

Abstract
The low-dielectric plasma membrane provides an energy barrier hindering transmembrane movement of charged particles. The positively charged, voltage-sensing fourth transmembrane domain (S4) of voltage-gated ion channels must surmount this energy barrier to initiate channel activation, typically necessitating both membrane depolarization and interaction with membrane lipid phospho-head groups (MLPHGs). In contrast, and despite containing S4, the KCNQ1 K+ channel α subunit exhibits predominantly constitutive activation when in complexes with transmembrane β subunits, MinK-related peptide (MiRP) 1 (KCNE2) or MiRP2 (KCNE3). Here, using a 2-electrode voltage clamp and scanning mutagenesis of channels heterologously expressed in Xenopus laevis oocytes, we discovered that 2 of the 8 MiRP2 extracellular domain acidic residues (D54 and D55) are important for KCNQ1-MiRP2 constitutive activation. Double-mutant thermodynamic cycle analysis revealed energetic coupling of D54 and D55 to R237 in KCNQ1 S4 but not to 10 other native or introduced polar residues in KCNQ1 S4 and surrounding linkers. MiRP2-D54 and KCNQ1-R237 also similarly dictated susceptibility to the inhibitory effects of MLPHG hydrolysis, whereas other closely situated polar residues did not. Thus, by providing negative charge near the plasma membrane extracellular face, MiRP2 uses a lipomimetic mechanism to constitutively stabilize the activated KCNQ1 voltage sensor.—Choi, E., Abbott, G. W. A shared mechanism for lipid- and β-subunit-coordinated stabilization of the activated K+ channel voltage sensor.
Keywords: KCNQ1, KCNE3, MinK-related peptide, colonic epithelium, activation gating
Voltage-gated potassium (Kv) channels facilitate repolarization of excitable cells such as cardiac myocytes and neurons by opening in response to membrane depolarization to allow K+ ion efflux down an electrochemical gradient. Kv channels are characterized by a voltage-sensing domain containing a positively charged fourth transmembrane domain (S4) helix. On membrane depolarization, S4 undergoes a conformational shift such that one or more of its basic residues moves from intracellular to extracellular exposure and positive charges in S4 interact with membrane lipid phospho-head groups (MLPHGs) at the extracellular side of the membrane, stabilizing S4 in its activated, transmembrane conformation (1, 2). This conformational shift in S4 serves to electromechanically communicate the membrane depolarization to the channel gate, which then opens.
In contrast, background or “leak” potassium channels are relatively voltage independent and exhibit a relatively high open probability even at hyperpolarized potentials, described as “constitutive” activation. They drive cells toward the potassium equilibrium potential (EK) and are important for excitable and nonexcitable cell types. The 2-pore domain K+ channels, encoded by KCNK genes, represent the most numerous family of K+ leak channels (3). In addition, the KCNQ1 Kv channel α subunit can, uniquely within the S4 superfamily, form leak channels by interaction with some MinK-related peptides, single-transmembrane domain ancillary subunits that interact with Kv channels to alter their functional properties (4). Specifically, MinK-related peptide (MiRP) 1 (encoded by KCNE2) and MiRP2 (encoded by KCNE3) form constitutively open, K+ leak channel complexes with KCNQ1 (5, 6). MinK (KCNE1) and MiRP4 (KCNE5) coassemble with KCNQ1 but, in contrast, modify but preserve its voltage-dependent activation; MiRP3 (KCNE4) is an inhibitory subunit to KCNQ1 (7,8,9,10). KCNQ1-MiRP1 complexes form in the apical membrane of parietal cells and are required for gastric acid secretion because they permit K+ efflux to balance K+ influx through the apical H+/K+-ATPase (11,12,13,14,15). KCNQ1 and MiRP2 colocalize in the basolateral membrane of colonic crypts and are thought to coassemble there to provide a K+ ion flux to regulate cAMP-stimulated chloride secretion (5, 11, 16).
The mechanisms by which MiRP1 and MiRP2 facilitate constitutive activation of KCNQ1 are important to elucidate because of the criticality of these complexes to global human health issues. Achlorhydria is a risk factor in Helicobacter pylori infection and human gastric cancer, the second largest cancer killer worldwide (17). Deregulated chloride secretion from the colon is a factor in secretory diarrhea, which kills 1.5–2.5 million children younger than age 5 annually worldwide (18).
Residues in the transmembrane domains of MinK and MiRP2 have been shown to be highly influential in the ability of these subunits to dictate KCNQ1 activation (19, 20) by interaction with KCNQ1 S6 (21, 22). In addition, the relative charge paucity of KCNQ1 S4 is also important in determining its susceptibility to loss of voltage dependence and facilitates stabilization of KCNQ1 S4 in the activated conformation by MiRP2 (23). Therefore, here we reasoned that negatively charged (acidic) residues in MiRP2 might stabilize KCNQ1 S4 in its activated conformation to promote constitutive activation. We describe the importance to KCNQ1-MiRP2 constitutive activation of two acidic residues in the extracellular domain of MiRP2 and propose that these charges participate in electrostatic interactions conducive to S4 activation even at hyperpolarized voltages and that they share a common mechanism and S4 charge partner with anionic MLPHGs.
MATERIALS AND METHODS
Molecular biology
Human MiRP2 and KCNQ1 mutants were constructed using the QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA), sequenced in their entirety to confirm correct sequence, and then subcloned into a pBluescript-based oocyte expression vector. cRNA transcripts were produced from NotI-linearized cDNA templates using the T3 mMessage mMachine kit (Ambion, Austin, TX, USA). cRNA was quantified by spectrophotometry, and its size integrity was verified by gel electrophoresis. Defolliculated stage V and VI oocytes from Xenopus laevis (Nasco, Fort Atkinson, WI, USA) were injected with 5 ng of KCNQ1 with or without 5 ng of MiRP2 or MinK cRNA.
Electrophysiology
Whole-oocyte, 2-electrode voltage-clamp (TEVC) recordings were performed at room temperature using an OC-725C amplifier (Warner Instruments, Hamden, CT, USA) and pClamp 9 software (Molecular Devices, Sunnyvale, CA, USA), 2–4 d after cRNA injection. Oocytes were bathed in a small-volume oocyte bath (Warner Instruments) and viewed with a dissection microscope. Bath solution was 96 mM NaCl, 4 mM KCl, 1 mM MgCl2, 0.3 mM CaCl2, and 10 mM HEPES (pH 7.4) (4 mM K+/96 mM Na+ solution). TEVC pipettes were 1–2 MΩ resistance when filled with 3 M KCl. Bacillus cereus sphingomyelinase (SMase) C (Sigma-Aldrich, St. Louis, MO, USA) was applied directly to the bath solution with the flow turned off, to an estimated final concentration of 10 ng/μl, and current was recorded during 3-s pulses from −80 to 0 mV (followed by a 1-s 30-mV tail pulse) every 10 s before and during application and after washout of SMase C. Currents were corrected for rundown by digital subtraction of recordings of each channel type using a similar protocol, including flow stop/start, but without SMase addition (n=4–6/group). A previous report suggested that part of the inhibitory effect of SMase on native cAMP-stimulated K+ currents in colonic epithelium arose from ceramide activation of c-Jun NH2-terminal kinase (JNK) (24). However, here we found that the JNK inhibitor SP600125 did not significantly alter the effect of SMase on KCNQ1-MiRP2 current when injected into oocytes (n=5, data not shown), suggesting that JNK activation does not contribute significantly to the functional effects of SMase on cloned KCNQ1-MiRP2 channels expressed in oocytes, under our experimental conditions. SP600125 stock solution was prepared with 1% DMSO and injected into oocytes at least 1 h before recording, to a final estimated concentration of 25 μM SP600125/0.1% DMSO.
Gibbs free energy calculations and thermodynamic cycle analysis
To calculate the macroscopic conductance-voltage relationship, normalized tail currents were plotted vs. prepulse voltage and fitted with single Boltzmann functions according to Eq. 1:
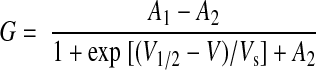 |
1 |
where G is the normalized macroscopic tail conductance, A1 is the initial conductance value when voltage is at −∞, A2 is the final conductance value when voltage is at +∞, V1/2 is the half-maximal voltage of activation, and Vs is the slope factor (Origin 6.1).
MiRP2 confers a constitutive component to KCNQ1 activation by stabilizing KCNQ1 S4 in the activated conformation (23). To determine the respective role of charged residues in MiRP2 and KCNQ1 in this S4 stabilization, the difference in Gibbs free energy (ΔG°) between the closed and constitutively open KCNQ1-MiRP2 channel was calculated here according to the Boltzmann equation:
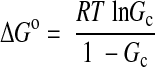 |
2 |
where R is the gas constant, T is the absolute temperature, and Gc is the fractional constitutive conductance. Gc was calculated by plotting mean normalized tail current vs. voltage and fitting with Eq. 1 where possible (Gc=A1) or otherwise Gc was assumed to equal G−140 mV, the fractional tail conductance after a −140 mV prepulse.
The change in ΔG° (ΔΔG°) caused by each mutation was calculated as
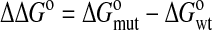 |
3 |
with se defined as
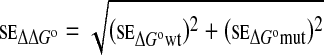 |
4 |
These values were used to determine whether mutations in MiRP2 and KCNQ1 S4 (and surrounding linkers) acted dependently on or independently of one another, using double-mutant thermodynamic cycle analysis as described previously (25, 26) to calculate the coupling energy (ΔΔG°coupling) for each pair of mutated residues:
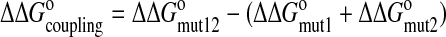 |
5 |
with se defined as
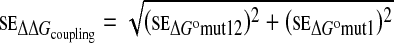 |
6 |
where ΔΔG°mut12 is the change in ΔG° for the double-mutant (mut12) compared with the wild-type channel. If the functional effects of two mutations (mut1 and mut2) are independent, ΔΔG°coupling is ∼0. If the effects are dependent, suggesting that the two mutated residues are coupled, the absolute value of ΔΔG°coupling is significantly >0.
Structural models
Homology modeling of MiRP2 (see Fig. 5F) was performed with the SwissModel program (27) using structural coordinates from NMR studies of MinK (28). Kv2.1 and associated lipid coordinates were taken from ref. 2; KCNQ1 S4 coordinates were from ref. 28. Molecular model images were prepared using DeepView (Swiss-pdb Viewer 4.0.1; http://spdbv.vital-it.ch/). Secondary structure predictions for MiRPs 1–4 (Fig. 1A) were performed using PHDsec (29), with structural assignments shown only for regions with an expected average correlation >0.69 (SUB_acc subset).
Figure 1.

MiRP2 D54 and D55 stabilize the open state of KCNQ1. A) Sequences of the predicted extracellular and transmembrane domains of the human KCNE family, aligned gap-free by their transmembrane domains. MK, MinK; M1–M4, MiRPs 1–4. Numbering is for human M2. Charged residues are highlighted. Green boxes, positioning of α-helices according to NMR structure determination for MK (28) or prediction using the PHD program for M1-M4 (29). Dashes, before start of methionine. B) Net extracellular domain charge for MinK (MK) and MiRPs 1–4 (M1–M4). C) Cartoons of voltage-dependent activation of homomeric KCNQ1 (left) vs. constitutive activation of MiRP2-KCNQ1 (right), indicating a hypothetical model for stabilization of KCNQ1 S4 in the activated state by some or all of the 8 MiRP2 extracellular domain acidic residues (blue minus symbols). Yellow, plasma membrane. For clarity, S4–S6 from only one KCNQ1 α subunit is shown. D) Exemplar current traces recorded in oocytes expressing KCNQ1 alone (Q1) or with wild-type (wt), EE44,45AA-MiRP2, DD54,55AA-MiRP2, or D54A-MiRP2 (M2) as indicated. Inset: currents were recorded by TEVC, using the standard voltage family protocol. Dashed line indicates 0 current level. E) Mean normalized conductance (G/Gmax, measured at arrow in D); channels and symbols as in D. n = 36 (Q1); 44 (+wt M2); 15 (E44,45A); 10 (D54,55A); 13 (D54A). Error bars = se. F) Mean ΔΔG° for the closed to open transition, calculated from macroscopic conductance plots as in E and for other single and multiple MiRP2 (M2) mutants with KCNQ1; n = 8–44. Error bars = se.
Figure 2.

Combined mutagenesis of charged residues in MiRP2 and KCNQ1 S4. A) Exemplar current traces recorded in oocytes coexpressing wild-type (wt) and mutant KCNQ1 and MiRP2 as indicated, recorded by TEVC using the protocol shown. Dashed line indicates 0 current level. B) Mean normalized conductance (G/Gmax, measured at arrow in A); for wild-type, D54A-MiRP2 and D55A-MiRP2 with wild-type, D242A-KCNQ1 or R237A-KCNQ1 as indicated; n = 7–44. C) Mean ΔΔG° for the closed to open transition, calculated from tail G/Gmax plots as in B and for other KCNQ1 single mutants with wild-type, D54A-MiRP2, and D55A-MiRP2 (M2); n = 7–44.
Figure 3.

Double-mutant thermodynamic cycle analysis for MiRP2 and KCNQ1 S4. A) Exemplar double-mutant thermodynamic cycles for D54A-MiRP2 with D242A-KCNQ1 or R237A-KCNQ1. B) Mean coupling energies (ΔΔG°coupling) for the closed to open transition, calculated from tail G/Gmax plots as in Fig. 2B, using ΔΔG° values from Fig. 2C and double-mutant cycles as in A; n = 7–44. Solid bars indicate values of ΔΔG°coupling > 1.5 kcal/mol. wt, wild-type.
Figure 4.

R237A-KCNQ1 activation is sensitive to the MiRP2 54,55 side chains. A) Exemplar current traces recorded in oocytes expressing R237A-KCNQ1 with DD54,55EE or NN MiRP2, using the protocol shown. Dashed line indicates 0 current level. B) Mean normalized conductance (measured at arrow in A) for currents generated by R237A-KCNQ1 alone (−) or with the MiRP2 variants shown; n = 7–46. wt, wild-type.
Figure 5.

R237-D54 interaction protects activated KCNQ1 S4 from the effects of membrane sphingomyelin hydrolysis. A) Hydrolysis of sphingomyelin by SMase. B) Cartoon of the Kv channel activated-S4-destabilizing effect of SMase. C) Exemplar traces before application and after washout of SMase for oocytes expressing KCNQ1 with MinK or MiRP2, using the voltage protocol shown. Dashed line indicates 0 current level. D, E) Rundown-normalized mean peak current at 0 mV before and during application and after washout of SMase, normalized to time 0 peak current, for oocytes expressing KCNQ1 and MiRP2 variants as indicated, using the protocol shown in C; n = 4–8/group. Red bar indicates SMase application. F) Hypothetical model indicating feasibility of R237-D54/55 interaction. Open and closed KCNQ1 S4 model coordinates are from ref. 28; MiRP2 was modeled (SwissModel) from the NMR structure of full-length MinK (28). Side chains are shown only for polar residues. MiRP2 Y60 and T80 indicate boundaries of proposed transmembrane domain (Fig. 1A). Inset: Kv2.1 S4 with nearby lipid molecules; coordinates from the crystal structure of Kv1.2/ Kv2.1 paddle chimera in lipid (2). ext, extracellular; int, intracellular.
RESULTS
The capacity for voltage-independent activation among the S4 superfamily is unique to KCNQ1 in complexes with MiRP1 (encoded by KCNE2) or MiRP2 (KCNE3) (5, 6), MinK (KCNE1) and MiRP4 (KCNE5) coassemble with KCNQ1 but preserve its voltage-dependent activation, and MiRP3 (KCNE4) inhibits KCNQ1 (7,8,9,10). MiRP2 is thought to stabilize the activated conformation of S4; it is unclear how this is achieved, but MiRP2 interacts with KCNQ1 S6 and may lie between S4 and S6 (21,22,23). Here, a sequence scan of the KCNE family revealed that the extracellular domains of MiRP1 and MiRP2 each carry a net charge of −3, compared with +2 (MinK), −1 (MiRP3), and +1 (MiRP4) (Fig. 1A, B). This suggested a model in which the greater net negative charge in MiRP1 and MiRP2 extracellular domains might energetically stabilize KCNQ1 S4 to facilitate constitutive activation in the same way that MLPHGs lower the energy barrier for S4 in Kv channel activation (Fig. 1C).
To test this model, we first calculated the difference in Gibbs free energy ΔG° (ΔΔG°) between the open and closed states for MiRP2 acidic residue-neutralized variants (Fig. 1D–F). Most MiRP2 single acidic residue alanine substitutions and the EE44,45AA double substitution had no or only minor effects on ΔΔG° (±<0.3 kcal/mol). In contrast, single-alanine substitutions at D54 and D55 produced ΔΔG° values of −0.85 and −0.7 kcal mol−1, respectively; these ΔΔG° values were not exceeded either with DD54,55AA-MiRP2 or by addition of further multiple acidic residue neutralizations. Further, ΔΔG° with charge-preserving DD54,55EE-MiRP2 was more similar to that of wild-type MiRP2 than to that of DD54,55AA-MiRP2, whereas the converse was observed with the size-preserving DD54,55NN-MiRP2 mutation (Fig. 1F). Thus, the negative charges at MiRP2 D54 and D55 specifically were each influential in KCNQ1-MiRP2 activation and acted by a common mechanism.
We next probed for energetic coupling between D54/55 and basic residues within KCNQ1 S4 and surrounding linkers, using double-mutant thermodynamic cycle analysis. We also tested Q234R- and H240R-KCNQ1, which restored Shaker-like S4 arginines where they were lacking in KCNQ1 S4. ΔΔG° was calculated using normalized tail currents, comparing the energy change associated with maximal open probability to the minimum open probability fitted from g/V plots (Fig. 2A, B). All mutant α-β subunit combinations were functional, and most produced negative ΔΔG° values; in contrast, channels formed with R231A- or R237A-KCNQ1 produced significant positive ΔΔG° values (Fig. 2C). Coupling energies (ΔΔG°coupling) for each mutant α-β subunit combination were calculated by comparing ΔΔG° values for double (α-β) mutants with the sum of ΔΔG° values for channels formed by the corresponding single α or β mutants with wild-type β or α partners (Fig. 3A). Interpretation of coupling energy analysis can yield reliable information regarding interaction of individual residues with one another, if two straightforward rules are followed: first, in general, only ΔΔG°coupling values > 1.5 kcal/mol can be interpreted as indicative of interaction. Second, even these values are only indicative of specific interactions if they are surrounded by closely positioned residues that give much lower ΔΔG°coupling values; otherwise, even high ΔΔG°coupling values may indicate whole-body helix movements or other broader conformational changes (26, 30). Here, we identified two pairings that satisfied both of these rules: D54-R237 and D55-R237 (Fig. 3B), suggesting that MiRP2 D54 and D55 each couple with KCNQ1-R237 to lower the energy barrier for the activated conformation of S4.
We previously found that homomeric R237A-KCNQ1 exhibited predominantly voltage-dependent activation but could be converted to a predominantly constitutively active channel by wild-type MiRP2 or MinK (23). This finding suggested that although there is a requirement for one of these β subunits for R237A-KCNQ1 constitutive activation, a net negative charge in the extracellular domain is not necessary, at least in the context of MinK. Here, focusing on the MiRP2-KCNQ1 D54/D55-R237 interaction, we examined the effects of coexpressing with R237A-KCNQ1 double-substituted MiRP2 variants with alanines, glutamic acids, or asparagines at positions 54 and 55 and also the pentA and hexA multiple-alanine mutants (Fig. 1F). One double mutation (DD54,55NN) showed a significant effect on constitutive current of channels formed by MiRP2 with R237A-KCNQ1, reducing it to <50% after a −120 mV prepulse (Fig. 4A, B); these findings were recapitulated using a longer hyperpolarizing prepulse to ensure channel closure before every voltage prepulse (Supplemental Fig. 1). Thus, although R237A-KCNQ1 constitutive activation does not require acidic residues at MiRP2 D54/55, it is still sensitive to the D54/55 side chain (just not in an exclusively charge- or size-specific manner), consistent with the proximity predicted from double mutant thermodynamic cycle analysis (Fig. 3).
The mechanism that these data suggest MiRP2 uses to stabilize the activated conformation of KCNQ1, i.e., provision of negative charge near the extracellular face for ion-pair formation with S4, is reminiscent of the interaction of MLPHGs with Kv2.1 and Kv1.3 S4 required for voltage-dependent activation (1). We therefore examined potential mechanistic commonalities between the two systems. The neutral B. cereus SMase C hydrolyzes the phosphodiester bond between the sphinogomyelin polar-head group and the lipid tail in X. laevis oocyte plasma membranes, resulting in removal of the charged phospholipid moiety thought to stabilize Kv2.1 and Kv1.3 S4, leaving an uncharged fatty acyl tail (ceramide) (31) (Fig. 5A, B). Here, we found that KCNQ1-MiRP2 was dramatically less sensitive than KCNQ1-MinK to the inhibitory effects of SMase (Fig. 5C, D). Strikingly, incorporation of the D54A substitution in MiRP2 reduced this protective effect of MiRP2 in channels with wild-type KCNQ1 (Fig. 5D); quantitatively similar results were observed for pentA-MiRP2 (data not shown) The requirement of KCNQ1 R237 charge for coordinating S4 stabilization by MiRP2 D54/D55 predicts that R237A substitution would have effects similar to those of D54A substitution on susceptibility of KCNQ1-MiRP2 to the effects of SMase. Accordingly, R237A substitution increased the susceptibility of KCNQ1-MiRP2 to the inhibitory effects of SMase (Fig. 5D). These data suggest commonalties in mechanism between S4 stabilization by MLPHGs and by MiRP2. We previously found that R231A-KCNQ1 channels are constitutively active even without MiRP2 (23). Here, we found that the R231A mutation partially protected channels formed with D54A-MiRP2 from the effects of SMase, resulting in less inhibition than with wild-type KCNQ1 (Fig. 5D, E). This effect was relatively specific for R231A because D242A, a KCNQ1 S4 variant that did not convert homomeric KCNQ1 to a constitutively active channel, did not have the same protective effect (Fig. 5E). Thus, constitutively stabilizing KCNQ1 S4 by R231A substitution reduced the necessity for either MLPHGs or MiRP2-D54.
DISCUSSION
Given the influence of KCNQ1 S4 charge on the effects of charge neutralization in the MiRP2 extracellular domain, the results of double-mutant cycle analysis, and the findings from SMase assays, we propose that MiRP2 D54 and D55 stabilize KCNQ1 S4 in its activated conformation, even at hyperpolarized voltages, by electrostatic interaction with KCNQ1 R237. A model of MiRP2 generated by SwissModel using the MinK NMR-derived structure (28) suggests that direct interaction between MiRP2 D54/D55 and R237 could be feasible with KCNQ1 S4 in its activated conformation (coordinates taken from ref. 28) (Fig. 5F). The stabilization of KCNQ1 S4 by D54/55 would thus be analogous to stabilization of Kv2.1 S4 by MLPHGs (1, 2) (Fig. 5F, inset). This model might require the MiRP2 extracellular domain to loop back slightly into the plasma membrane (Fig. 5F), but not necessarily, because the membrane is likely to be highly deformed around S4 (Fig. 5F, inset).
PHD (29) structure predictions (Fig. 1A) place MiRP2 D54 and D55 in an aperiodic stretch between the transmembrane domain and a membrane-distal α-helical subdomain conserved in MiRP1 and MiRP4; this membrane-distal α-helical subdomain is also apparent in the NMR-determined MinK structure (28). It is noteworthy that MiRP4 also contains a membrane-proximal pair of aspartic acid residues that could potentially interact with KCNQ1 S4 (Fig. 1A). However, the net charge of the MiRP4 extracellular domain is +1, compared with −3 for MiRP1 and MiRP2 (Fig. 1B). Given this difference, we speculate that basic residues in the membrane-distal portion of MiRP4 form salt bridges with one or both of the membrane-proximal MiRP4 aspartic acids, diminishing their availability to stabilize S4. MiRP4 contains 7 extracellular basic residues, compared with only 4 and 5 in MiRP1 and MiRP2, respectively; the acidic residue count is 6 (MiRP4), 7 (MiRP1), and 8 (MiRP2). This speculation notwithstanding, Kv α-MiRP subunit complexes are likely to be tightly-packed complexes, and the effects of point mutations in either subunit type and interpretation of these effects are therefore contextual, subject to allosteric influence, and not always open to extrapolation to the entire gene family. This last caveat is particularly likely for the extracellular domain, where sequence identity between the MiRPs is lower than that for the transmembrane and membrane-proximal intracellular domains (Fig. 1A). Thus, the role we assign here to MiRP2 extracellular domain acidic residues may not be applicable to other MiRPs for reasons other than their net charge balance.
The findings of several previous studies suggest that the capacity of MiRP2 and MiRP1 to convert KCNQ1 to a constitutively active channel is physiologically important, particularly in nonexcitable, polarized epithelial cells. Constitutively active KCNQ1-MiRP2 channels are thought to form in the basolateral membrane of colonic crypts and airway epithelia, where they must remain open at membrane potentials negative to EK to facilitate membrane hyperpolarization and thus provide a driving force for luminal Cl− ion secretion (5, 16). The KCNQ1-MiRP1 channel is essential for gastric acid secretion because it provides a luminal K+-recycling conduit that supports the function of the apical H+/K+-ATPase; deletion of kcne2, the gene that encodes MiRP1, causes achlorhydria (lack of gastric acid secretion) in mice (5, 11, 13,14,15). Recently, we also discovered that KCNQ1-MiRP1 plays an important role in thyroid hormone biosynthesis and is expressed in the basolateral membrane of thyrocytes (32). Deletion of kcne2 (MiRP1) in mice impairs thyroid I− accumulation, causing hypothyroidism, especially during gestation and in pups born to kcne2−/− dams (32). KCNQ1-MiRP1 may facilitate Na+/I− symporter activity in thyrocytes, probably by supporting the function of the basolateral Na+/K+-ATPase, but this is not yet known.
In summary, our data suggest that the unusual but physiologically and pathophysiologically important gating mechanism of KCNQ1-MiRP2 is facilitated by provision of voltage sensor stabilization by MiRP2, reminiscent of that reported for MLPHGs and S4 of Kv channels. The current findings also demonstrate that S4 charges sense not only membrane potential but also the charge status of membrane lipids themselves and suggest the intriguing possibility that MiRP2 plays a role in protecting KCNQ1 (and potentially other K+ channel α subunits) from sphingomyelinase attack in vivo.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Gianina Panaghie (Weill Medical College, Cornell University) for construction of mutant KCNQ1 cDNAs, and Kerry Purtell for expert technical assistance. G.W.A. is supported by the National Institutes of Health (R01 HL079275) and the American Heart Association (0855756D).
References
- Xu Y, Ramu Y, Lu Z. Removal of phospho-head groups of membrane lipids immobilizes voltage sensors of K+ channels. Nature. 2008;451:826–829. doi: 10.1038/nature06618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S B, Tao X, Campbell E B, MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- Goldstein S A, Bockenhauer D, O'Kelly I, Zilberberg N. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat Rev Neurosci. 2001;2:175–184. doi: 10.1038/35058574. [DOI] [PubMed] [Google Scholar]
- McCrossan Z A, Abbott G W. The MinK-related peptides. Neuropharmacology. 2004;47:787–821. doi: 10.1016/j.neuropharm.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Schroeder B C, Waldegger S, Fehr S, Bleich M, Warth R, Greger R, Jentsch T J. A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature. 2000;403:196–199. doi: 10.1038/35003200. [DOI] [PubMed] [Google Scholar]
- Tinel N, Diochot S, Borsotto M, Lazdunski M, Barhanin J. KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. EMBO J. 2000;19:6326–6330. doi: 10.1093/emboj/19.23.6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. KVLQT1 and IsK (minK) proteins associate to form the IKS cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Sanguinetti M C, Curran M E, Zou A, Shen J, Spector P S, Atkinson D L, Keating M T. Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKS potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Angelo K, Jespersen T, Grunnet M, Nielsen M S, Klaerke D A, Olesen S P. KCNE5 induces time- and voltage-dependent modulation of the KCNQ1 current. Biophys J. 2002;83:1997–2006. doi: 10.1016/S0006-3495(02)73961-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunnet M, Jespersen T, Rasmussen H B, Ljungstrom T, Jorgensen N K, Olesen S P, Klaerke D A. KCNE4 is an inhibitory subunit to the KCNQ1 channel. J Physiol. 2002;542:119–130. doi: 10.1113/jphysiol.2002.017301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedek K, Waldegger S. Colocalization of KCNQ1/KCNE channel subunits in the mouse gastrointestinal tract. Pflügers Arch. 2001;442:896–902. doi: 10.1007/s004240100609. [DOI] [PubMed] [Google Scholar]
- Grahammer F, Herling A W, Lang H J, Schmitt-Graff A, Wittekindt O H, Nitschke R, Bleich M, Barhanin J, Warth R. The cardiac K+ channel KCNQ1 is essential for gastric acid secretion. Gastroenterology. 2001;120:1363–1371. doi: 10.1053/gast.2001.24053. [DOI] [PubMed] [Google Scholar]
- Heitzmann D, Grahammer F, von Hahn T, Schmitt-Graff A, Romeo E, Nitschke R, Gerlach U, Lang H J, Verrey F, Barhanin J, Warth R. Heteromeric KCNE2/KCNQ1 potassium channels in the luminal membrane of gastric parietal cells. J Physiol. 2004;561:547–557. doi: 10.1113/jphysiol.2004.075168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrecht N W, Yakubov I, Scott D, Sachs G. Identification of the K efflux channel coupled to the gastric H-K-ATPase during acid secretion. Physiol Genomics. 2005;21:81–91. doi: 10.1152/physiolgenomics.00212.2004. [DOI] [PubMed] [Google Scholar]
- Roepke T K, Anantharam A, Kirchhoff P, Busque S M, Young J B, Geibel J P, Lerner D J, Abbott G W. The KCNE2 potassium channel ancillary subunit is essential for gastric acid secretion. J Biol Chem. 2006;281:23740–23747. doi: 10.1074/jbc.M604155200. [DOI] [PubMed] [Google Scholar]
- Bleich M, Warth R. The very small-conductance K+ channel KvLQT1 and epithelial function. Pflügers Arch. 2000;440:202–206. doi: 10.1007/s004240000257. [DOI] [PubMed] [Google Scholar]
- Ries L A G, Melbert D, Krapcho M, Stinchcomb D G, Howlader M, Horner M J, Mariotto A, Miller B A, Feuer E J, Altekruse S F, Lewis D R, Clegg L, Eisner M P, Reichman M, Edwards B K. Bethesda, MD: National Cancer Institute; SEER Cancer Statistics Review, 1975–2005. 2008 [Google Scholar]
- Kosek M, Bern C, Guerrant R L. The global burden of diarrhoeal disease, as estimated from studies published between 1992 and 2000. Bull World Health Organ. 2003;81:197–204. [PMC free article] [PubMed] [Google Scholar]
- Melman Y F, Domenech A, de la Luna S, McDonald T V. Structural determinants of KvLQT1 control by the KCNE family of proteins. J Biol Chem. 2001;276:6439–6444. doi: 10.1074/jbc.M010713200. [DOI] [PubMed] [Google Scholar]
- Melman Y F, Krumerman A, McDonald T V. A single transmembrane site in the KCNE-encoded proteins controls the specificity of KvLQT1 channel gating. J Biol Chem. 2002;277:25187–25194. doi: 10.1074/jbc.M200564200. [DOI] [PubMed] [Google Scholar]
- Melman Y F, Um S Y, Krumerman A, Kagan A, McDonald T V. KCNE1 binds to the KCNQ1 pore to regulate potassium channel activity. Neuron. 2004;42:927–937. doi: 10.1016/j.neuron.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Panaghie G, Tai K K, Abbott G W. Interaction of KCNE subunits with the KCNQ1 K+ channel pore. J Physiol. 2006;570:455–467. doi: 10.1113/jphysiol.2005.100644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaghie G, Abbott G W. The role of S4 charges in voltage-dependent and voltage-independent KCNQ1 potassium channel complexes. J Gen Physiol. 2007;129:121–133. doi: 10.1085/jgp.200609612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saslowsky D E, Tanaka N, Reddy K P, Lencer W I. Ceramide activates JNK to inhibit a cAMP-gated K+ conductance and Cl− secretion in intestinal epithelia. FASEB J. 2009;23:259–270. doi: 10.1096/fj.08-116467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H R, Kuo C C. The activation gate and gating mechanism of the NMDA receptor. J Neurosci. 2008;28:1546–1556. doi: 10.1523/JNEUROSCI.3485-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan R, Lewis J H, MacKinnon R. Spatial localization of the K+ channel selectivity filter by mutant cycle-based structure analysis. Neuron. 1996;16:131–139. doi: 10.1016/s0896-6273(00)80030-6. [DOI] [PubMed] [Google Scholar]
- Schwede T, Kopp J, Guex N, Peitsch M. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C, Tian C, Sonnichsen F D, Smith J A, Meiler J, George A L J, Vanoye C G, Kim H J, Sanders C R. Structure of KCNE1 and implications for how it modulates the KCNQ1 potassium channel. Biochemistry. 2008;47:7999–8006. doi: 10.1021/bi800875q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost B. PHD: predicting one-dimensional protein structure by profile-based neural networks. Methods Enzymol. 1996;266:525–539. doi: 10.1016/s0076-6879(96)66033-9. [DOI] [PubMed] [Google Scholar]
- Hidalgo P, MacKinnon R. Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science. 1995;268:307–310. doi: 10.1126/science.7716527. [DOI] [PubMed] [Google Scholar]
- Coll O, Morales A, Fernandez-Checa J C, Garcia-Ruiz C. Neutral sphingomyelinase-induced ceramide triggers germinal vesicle breakdown and oxidant-dependent apoptosis in Xenopus laevis oocytes. J Lipid Res. 2007;48:1924–1935. doi: 10.1194/jlr.M700069-JLR200. [DOI] [PubMed] [Google Scholar]
- Roepke T K, King E C, Reyna-Neyra A, Paroder M, Purtell K, Koba W, Fine E, Lerner D J, Carrasco N, Abbott G W. Kcne2 deletion uncovers its crucial role in thyroid hormone biosynthesis. Nat Med. 2009;15:1186–1194. doi: 10.1038/nm.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.