonitrile to precipitate proteins. Samples and standards were centrifuged at 10,000g for 20 min. The supernatant was aspirated and stored at −80°C until LC/MS/MS analysis.
LC/MS/MS Assay for GHB, GABA, and Glutamate
GHB in plasma, aCSF, and brain were quantified using a validated LC/MS/MS assay that was described previously (Raybon and Boje, 2007) with minor modifications. The LC/MS/MS method for GHB was extended to simultaneously quantify GABA and glutamate in discrete brain regions. The LC/MS/MS system consisted of an Agilent 1100 series HPLC consisting of an online degasser, binary pump, and autosampler (Agilent Technologies, Santa Clara, CA) connected to a PE Sciex API 3000 triple-quadruple tandem mass spectrometer equipped with a turbo ion spray (Applied Biosystems, Foster City, CA). Seven microliters of sample was injected on an Xterra MS C18 column (250 × 2.1 mm i.d., 5-μm particle size; Waters, Milford, MA). Mobile phase A consisted of 0.1% formic acid in HPLC-grade water with 5% acetonitrile, whereas mobile phase B consisted of 0.1% formic acid in acetonitrile with 5% HPLC-grade water. A gradient elution with a flow rate of 0.2 ml/min was used to separate compounds: 100% to 90% A over 5 min, 90% to 10% A over 2.5 min, and 10% to 100% A over 4.5 min. The retention times were 2.60, 2.75, and 3.6 min for GABA, glutamate, and GHB, respectively. The mass spectrometer was operated in positive ionization mode with multiple reaction monitoring. Compound-specific mass spectrometer parameters are listed in Table 1. The peak area ratios of the analyte and internal standard were determined using Analyst version 1.4.2 (Applied Biosystems).
TABLE 1.
Mass spectrometer conditions for MRM of GHB, GABA, and glutamate
| Parameter (units) | GHB | GABA | Glutamate |
|---|---|---|---|
| Q1/Q3 | 105.2/87.2 | 104.2/87.0 | 148.0/84.0 |
| Declustering potential (V) | 20 | 20 | 22 |
| Focusing potential (V) | 125 | 100 | 150 |
| Collision energy | 20 | 18 | 22 |
| Collision cell exit potential (V) | 10 | 8 | 12 |
Data and Statistical Analysis
Plasma area under the curve (AUC) was calculated by the log-linear trapezoidal method in WinNonlin version 5.2 (Pharsight, Mountain View, CA). Plasma and brain extracellular fluid (ECF) GHB concentrations at RRR were determined by noncompartmental analysis from time course data. Statistical analyses were performed in GraphPad Prism version 4.0 (GraphPad Software Inc., San Diego, CA). Significant differences between means were determined by one-way analysis of variance followed by a Tukey's multiple comparison. P < 0.05 was considered statistically significant. Nonlinear regression analysis in GraphPad Prism version 4.0 (GraphPad Software Inc.) was used to evaluate the relationship between sedative/hypnotic effect and exposure.
Retrodialysis recovery of drug was used to calculate the relative in vivo recovery of GHB from the dialysis probe. The recovery of GHB from the probe was calculated using the equation below:
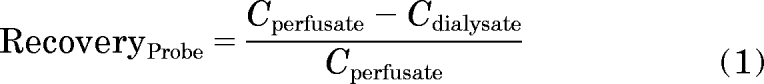 |
where Cperfusate is the perfusate GHB concentration (inlet) and Cdialysate is the dialysate GHB concentration (outlet). To determine GHB concentrations in frontal cortex ECF, dialysate GHB concentrations after intravenous administration of GHB were divided by the experimentally determined GHB probe recovery.
Results
LC/MS/MS Assay for GHB, GABA, and Glutamate.
The lower limits of quantitation for GHB were 1 μg/ml, 0.05 μg/ml, and 8 μg/g in plasma, CSF, and brain, respectively. Endogenous GHB was undetectable in untreated matrix samples and therefore was not included in the calculation of GHB concentration. The standard curve ranges were 1 to 500 μg/ml, 0.05 to 10 μg/ml, and 8 to 800 μg/g in plasma, CSF, and brain, respectively, based on regression analysis (r2 > 0.99) of peak area ratios (GHB/GHB-d6) versus GHB concentration. GABA and glutamate had lower limits of quantification of 0.05 and 0.5 μg/ml in all the brain regions. The standard curve ranges were 0.05 to 10 μg/ml and 0.5 to 15 μg/ml for GABA and glutamate based on regression analysis of peak ratios (GABA/GABA-d6 and glutamate/GABA-d6) versus GABA or glutamate concentration. Accuracy and precision for GHB (all matrices), GABA, and glutamate were 90 to 105 and 0.5 to 1.5%, respectively.
Dose-Dependent Effects of GHB on Sedative/Hypnotic Effect.
The sedative/hypnotic effect of GHB increases in a dose-dependent manner (Fig. 1) with a significant increase in sleep time observed in animals treated with 400, 600, and 1000 mg/kg compared with the 200 mg/kg dose group. The observed increases in sedative/hypnotic effect are correlated to increases in GHB exposure with increasing dose. Figure 2 illustrates the relationship between sedative/hypnotic effect (sleep time) and overall plasma exposure (AUC). Sleep time was only observed in 2 of 10 rats administered 200 mg/kg i.v. GHB, indicating that there is a threshold exposure required to observe sedative/hypnotic effects of GHB. In addition, the data presented in Fig. 2 correspond to exposure data from studies where GHB was concomitantly administered with lactate or luteolin (Wang et al., 2008a,b), which resulted in increased renal clearance and decreased plasma GHB AUCs. This relationship further shows that any reductions in plasma GHB exposure would correlate with a reduction in sedative/hypnotic effect.
Fig. 1.

Relationship between GHB dose and sedative/hypnotic effect. Animals were administered GHB by intravenous bolus (200, 400, 600, or 1000 mg/kg). Data are presented as mean ± S.D. (n = 7–10/dose). Sleep time represents the difference between RRR and LRR. *, P < 0.05 compared with 200 mg/kg dose.
Fig. 2.

TK/TD relationship between GHB-induced sleep time (sedative/hypnotic effect) and plasma AUC. Each data point represents an individual animal. Data were obtained from the current TK/TD study involving intravenous administration of GHB (200–1000 mg/kg). Plasma AUC was calculated by noncompartmental analysis. The solid line is the fitted line based on linear regression, whereas the dashed lines represent the 95% confidence intervals.
Concentration Dependence of Sedative/Hypnotic Effect.
To further investigate the relationship between GHB concentration and sedative/hypnotic effect, GHB concentrations were assessed in plasma, CSF, and brain (total and unbound) at RRR (offset of effect). Figure 3, A and B, illustrates the plasma GHB concentrations at RRR after administration of 200 to 1000 mg/kg i.v. GHB and calculated based on interpolation from the concentration-time profiles [Fig. 3A; TK/TD study] or directly measured at RRR (Fig. 3B; TK/TD at offset of effect study). Mean plasma GHB concentrations were between 300 and 400 μg/ml at RRR, independent of the dose administered, showing that there is a direct relationship between plasma GHB concentration and offset of sedative/hypnotic effect. In contrast, a significant dose-dependent increase in CSF GHB concentration was observed at RRR, indicating that CSF concentrations do not correlate with effect (Fig. 4). Consistent with the observed results in plasma, GHB concentrations in whole brain and discrete brain regions correlated with RRR (Fig. 5). Frontal cortex and hippocampus GHB concentrations at RRR were slightly higher than observed in whole brain, consistent with previous findings of enriched endogenous GHB concentrations in these regions. Figure 6 illustrates the unbound GHB concentration in the frontal cortex ECF at RRR, as measured by microdialysis. Unbound GHB concentrations correlated with RRR, which is consistent with total GHB concentrations in the frontal cortex.
Fig. 3.

Dependence of RRR on plasma GHB concentration. A, plasma GHB concentrations at RRR were calculated by noncompartmental analysis from the GHB pharmacokinetic profiles. Rats were administered GHB by intravenous bolus (200, 400, 600, or 1000 mg/kg), and plasma samples were obtained for up to 6 h after dose. B, plasma GHB concentrations were obtained by destructive sampling at RRR. Rats were administered GHB by intravenous bolus (400, 600, or 800 mg/kg). Data are presented as mean ± S.D. (n = 7–10/dose, except 200 mg/kg where n = 2). There were no statistically significant differences.
Fig. 4.

TK/TD relationships between GHB cerebrospinal fluid concentrations and RRR. GHB (400, 600, or 800 mg/kg) was administered by intravenous bolus. When animals regained their righting reflex, CSF was obtained from the cisterna magna. Data are presented as mean ± S.D. (n = 7–8/dose). *, P < 0.05 compared with 400 mg/kg dose.
Fig. 5.

TK/TD relationships between GHB concentrations in whole brain and discrete brain regions and RRR. GHB (400, 600, or 800 mg/kg) was administered by intravenous bolus. When animals regained their righting reflex, brain regions [whole brain (right hemisphere) (A), frontal cortex (B), striatum (C), and hippocampus (D)] were obtained following exsanguination of anesthetized animals. Data are presented as mean ± S.D. (n = 7–8 for whole brain and n = 3–4 for discrete brain regions).
Fig. 6.

TK/TD relationships between GHB concentrations in frontal cortex ECF and RRR. GHB (400, 600, or 800 mg/kg) was administered by intravenous bolus. Dialysate samples were collected in 20-min intervals for 6 h after dose. Frontal cortex ECF GHB concentration at RRR was determined by noncompartmental analysis. Data are presented as mean ± S.D. (n = 3/dose). There were no statistically significant differences.
Effect of GHB on GABA and Glutamate in Discrete Brain Regions.
Table 2 details the percentage change from baseline of GABA and glutamate at offset of the sedative/hypnotic effect (RRR). Data were calculated from time course data based on mean RRR for the specific dose. The brain concentrations of neurotransmitters do not correlate with the offset of TD effect in any of the discrete brain regions assessed. In addition, the changes in neurotransmitter concentrations were evaluated over 5 h after dose. Figure 7 represents the time course of percentage changes of GABA in the frontal cortex. Whereas both GABA and glutamate levels deviate from baseline in the frontal cortex, a dose-response trend was not observed over the dose range used in this study. These results were consistent with the results observed in the striatum and hippocampus of the same group of rats (data not shown).
TABLE 2.
Mean percentage changes in GABA and glutamate in discrete brain regions at RRR after administration of 400, 600, or 800 mg/kg i.v. GHB
Percentage changes were calculated based on mean RRR for a given dose group.
| Dose | GABA |
Glutamate |
||||
|---|---|---|---|---|---|---|
| Frontal Cortex | Striatum | Hippocampus | Frontal Cortex | Striatum | Hippocampus | |
| % baseline | ||||||
| 400 mg/kg | 92.9 | 97.8 | 93.4 | 78.4 | 79.9 | 93.9 |
| 600 mg/kg | 94.9 | 119.0 | 91.6 | 92.5 | 87.1 | 97.8 |
| 800 mg/kg | 102.5 | 98.3 | 91.8 | 99.7 | 66.7 | 81.8 |
Fig. 7.

Time course of GABA changes from baseline in frontal cortex after administration of intravenous GHB at doses of 400 (dotted line), 600 (dashed line), or 800 (solid line) mg/kg. Results are presented as a percentage of the mean baseline GABA level in GHB-naive animals (n = 3/time point).
Discussion
Given the lack of available therapeutic strategies for the effective treatment of GHB overdoses, it is important to elucidate the mechanisms contributing to the observed TD effects of GHB. Further understanding of these mechanisms and the relationships between systemic GHB concentrations and sedative/hypnotic effect will facilitate the development of therapeutic strategies aimed at reducing toxic effects of GHB. The present study shows the TD relationship between plasma and brain (whole and discrete brain regions) concentrations and offset of sedative/hypnotic effect. Furthermore, we identified a threshold GHB plasma exposure for its sedative/hypnotic effect (total sleep time). In addition, the absence of a direct relationship between the neurotransmitters GABA and glutamate and offset of TD effect was observed.
The results of the present study are consistent with literature reports investigating the dose- and concentration-response relationships for GHB. Consistent with our observations, Van Sassenbroek et al. (2001) showed that plasma GHB concentrations at RRR in rats treated with 300 mg/kg (5-min intravenous infusion) were 452 ± 35 μg/ml. Furthermore, a threshold for sedative/hypnotic effect was observed at 150 mg/kg i.v. GHB, consistent with the lack of effect observed with 200 mg/kg i.v. in the present study (Van Sassenbroeck et al., 2001). In addition, brain GHB concentrations have been reported to parallel plasma concentrations and show similar relationships with offset of sedative/hypnotic effect (Snead et al., 1976; Snead, 1978). The relationship between plasma and brain GHB concentrations (and total exposure) is not limited to the sedative/hypnotic effect; literature reports have illustrated exposure- and concentration-effect for electroencephalographic effects (Snead et al., 1976; Snead, 1978, 1991), time to complete fine motor task (Goodwin et al., 2009), and ataxia (Goodwin et al., 2009). Interestingly, GHB exhibits complex dose-response relationships with body temperature, suggesting there are multiple pathways contributing to the observed toxicological effects of GHB (Snead, 1990).
Previous studies have identified a number of potential mechanisms of action for GHB-induced toxicological effects, including direct binding to the GABAB receptor, metabolic conversion of GHB to GABA, or GHB-mediated stimulation of GABA release (Hechler et al., 1997; Maitre, 1997). The results of the present study extend the findings to show the relationship between GHB concentration and sedative/hypnotic effect (offset of effect and total sleep time). Carai et al. (2001) used receptor-specific antagonists SCH 50911 (GABAB receptor-specific) and 6,7,8,9-tetrahydro-5-(H)-benzocycloheptene-5-ol-4-ylideneacetic acid (NCS-382) (GHB receptor-specific) to show that the GHB-induced sedative/hypnotic effect resulted from activation of the GABAB receptor and not the GHB receptor. In addition, studies conducted in GABAB1(−/−) mice did not show any sedative effects after administration of 1000 mg/kg i.p. GHB (Kaupmann et al., 2003), suggesting that activation of the GABAB receptor is responsible for the toxicological action of GHB. Furthermore, GHB-associated hypothermia was also absent in GABAB1(−/−) mice treated with high doses of GHB (Kaupmann et al., 2003).
Although these results confirm the involvement of the GABAB receptor, they do not distinguish between direct and indirect interactions of GHB with the receptor. To investigate the effects of GHB on GABA formation (indirect interaction), we evaluated changes in GABA levels after the administration of a range of GHB doses. Literature reports indicate that 0.5 to 2% GHB is converted to GABA (Hechler et al., 1997; Gobaille et al., 1999); however, there is a delay in the formation of GABA of 160 min after the administration of 416 mg/kg i.p. GHB (Gobaille et al., 1999). The delay in formation of GABA is inconsistent with the rapid onset of pharmacological and toxicological effects of GHB after GHB administration. We observed initial decreases in total GABA levels in the frontal cortex and hippocampus at all doses, but GABA levels did not correlate with offset of the sedative/hypnotic effect in any brain region evaluated. Within the brain, GHB is metabolized to succinic acid, which is thought to inhibit the formation of GABA from glutamate. It is possible that at the high doses of GHB administered in this study [brain concentrations are approximately five times higher than used by Hechler et al. (1997)], a sufficient amount of succinic acid is formed within the brain to cause the observed decreases in GABA levels. These results suggest that at high GHB doses the formation of GABA does not contribute to the observed sedative/hypnotic effects.
Another potential indirect interaction with the GABAB receptor is the GHB-mediated stimulation of GABA release (Gobaille et al., 1999). Gobaille et al. (1999) showed alterations in GABA release in the rat frontal cortex using intracerebral microdialysis. However, in their study, GHB concentrations in the dialysate were not directly measured, so concentration-effect relationships cannot be determined for the observed responses. Linear extrapolations of GHB brain concentrations from previous studies may be inaccurate because of the nonlinearity of GHB pharmacokinetics. Interestingly, GHB had divergent effects on GABA release, dependent on the administered dose; the low dose decreased GABA release, whereas the high dose increased GABA release (Gobaille et al., 1999). In addition, decreased release of GABA after low-dose GHB has been observed in the thalamus (Banerjee and Snead, 1995). To confirm the influence of high doses of GHB on GABA release, GHB and GABA should be studied simultaneously using intracerebral microdialysis or equilibrium dialysis. These endpoints could then be further correlated to the sedative/hypnotic effect used in the present study.
An interesting finding of the present study is the lack of a relationship between sedative/hypnotic effect and CSF GHB concentrations. In contrast to the observed results in plasma and brain, there was a significant dose-dependent increase in CSF GHB concentrations at RRR. CSF concentrations have been commonly used as surrogate markers for unbound brain concentrations (Liu et al., 2006). However, studies examining transporter expression at the blood-CSF barrier (BCSFB) have shown the expression of many efflux transporters (Kusuhara and Sugiyama, 2004), suggesting that CSF concentrations may not be reflective of unbound brain concentrations in all situations (de Lange and Danhof, 2002; Lin, 2008). Drugs that are lipophilic and pass the blood-brain barrier (BBB) and BCSFB by passive diffusion typically have CSF concentrations that correlate well with unbound concentrations in the brain (de Lange and Danhof, 2002). In contrast, hydrophilic drugs are primarily transported transcellularly and may require an active transport mechanism to pass the BBB or BCSFB (de Lange and Danhof, 2002). For such drugs, CSF concentrations may not correlate with unbound concentrations in the brain because the CSF functions as a slow equilibrium compartment (Lin, 2008). It appears that the lack of correlation between sedative/hypnotic effect and GHB CSF concentrations is probably caused by its hydrophilic nature and need for an influx transporter at the BBB. The present study also showed regional differences in GHB brain concentrations, which further complicates the relationship between CSF and unbound brain concentrations. An additional possibility for the lack of a relationship between CSF and brain concentrations is the saturation of an efflux transporter on the BCSFB, which would result in increasing CSF concentrations with increasing dose, consistent with the observed results for GHB. Further studies are required to assess the influence of efflux transporters on GHB disposition. GHB is a known substrate of the monocarboxylate transporters (isoforms 1–4) (Wang et al., 2008a); however, there is a lack of information in the literature with respect to the affinity of GHB for efflux transporters. To clarify the observed relationships, we conducted a study to directly assess unbound GHB concentrations at RRR in the frontal cortex using intracerebral microdialysis. The observed relationship between frontal cortex ECF GHB concentrations and RRR confirms that CSF GHB concentrations are not an accurate marker of unbound concentrations in the brain. However, the mechanism(s) contributing to the disparity between CSF and unbound brain GHB concentrations needs to be further explored.
Current therapeutic strategies for the treatment of GHB overdose involve supportive care, such as intubation and mechanical ventilation to overcome respiratory depression (Mason and Kerns, 2002). The observed relationships between GHB concentration and sedative/hypnotic effect suggest that the use of strategies that decrease GHB concentrations in the plasma and/or brain would result in reduced sedative/hypnotic effect. One such strategy involves the inhibition of active renal reabsorption of GHB mediated by the MCTs, thereby increasing the renal clearance of GHB (Morris et al., 2005; Wang et al., 2008a). Coadministration of l-lactate (an endogenous MCT substrate) and GHB results in increased GHB renal clearance and decreased plasma concentrations (Wang et al., 2008a); however, the time course of GHB in the brain was not assessed in that study. GHB brain ECF has been shown to correlate with GHB sedative/hypnotic effects (Kapadia et al., 2007; Raybon and Boje, 2007), and future studies evaluating the effectiveness of this therapeutic strategy should evaluate this concentration-effect relationship in the presence of MCT inhibitors, such as l-lactate and luteolin. Alternatively, the use of GABAB receptor antagonists has been proposed for the treatment of acute GHB overdoses (Jensen and Mody, 2001). Administration of the GABAB receptor antagonists SCH 50911 and 3-aminopropyl-cyclohexylmethylphosphinic acid (CGP 46381) before GHB resulted in a decreased sedative/hypnotic effect (Carai et al., 2001). The advantage of the strategy using l-lactate to increase GHB renal clearance is ease of translation of this strategy to the clinic because lactate preparations are already available clinically. In addition, the potential exists for lactate to be used in combination with a GABAB antagonist as they work via distinct targets. This approach may lead to synergistic effects improving on the affects of the individual components. However, all strategies should be investigated further to optimize a detoxification strategy.
In summary, we have shown the exposure- and concentration-response relationships for GHB-induced sedative/hypnotic effect. The offset of sedative/hypnotic effect correlated to GHB concentrations in plasma and brain (whole and discrete brain regions) over a range of GHB doses. In addition, we have shown the absence of a relationship between GABA and glutamate changes at the offset of sedative/hypnotic effect. These results support the use of a therapeutic detoxification strategy that improves GHB clearance and reduces exposure, thereby decreasing the sedative effects of GHB.
This work was supported in part by the National Institutes of Health National Institute of Drug Abuse [Grant DA023223]; and a fellowship from Pfizer Global Research and Development.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
doi:10.1124/jpet.109.165381.
- GHB
- γ-hydroxybutyric acid
- GABA
- γ-aminobutyric acid
- LRR
- loss of righting reflex
- RRR
- return to righting reflex
- MCT
- monocarboxylate transporter
- TK
- toxicokinetic(s)
- CSF
- cerebrospinal fluid
- TD
- toxicodynamic(s)
- GHB-d6
- deuterated GHB
- aCSF
- artificial cerebrospinal fluid
- HPLC
- high-performance liquid chromatography
- LC/MS/MS
- liquid chromatography/tandem mass spectrometry
- AUC
- area under the curve
- ECF
- extracellular fluid
- NCS-382
- 6,7,8,9-tetrahydro-5-(H)-benzocycloheptene-5-ol-4-ylideneacetic acid
- BCSFB
- blood-CSF barrier
- BBB
- blood-brain barrier
- CGP 46381
- 3-aminopropyl-cyclohexylmethylphosphinic acid
- SCH 50911
- (2S)(+)5,5-dimethyl-2-morpholineacetic acid.
References
- Arena and Fung, 1980.Arena C, Fung HL. (1980) Absorption of sodium gamma-hydroxybutyrate and its prodrug gamma-butyrolactone: relationship between in vitro transport and in vivo absorption. J Pharm Sci 69:356–358 [DOI] [PubMed] [Google Scholar]
- Banerjee and Snead, 1995.Banerjee PK, Snead OC., 3rd (1995) Presynaptic gamma-hydroxybutyric acid (GHB) and gamma-aminobutyric acidB (GABAB) receptor-mediated release of GABA and glutamate (GLU) in rat thalamic ventrobasal nucleus (VB): a possible mechanism for the generation of absence-like seizures induced by GHB. J Pharmacol Exp Ther 273:1534–1543 [PubMed] [Google Scholar]
- Bouw and Hammarlund-Udenaes, 1998.Bouw MR, Hammarlund-Udenaes M. (1998) Methodological aspects of the use of a calibrator in in vivo microdialysis-further development of the retrodialysis method. Pharm Res 15:1673–1679 [DOI] [PubMed] [Google Scholar]
- Carai et al., 2001.Carai MA, Colombo G, Brunetti G, Melis S, Serra S, Vacca G, Mastinu S, Pistuddi AM, Solinas C, Cignarella G, et al. (2001) Role of GABA(B) receptors in the sedative/hypnotic effect of gamma-hydroxybutyric acid. Eur J Pharmacol 428:315–321 [DOI] [PubMed] [Google Scholar]
- Carter et al., 2006.Carter LP, Chen W, Coop A, Koek W, France CP. (2006) Discriminative stimulus effects of GHB and GABA(B) agonists are differentially attenuated by CGP35348. Eur J Pharmacol 538:85–93 [DOI] [PubMed] [Google Scholar]
- Carter et al., 2003.Carter LP, Flores LR, Wu H, Chen W, Unzeitig AW, Coop A, France CP. (2003) The role of GABAB receptors in the discriminative stimulus effects of gamma-hydroxybutyrate in rats: time course and antagonism studies. J Pharmacol Exp Ther 305:668–674 [DOI] [PubMed] [Google Scholar]
- Carter et al., 2009.Carter LP, Koek W, France CP. (2009) Behavioral analyses of GHB: receptor mechanisms. Pharmacol Ther 121:100–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange and Danhof, 2002.de Lange EC, Danhof M. (2002) Considerations in the use of cerebrospinal fluid pharmacokinetics to predict brain target concentrations in the clinical setting: implications of the barriers between blood and brain. Clin Pharmacokinet 41:691–703 [DOI] [PubMed] [Google Scholar]
- de Lange et al., 2000.de Lange EC, de Boer AG, Breimer DD. (2000) Methodological issues in microdialysis sampling for pharmacokinetic studies. Adv Drug Deliv Rev 45:125–148 [DOI] [PubMed] [Google Scholar]
- Ferrara et al., 1992.Ferrara SD, Zotti S, Tedeschi L, Frison G, Castagna F, Gallimberti L, Gessa GL, Palatini P. (1992) Pharmacokinetics of gamma-hydroxybutyric acid in alcohol dependent patients after single and repeated oral doses. Br J Clin Pharmacol 34:231–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung et al., 2004.Fung HL, Haas E, Raybon J, Xu J, Fung SM. (2004) Liquid chromatographic-mass spectrometric determination of endogenous gamma-hydroxybutyrate concentrations in rat brain regions and plasma. J Chromatogr B Analyt Technol Biomed Life Sci 807:287–291 [DOI] [PubMed] [Google Scholar]
- Gallimberti et al., 2000.Gallimberti L, Spella MR, Soncini CA, Gessa GL. (2000) Gamma-hydroxybutyric acid in the treatment of alcohol and heroin dependence. Alcohol 20:257–262 [DOI] [PubMed] [Google Scholar]
- Gobaille et al., 1999.Gobaille S, Hechler V, Andriamampandry C, Kemmel V, Maitre M. (1999) gamma-Hydroxybutyrate modulates synthesis and extracellular concentration of gamma-aminobutyric acid in discrete rat brain regions in vivo. J Pharmacol Exp Ther 290:303–309 [PubMed] [Google Scholar]
- Gold and Roth, 1977.Gold BI, Roth RH. (1977) Kinetics of in vivo conversion of gamma-[3H]aminobutyric acid to gamma-[3H]hydroxybutyric acid by rat brain. J Neurochem 28:1069–1073 [DOI] [PubMed] [Google Scholar]
- Goodwin et al., 2009.Goodwin AK, Brown PR, Jansen EE, Jakobs C, Gibson KM, Weerts EM. (2009) Behavioral effects and pharmacokinetics of gamma-hydroxybutyrate (GHB) precursors gamma-butyrolactone (GBL) and 1,4-butanediol (1,4-BD) in baboons. Psychopharmacology (Berl) 204:465–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hechler et al., 1997.Hechler V, Ratomponirina C, Maitre M. (1997) gamma-Hydroxybutyrate conversion into GABA induces displacement of GABAB binding that is blocked by valproate and ethosuximide. J Pharmacol Exp Ther 281:753–760 [PubMed] [Google Scholar]
- Jensen and Mody, 2001.Jensen K, Mody I. (2001) L-type Ca2+ channel-mediated short-term plasticity of GABAergic synapses. Nat Neurosci 4:975–976 [DOI] [PubMed] [Google Scholar]
- Kapadia et al., 2007.Kapadia R, Böhlke M, Maher TJ. (2007) Detection of gamma-hydroxybutyrate in striatal microdialysates following peripheral 1,4-butanediol administration in rats. Life Sci 80:1046–1050 [DOI] [PubMed] [Google Scholar]
- Kaupmann et al., 2003.Kaupmann K, Cryan JF, Wellendorph P, Mombereau C, Sansig G, Klebs K, Schmutz M, Froestl W, van der Putten H, Mosbacher J, et al. (2003) Specific gamma-hydroxybutyrate-binding sites but loss of pharmacological effects of gamma-hydroxybutyrate in GABA(B)(1)-deficient mice. Eur J Neurosci 18:2722–2730 [DOI] [PubMed] [Google Scholar]
- Koek et al., 2006.Koek W, Chen W, Mercer SL, Coop A, France CP. (2006) Discriminative stimulus effects of gamma-hydroxybutyrate: role of training dose. J Pharmacol Exp Ther 317:409–417 [DOI] [PubMed] [Google Scholar]
- Koek and France, 2008.Koek W, France CP. (2008) Cataleptic effects of gamma-hydroxybutyrate (GHB) and baclofen in mice: mediation by GABA(B) receptors, but differential enhancement by N-methyl-d-aspartate (NMDA) receptor antagonists. Psychopharmacology (Berl) 199:191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusuhara and Sugiyama, 2004.Kusuhara H, Sugiyama Y. (2004) Efflux transport systems for organic anions and cations at the blood-CSF barrier. Adv Drug Deliv Rev 56:1741–1763 [DOI] [PubMed] [Google Scholar]
- Lettieri and Fung, 1979.Lettieri JT, Fung HL. (1979) Dose-dependent pharmacokinetics and hypnotic effects of sodium gamma-hydroxybutyrate in the rat. J Pharmacol Exp Ther 208:7–11 [PubMed] [Google Scholar]
- Lin, 2008.Lin JH. (2008) CSF as a surrogate for assessing CNS exposure: an industrial perspective. Curr Drug Metab 9:46–59 [DOI] [PubMed] [Google Scholar]
- Liu et al., 2006.Liu X, Smith BJ, Chen C, Callegari E, Becker SL, Chen X, Cianfrogna J, Doran AC, Doran SD, Gibbs JP, et al. (2006) Evaluation of cerebrospinal fluid concentration and plasma free concentration as a surrogate measurement for brain free concentration. Drug Metab Dispos 34:1443–1447 [DOI] [PubMed] [Google Scholar]
- Maitre, 1997.Maitre M. (1997) The gamma-hydroxybutyrate signalling system in brain: organization and functional implications. Prog Neurobiol 51:337–361 [DOI] [PubMed] [Google Scholar]
- Mamelak, 2009.Mamelak M. (2009) Narcolepsy and depression and the neurobiology of gammahydroxybutyrate. Prog Neurobiol 89:193–219 [DOI] [PubMed] [Google Scholar]
- Mamelak et al., 1986.Mamelak M, Scharf MB, Woods M. (1986) Treatment of narcolepsy with gamma-hydroxybutyrate. A review of clinical and sleep laboratory findings. Sleep 9:285–289 [DOI] [PubMed] [Google Scholar]
- Mason and Kerns, 2002.Mason PE, Kerns WP., 2nd (2002) Gamma hydroxybutyric acid (GHB) intoxication. Acad Emerg Med 9:730–739 [DOI] [PubMed] [Google Scholar]
- Mathivet et al., 1997.Mathivet P, Bernasconi R, De Barry J, Marescaux C, Bittiger H. (1997) Binding characteristics of gamma-hydroxybutyric acid as a weak but selective GABAB receptor agonist. Eur J Pharmacol 321:67–75 [DOI] [PubMed] [Google Scholar]
- Morris et al., 2005.Morris ME, Hu K, Wang Q. (2005) Renal clearance of gamma-hydroxybutyric acid in rats: increasing renal elimination as a detoxification strategy. J Pharmacol Exp Ther 313:1194–1202 [DOI] [PubMed] [Google Scholar]
- Okun et al., 2001.Okun MS, Boothby LA, Bartfield RB, Doering PL. (2001) GHB: an important pharmacologic and clinical update. J Pharm Pharm Sci 4:167–175 [PubMed] [Google Scholar]
- Palatini et al., 1993.Palatini P, Tedeschi L, Frison G, Padrini R, Zordan R, Orlando R, Gallimberti L, Gessa GL, Ferrara SD. (1993) Dose-dependent absorption and elimination of gamma-hydroxybutyric acid in healthy volunteers. Eur J Clin Pharmacol 45:353–356 [DOI] [PubMed] [Google Scholar]
- Paxinos and Watson, 1986.Paxinos G, Watson C. (1986) The Rat Brain in Stereotaxic Coordinates, Academic Press, New York [Google Scholar]
- Raybon and Boje, 2007.Raybon JJ, Boje KM. (2007) Pharmacokinetics and pharmacodynamics of gamma-hydroxybutyric acid during tolerance in rats: effects on extracellular dopamine. J Pharmacol Exp Ther 320:1252–1260 [DOI] [PubMed] [Google Scholar]
- Smith et al., 2006.Smith MA, Gergans SR, Lyle MA. (2006) The motor-impairing effects of GABA(A) and GABA(B) agonists in gamma-hydroxybutyrate (GHB)-treated rats: cross-tolerance to baclofen but not flunitrazepam. Eur J Pharmacol 552:83–89 [DOI] [PubMed] [Google Scholar]
- Snead, 1978.Snead OC., 3rd (1978) Gamma hydroxybutyrate in the monkey. I. Electroencephalographic, behavioral, and pharmacokinetic studies. Neurology 28:636–642 [DOI] [PubMed] [Google Scholar]
- Snead, 1990.Snead OC., 3rd (1990) gamma-Hydroxybutyric acid-induced seizures bear no relation to core temperature. Epilepsia 31:253–258 [DOI] [PubMed] [Google Scholar]
- Snead, 1991.Snead OC., 3rd (1991) The gamma-hydroxybutyrate model of absence seizures: correlation of regional brain levels of gamma-hydroxybutyric acid and gamma-butyrolactone with spike wave discharges. Neuropharmacology 30:161–167 [DOI] [PubMed] [Google Scholar]
- Snead et al., 1976.Snead OC, 3rd, Yu RK, Huttenlocher PR. (1976) Gamma hydroxybutyrate. Correlation of serum and cerebrospinal fluid levels with electroencephalographic and behavioral effects. Neurology 26:51–56 [DOI] [PubMed] [Google Scholar]
- Tedeschi et al., 2003.Tedeschi L, Carai MA, Frison G, Favretto D, Colombo G, Ferrara SD, Gessa GL. (2003) Endogenous gamma-hydroxybutyric acid is in the rat, mouse and human gastrointestinal tract. Life Sci 72:2481–2488 [DOI] [PubMed] [Google Scholar]
- Van Sassenbroeck et al., 2001.Van Sassenbroeck DK, De Paepe P, Belpaire FM, Rosseel MT, Martens P, Boon PA, Buylaert WA. (2001) Relationship between gamma-hydroxybutyrate plasma concentrations and its electroencephalographic effects in the rat. J Pharm Pharmacol 53:1687–1696 [DOI] [PubMed] [Google Scholar]
- Wang et al., 2008a.Wang Q, Wang X, Morris ME. (2008a) Effects of L-lactate and D-mannitol on gamma-hydroxybutyrate toxicokinetics and toxicodynamics in rats. Drug Metab Dispos 36:2244–2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang et al., 2008b.Wang X, Wang Q, Morris ME. (2008b) Pharmacokinetic interaction between the flavonoid luteolin and gamma-hydroxybutyrate in rats: potential involvement of monocarboxylate transporters. AAPS J 10:47–55 [DOI] [PMC free article] [PubMed] [Google Scholar]