

Abstract
Little is known about which components of the axonal cytoskeleton might break during rapid mechanical deformation, such as occurs in traumatic brain injury. Here, we micropatterned neuronal cell cultures on silicone membranes to induce dynamic stretch exclusively of axon fascicles. After stretch, undulating distortions formed along the axons that gradually relaxed back to a straight orientation, demonstrating a delayed elastic response. Subsequently, swellings developed, leading to degeneration of almost all axons by 24 h. Stabilizing the microtubules with taxol maintained the undulating geometry after injury but greatly reduced axon degeneration. Conversely, destabilizing microtubules with nocodazole prevented undulations but greatly increased the rate of axon loss. Ultrastructural analyses of axons postinjury revealed immediate breakage and buckling of microtubules in axon undulations and progressive loss of microtubules. Collectively, these data suggest that dynamic stretch of axons induces direct mechanical failure at specific points along microtubules. This microtubule disorganization impedes normal relaxation of the axons, resulting in undulations. However, this physical damage also triggers progressive disassembly of the microtubules around the breakage points. While the disintegration of microtubules allows delayed recovery of the “normal” straight axon morphology, it comes at a great cost by interrupting axonal transport, leading to axonal swelling and degeneration.—Tang-Schomer, M. D., Patel, A. R,, Baas, P. W., Smith, D. H. Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration.
Keywords: traumatic brain injury, traumatic axonal injury, taxol, micropattern
Due to their viscoelastic nature, axons are supple during normal daily activities and can accommodate substantial mechanical deformation, such as stretching (1, 2). However, axons are thought to become brittle under very rapid or “dynamic” mechanical loading conditions, predisposing them to mechanical damage. This viscoelastic response may be critical in traumatic brain injury (TBI), where axons in the white matter are exposed to high strain rates as the brain is rapidly deformed (3, 4). Indeed, diffuse axonal injury (DAI), identified as damage or dysfunction of axons throughout the white matter, is the most common and important pathology in TBI (5,6,7,8). Although dynamic deformation of axons during trauma rarely leads to primary disconnection (7,8,9), immediate structural damage has been found, manifested as delayed elasticity (10). In turn, this primary damage to unidentified cytoskeletal components may play an important role in the well-characterized interruption of axonal transport and protein accumulation in swellings that lead to secondary disconnection and degeneration following traumatic axonal injury (11, 12).
While there has been extensive characterization of cytoskeletal disruption after axon trauma, it has been difficult to determine whether the damage is directly due to mechanical disruption or results from biochemical processing (13, 14). For example, while a reduced spacing or compaction of neurofilaments has been found shortly after injury, this is thought to be due to proteolysis or dephosphorylation of the neurofilament sidearms (15, 16). Likewise, a reduction in the number of axon microtubules has also been found after mechanical trauma (14, 15, 17). However, this loss was observed >1 h after injury and thought to be mediated by posttraumatic influx of calcium (14, 15). Nonetheless, the structure and stability of microtubules may render them especially vulnerable to mechanical disruption.
Representing the stiffest structural components of axons (18), microtubules are composed of regions of either stable or dynamic tubulin polymers (19). Dynamic regions inherently display instability during the exchange of tubulin with a diffusive monomer pool, allowing rapid changes in the length of individual microtubules during axonal remodeling, such as in development and regeneration (20). However, this inherent instability of microtubules may prove a liability on mechanical trauma to axons. Notably, microtubules along the middle lengths of axons are rather stable compared with those in the terminal regions and can resist depolymerization from calcium or pharmacological stimulations (21).
In this study, we developed a micropatterned cell culture system to examine the immediate and evolving effects of dynamic stretch applied exclusively to axons. This patterned culture was comprised of laddered parallel fascicles of axons (2 mm in length) spanning 2 discrete populations of cortical neurons. With a silicone membrane on the bottom of the culture wells, dynamic stretch could be induced exclusively to the axon-only region using loading conditions akin to in vivo traumatic axonal injury. We found the first direct evidence that microtubules represent one of the “weakest links” of the cytoskeleton in axons undergoing rapid mechanical stretch. Furthermore, traumatic microtubule damage appears to trigger a unique sequence of events that can end in axonal degeneration.
MATERIALS AND METHODS
Isolated axon culture in micropatterned channels
The isolated axon culture system consists of a molded elastomeric stamp placed against a deformable silicone membrane (0.005-inch thickness; Specialty Manufacturing, St. Paul, MN, USA; Fig. 1). Microchannels (0.4×2 mm×0.2 mm deep) were fabricated onto the surface of the stamp by casting polydimethylsiloxane (Sylgard 184; Dow Corning, Midland, MI, USA) from patterned lithographic masters, as described previously (22). Primary cortical neurons from embryonic day 18 Sprague-Dawley rats (Charles River, Wilmington, MA, USA) were plated on the micropatterned culture platform precoated with 20 μg/ml poly-l-lysine. At the time of cell plating, the microchannels were filled with sterile water; this strategy was found to effectively prevent cell bodies from entering the channels. The cells were plated at a density of 375,000–500,000 cells/cm2 and cultured in NeuroBasal medium (Invitrogen, Carlsbad, CA, USA) supplemented with B-27 neural supplement (Invitrogen) and 5% fetal bovine serum (HyClone, Logan, UT, USA). Over 24 h, the channels were sufficiently perfused with culture medium and provided directional cues for enhanced axon growth. By 3–4 d in culture, axon processes started to enter the microchannels, and by 7–10 d they had traversed the 2-mm length to integrate with neurons on the other side.
Figure 1.

Dynamic mechanical stretch of isolated cortical axons. A, B) Schematic illustration of axonal stretch injury model. Axon-only region of the elastic membrane overlaps with a 2- × 15-mm slit at the bottom of an airtight chamber. A controlled air pulse deflects the elastic membrane downward, thus inducing a tensile elongation exclusively of axons. C) Phase-contrast photomicrograph of the axon-only region, formed by a silicone stamp that creates microchannels permitting only axon outgrowth across the channels. D) Fluorescence microscopic confirmation that the neurites in the microchannels were axons demonstrated by immunoreactivity to neurofilament protein (NF, green), while immunoreactivity to microtubule-associated protein 2 (MAP2, red), a specific marker for the dendrites, was found only outside the channels. Scale bar = 100 μm.
Chemicals
Experiments were performed at 10–12 d in vitro in controlled saline solution (CSS; 120 mM NaCl, 5.4 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 15 mM glucose, and 25 mM HEPES, pH 7.4). The wells were treated 30 min before stretch with either CSS, taxol (0.01–1 μM; Sigma, Ronkonkoma, NY, USA), a microtubule stabilizer, or nocodazole (0.01–50 μM), a microtubule destabilizer by competing for free tubulin (23). Other than combinatorial drug treatments when nocodazole dosages exceeded 1 μM with the presence of taxol, the dosages used for single-drug treatment were magnitudes smaller (≤1 μM) compared with those known to disrupt microtubules (10–100 μM; ref. 24).
In vitro dynamic stretch injury of axons
The culture wells were placed in a sealed device in an orientation that placed the region containing the cultured axons directly above the machined 2- × 15-mm slit of a bottom metal plate (Fig. 1A, B), as described previously (10, 25, 26). A controlled air pulse was used to rapidly change the pressure in the chamber and deflect downward the portion of the substrate containing the cultured axons. This deflection induced a tensile elongation of the cultured axons. The measurement of nominal uniaxial strain (ε) is calculated by determining the centerline membrane deflection (δ) relative to the slit width (w) and substituting into the geometric relationship:
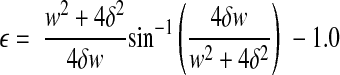 |
Membrane deflection δ was determined by z-distance change of the center of the focal plane at a specific chamber pressure from that of 0 pressure (the default ambient pressure). Once the pressure-strain relationship was determined, the pressure values were applied in a controlled fashion to axonal cultures for desired strain levels.
Live-cell imaging and morphological analysis
Fluo-4 AM ester (2 μM; Molecular Probes, Carlsbad, CA, USA) was used to visualize live axons (25). Fluorescence microscopy was performed on a Nikon TE300 inverted microscope with a CCD camera (Cooke Corporation, Romulus, MI, USA). Fluorescence images were collected using the Camware v2.15 software. Three to 5 microscopic fields (150×113 μm with average ∼50 axons) corresponding to different, nonoverlapping microchannels (total 25 microchannels/well) in the axon-only region were randomly chosen. Images were acquired with an ×60 oil lens (at a final view of ×600) and analyzed with NIH ImageJ (U.S. National Institutes of Health, Bethesda, MD, USA). For each observation, ≥5 independent trials (wells) were performed and analyzed with the Student’s t test.
We used NIH ImageJ to quantify axonal alterations. The contour of axonal process was traced, and the ratio of poststretch to prestretch length was determined. Undulations of single axons were easily identified as bending distortions with a width of 5–8 μm and amplitude of 3–6 μm. Length changes of individual undulations were similarly quantified as that of the total length of the axon. The density of undulations was expressed as number of undulations per axon of ∼150 μm length in a microscopic field (at a final view of ×600).
Immunocytochemical examination
Fixed cells (4% paraformaldehyde, 20 min) were permeabilized (0.1% Triton X-100 in phosphate saline solution, 20 min) and incubated in a primary antibody solution (1:500 to 1:1000 dilution) overnight at 4°C, followed with a secondary antibody (1:200 to 1:500 dilution) incubation for 1 h at room temperature, and then they were extensively washed. Fluorescence images were acquired with an ×60 oil-objective lens.
Monoclonal microtubule-associated protein 2 (MAP2; 1:1000; Sigma) and rabbit polyclonal anti-neurofilament 68 kDa (NF; 1:500; Covance Research Products, Princeton, NJ, USA) antibodies were used to differentiate cell bodies and axons, respectively. Monoclonal anti-βIII-tubulin antibody (1:500; Sigma) and goat anti-mouse Alexa 488 and goat anti-rabbit Alexa 564 (1:200; Invitrogen) secondary antibodies were used.
Electron microscopic examination
We used standard transmission electron microscopic (TEM) techniques. Specifically, cells were fixed with a mixture of 2% paraformaldehyde and 2.5% glutaraldehyde overnight, washed with 0.1 M sodium cacodylate buffer in three 10-min rinses, postfixed with 2% osmium plus 1.5% potassium ferricyanide, followed with 3 buffer washes and 2 diH2O washes. To enhance the intensity of cell membranes, cells were stained with 2% uranyl acetate for 20 min, followed with 2 diH2O washes. Serial dehydration was performed with 3 min incubation in 50–100% EtOH. Cells were embedded in resin, separated from the silicone membrane, and reembedded. For each sample, both longitudinal and cross-sections of 60 nm were obtained (imaged at ×2–9×104). Axons in each cross-section (covering the area of 1 microchannel) were examined for the presence of a typical 24-nm-wide circular profile of microtubules.
RESULTS
Isolated axonal culture in micropatterned channels
In the present study, dynamic tensile elongation was applied exclusively to integrated axons spanning 2 populations of cortical neurons (Fig. 1A, B). As a new adaptation from previous studies (10, 25, 26), micropatterned channels were incorporated on the elastic substrate via soft lithography techniques (22). Compartmentalization of axon outgrowth was confined within these channels, producing 2-mm regions of axons arranged in a uniaxial fashion (Fig. 1C). Confirmation that the processes in the gap region of the microchannels contained only axons was demonstrated by their immunoreactivity to neurofilament protein, while immunoreactivity for the dendritic marker MAP-2 was only found outside of the channels in the cell body region (Fig. 1D).
Evidence of primary mechanical damage to the axonal cytoskeleton after dynamic stretch injury
As the elastic substrate was extended under controlled mechanical loading conditions, axons remained attached and experienced strains that corresponded to the underlying substrate. Under slow loading rates of stretch (with a duration of minutes), the axons easily tolerated stretching up to twice their original length (a strain of 100%) and returned back to their prestretch length with no evidence of damage. Under dynamic loading conditions, i.e., with strain pulse duration of <50 ms, axons were consistently found to display multiple regions of undulating distortions along their length immediately after stretch, consistent with our previous observations (10, 25, 26). To examine whether these undulations were a direct response to the dynamic tensile stretch, we characterized axonal length changes at a fixed strain rate of 44 s−1 and varying strain levels (corresponding to durations of 1–30 ms; Fig. 2). The magnitude and density of these undulations increased in proportion to an increase in the mechanical loading strain (Fig. 2B, C). For example, at a strain of 75%, the total poststretch length of axons in the gap region increased by ∼8%, with a remarkable ∼65% localized length increase in the region of a typical undulation.
Figure 2.

Degree of axonal undulation is proportional to the applied mechanical strain. A) Fluorescence photomicrograph of axons loaded with fluo-4 displaying multiple undulations (arrow) immediately after stretch (1–30 ms duration, with a fixed strain rate of 44 s−1), the extent of which is proportional to the applied mechanical strain levels. Scale bars = 5 μm. B) Quantitative analysis of total length of axons (normalized to prestretch lengths): 30% strain: CSS (see Materials and Methods), n = 18, taxol, n = 7; 50% strain: CSS, n = 18, taxol, n = 6; 75% strain: CSS, n = 15, taxol, n = 18. n = total number of axons examined. C) Quantitative analysis of undulation densities, defined as number of undulations per axon process of ∼150 μm length: 30% strain: CSS, n = 6, taxol, n = 3; 50% strain: CSS, n = 6, taxol, n = 3; 75% strain: CSS, n = 5, taxol, n = 6. n = total number of microscopic fields examined (field of 150 μm × 113 μm with average ∼50 axons). ++P < 0.01 vs. control CSS; Student’s t test.
Linking microtubule stability with morphological alterations of axons after dynamic stretch injury
To examine the involvement of microtubules in stretch-induced axon shape changes, the axons were treated with taxol or nocodazole at 30 min before stretch. Notably, neither drug treatments (0.01–1 μM in CSS) utilized induced overt morphological changes in noninjured axons (Fig. 2A and Fig. 3A).
Figure 3.

Microtubule destabilization and axon relaxation after dynamic stretch injury. A) Phase-contrast and fluorescence (fluo-4) photomicrographs demonstrating that axons without treatment (CSS) displayed multiple undulations after stretch and gradually resumed their original straight orientation within 40 min. However, axons pretreated with taxol (T; 1 μM) maintained their undulations for at least 3 h, while removal of taxol at 40 min poststretch allowed relaxation of axonal undulations by ∼3 h postinjury. In contrast, axons pretreated with nocodazole (N; 0.1 μM) relaxed to their prestretch orientation immediately after injury. Scale bars = 5 μm. B) Quantitative analysis of the length of individual undulations over time demonstrated that only pretreatment with taxol (1 μM) maintained the undulations, while a lower pretreatment dose (10 nM) or poststretch treatment (1 μM) had no effect. Values of data points are listed in Supplemental Table S1.
For axons treated with taxol and subsequently stretched, there were increases of undulation length and density at all strain levels tested, compared with nontreated stretched axons (Fig. 2B, C). At 30 and 50% levels, the differences were statistically significant. These increases appeared to be related to a greatly reduced rate of relaxation of the axons compared with untreated axons (Fig. 3). Indeed, the taxol-treated axons were essentially fixed in the dysmorphic undulating geometry for at least 3 h after dynamic stretch at the highest levels of strain (75% strain). These effects were dose dependent and were not found with the lowest dose of 10 nM. In addition, the effects were dependent on preinjury treatment, since no postinjury dose of taxol (1–10 μM) blocked relaxation of undulations.
Preinjury treatment with nocodazole (0.1 μM) had the opposite effect of taxol treatment. There was an almost immediate relaxation of nocodazole-treated axons, with <2 undulations noted among 50 axon processes of a length of ∼150 μm (width of a microscopic view), compared with ∼60 undulations/50 axon processes in CSS after dynamic stretch at 75% strain (Fig. 4). When the combination treatment of nocodazole and taxol was examined, there was a dose-dependent mutually opposing effect on undulation relaxation. For example, 50 μM nocodazole and 1 μM taxol resulted in ∼14 undulations/50 axon processes, compared with ∼45 undulations/50 axon processes with 1 μM nocodazole plus 1 μM taxol treatment, and ∼70 undulations/50 axon processes with 1 μM taxol treatment only (Fig. 4). Despite the different time course of undulation relaxation, the initial length distention after stretch at 75% strain was not significantly different, i.e., with a ratio of ∼1.6 (Fig. 3).
Figure 4.

Quantitative analysis of undulation densities. CSS (see Materials and Methods), n = 5; nocodazole (N; 0.1 μM), n = 9; N (1 μM), n = 3; taxol (T; 1 μM), n = 3; T (1 μM) + N (1 μM), n = 3; T (1 μM) + N (50 μM), n = 8. n = total number of microscopic fields examined (field of 150×113 μm with average ∼50 axons). ++P < 0.01 vs. control CSS; Student’s t test.
Role of microtubule destabilization in the progression to axonal degeneration after dynamic stretch injury
We further examined the involvement of microtubules in trauma-induced axonal degeneration after dynamic stretch at 75% strain. Although the straight geometry of the stretched axons was gradually restored, these axons were mechanically injured, manifested by development of focal swellings in hours and eventual degeneration (Fig. 5). With the use of fluorescence live imaging of axons loaded with fluo-4, axonal swellings were found to develop by 1 h and gradually enlarged in size within 3 h (Fig. 5A). Simultaneous phase-contrast imaging confirmed that these swellings were volumetric enlargements. In addition, the same axonal swellings identified with live imaging were found to be associated with tubulin accumulation by immunocytochemical staining (Fig. 5B).
Figure 5.

Microtubule destabilization and axon degeneration after dynamic stretch injury. A) Simultaneous live imaging of fluo-4 fluorescence (top) and phase-contrast (bottom) photomicrographs show development of axonal swellings after stretch injury by 1 h and gradually enlarged in size. B) Axonal swellings shown with live imaging (left) correspond with β-tubulin immunoreactivity of the same axons following fixation (right). C) β-Tubulin immunostaining of fixed cultures shows axon degeneration over hours. In control injured axons (CSS), there was a progression of axonal swellings (arrows) leading to a complete loss of axons by 24 h. However, pretreatment with taxol (1 μM) substantially reduced the appearance of swellings after injury and prevented degeneration, with 50% of the axons remaining intact by 24 h postinjury. In contrast, pretreatment with nocodazole (0.1 μM) rapidly accelerated degeneration, with almost complete loss of axons by 3 h. Each photomicrograph represents ≥5 independent cultures. Scale bars = 5 μm.
Taxol treatment was found to greatly delay axonal degeneration after stretch injury (Fig. 5C). Specifically, axons pretreated with taxol (1 μM) remained mostly intact with little swelling at 3 h after injury, compared with extensive axonal swellings in untreated axons. Remarkably, by 24 h, >50% of axons pretreated with taxol still remained intact, compared with a complete loss of structure of injured axons in CSS. In addition, pretreated axons with immediate removal of taxol were also largely preserved at 24 h postinjury. In contrast, most of the axons pretreated with nocodazole (0.1 μM) were lost within 1–3 h postinjury. Notably, neither drug treatment induced detectable loss of noninjured axons at 24 h postinjury.
Ultrastructural analysis of axons reveals dynamic stretch injury-induced microtubule breaking and progressive loss
TEM examination of the longitudinal sections (60-nm thick) of axons demonstrated ultrastructural alterations of the axonal microtubule lattice induced by dynamic stretch injury (Fig. 6). The microtubules were easily identified as dark 24-nm-wide filamentous structures that traversed the main axis of an axon. Immediately after injury (∼1 min), microtubules appeared to remain unchanged in straight segments. However, major alterations of microtubule configuration were found in the undulations (Fig. 6A). Specifically, the continuity of microtubules was lost at the peak of the undulations. At the breakage points, microtubules displayed conspicuous free ends that appeared frayed, similar to a shorting microtubule undergoing catastrophic depolymerization (27). Some portions of microtubules were twisted with a spiral configuration. Different degrees of axon undulations from multiple axons demonstrated progressive phases of undulation relaxation and acute formation of axonal swellings immediately after stretch injury (Fig. 6B). Collapse of the undulation appeared to be associated with localized microtubule loss within the distortions. In particular, few microtubules remained continuous within the swelling, in a sharp contrast to the apparently intact microtubule lengths in the adjacent straight axonal segment.
Figure 6.

Microtubule breaking immediately after dynamic stretch injury. TEM (×2–9 × 104) of 60-nm-thick longitudinal sections of axons demonstrate microtubule disruptions observed immediately after injury. A) In straight segments of axons, microtubules traversed the main axis of axons without regions of interruptions. Within undulations, microtubules demonstrated uniformly open spacing at the peak, displaying frayed free ends (asterisk). Remaining portions of microtubules were disorganized and twisted, with a curling configuration (arrow). B) Demonstration of progressive phases of undulation relaxation and acute formation of axonal swellings after stretch injury. C) Taxol (1 μM)-pretreated axons demonstrated similar microtubule disruptions in axons as control injured axons immediately after stretch injury (top). In contrast, nocodazole (Noc; 0.1 μM)-pretreated axons demonstrated almost complete loss of microtubules within the undulation (bottom). Scale bars = 500 nm.
Similar breakage of microtubules in undulations was found with drug-treated axons postinjury (Fig. 6C). Immediately after stretch injury, there were no notable changes of microtubule breakage ratio between taxol (1 μM)-treated and control injured axons. This ultrastructural evidence supported previous observation of the similar undulation magnitude and density at 75% strain level between the 2 groups (Figs. 2 and 4), suggesting that 1 μM taxol had no effect on immediate mechanical breakage of microtubules. In contrast, there was a great loss of microtubules within undulations of nocodazole (0.1 μM)-treated axons. This ultrastructural evidence provided the explanation for the rapid relaxation of axon undulation with nocodazole treatment (Fig. 3).
Quantitative TEM analysis of axon cross-sections revealed progressive loss of microtubules in injured axons and its modulation by taxol or nocodazole treatment (Fig. 7). More than 99% taxol pretreated axons survived, compared with ∼67% nontreated axons and ∼53% nocodazole pretreated axons, immediately after injury. By 3 h, 90% taxol pretreated axons still contained their normal complement of microtubules, compared with ∼58% nontreated and ∼30% nocodazole-pretreated axons.
Figure 7.

Progressive loss of microtubules after dynamic stretch injury. A) TEM cross-sections (60-nm thickness, imaged at ×2–9×104) of axons demonstrate progressive microtubule loss. Axons without injury (sham), axons in CSS (control), or axons pretreated with taxol (1 μM) or nocodazole (0.1 μM) had similar numbers of microtubules (arrow). After stretch injury, control axons gradually diminished their microtubules. However, axons pretreated with taxol maintained their number of microtubules over time, and axons pretreated with nocodazole accelerated their loss of microtubules. B) Quantitative analysis showed relative proportions of surviving axons with cross-sections containing ≥1 microtubule. Refer to numeric values in Supplemental Table S2. Scale bars = 500 nm.
DISCUSSION
Widespread axon swelling and degeneration are the hallmarks of pathological consequence of traumatic brain injury. However, little is known about how direct mechanical trauma induces these changes. Using real-world mechanical loading conditions to induce dynamic stretch injury of isolated axons, we found that primary microtubule damage plays a central role in evolving axonal pathology after trauma. Indeed, our data show that dynamic stretch ruptures axonal microtubules at specific points triggering depolymerization of the microtubules. With immediate localized microtubule disconnection triggering a subsequent total loss of microtubules, axon transport is progressively derailed, leading to mounting swelling, protein accumulation, and, ultimately, axon degeneration.
Using an adaptation of micropatterned system, we established pristine 2-mm-long axon tracks that span 2 populations of cortical neurons. The design of laddered microchannels provided directional cues for the growth of parallel fascicles of axons while restricting entry of neurons. This geometry ensured the capacity for uniaxial stretch of the axons, which is thought to be an important aspect of axonal trauma in vivo (28).
By applying tensile stretching exclusively to axons, we found that the rate of mechanical loading plays an important role in the extent of injury. Under slow loading rates of stretch, with duration of minutes, the axons could easily tolerate extending to twice their original length. However, under dynamic loading conditions with strain pulse duration of less than <50 ms, multiple pathological changes were observed. Notably, these injury parameters are similar to the mechanical forces known to induce DAI in humans and animal models during head rotational acceleration (3, 4, 28).
A major morphological change observed immediately following dynamic stretch of axons was the formation of undulations along the axon lengths. As an induced form of “delayed elasticity,” these undulations occurred during 20-ms stretch and only gradually relaxed back to their prestretch length and straight morphology within ∼40 min poststretch. It has been found previously that neurites demonstrate <1 min lag during normal viscoelastic elongation/retraction (1, 29). However, the prolonged delayed recovery of axon shape in the present study demonstrates that there was an immediate major mechanical disruption of the axonal cytoskeleton during stretch that restricted normally rapid relaxation. Although the straight geometry of the stretch-injured axons was gradually restored, accumulations of transported proteins subsequently formed in swellings (10), similar in appearance to axonal pathology found in DAI (7, 30). Within hours, the axons began to degenerate and almost none remained by 24 h postinjury.
By manipulating microtubule stability, we found that microtubule damage and disorganization are the primary sources for the delayed elasticity and subsequent degeneration of axons after dynamic stretch injury. Application of the microtubule-stabilizing agent taxol before injury greatly reduced the rate of relaxation of the axons after injury, essentially fixing the dysmorphic undulating geometry in place for several hours. Paradoxically, despite inhibiting restoration of apparently “normal” straight morphology, taxol treatment greatly reduced axon degeneration. By 24 h after injury, taxol treatment preserved more than half of the axons compared with almost complete obliteration of axons in nontreated injured cultures. This apparent protective stabilizing effect was related to the availability of taxol very close to the time of injury, since application only 5 min after injury had little effect on slowing relaxation of the axon undulations. Notably, acute application of taxol before injury is sufficient for the protective effect, since pretreated axons with immediate removal of taxol were still largely preserved at 24 h postinjury. These dramatic protective effects of taxol treatment suggest a novel therapeutic strategy for this class of compounds for traumatic axonal injury. However, it will be important to establish whether there is a postinjury therapeutic window of opportunity for microtubule stabilization to prevent subsequent depolymerization.
Compared with taxol treatment, a completely opposite paradoxical effect was found with the application of nocodazole, an agent that promotes microtubule instability by competing for free tubulin (23). Preinjury nocodazole treatment resulted in an extremely rapid relaxation of axonal shape to a straight geometry within minutes after injury. Despite this rapid restoration of a normal-appearing shape, nocodazole-treated axons subsequently displayed greatly accelerated swelling and degeneration compared with nontreated injured axons. In addition, the combination of taxol and nocodazole created a dosage-dependent opposing effect on undulations. Similar to taxol, the effect of nocodazole solely depended on the availability of the drug at the time of injury. In contrast, nocodazole applied at submicromolar concentrations used in this study had a minimal effect on microtubules of uninjured axons for ≥24 h. Therefore, the action of nocodazole depended on dynamic stretch injury-induced damage, i.e., the normally stable microtubules along the axon length became much more vulnerable and succumbed to drug-promoted destabilization.
To directly identify ultrastructural alterations of the axonal microtubule lattice induced by stretch injury, TEM was used to examine longitudinal sections of axons. Immediately after injury, the straight regions of the axons displayed the normal parallel configuration of microtubules with no interruptions. However, open spacing of individual microtubules was uniformly found at the peaks of the undulations. This spacing was bordered by liberated free ends of microtubules that appeared frayed and/or curled at the disconnection points. In addition, rather than a parallel arrangement, the collective microtubule polymers appeared twisted and tortuous, suggesting severe disruption near the breakage points.
The highly specific structural damage within axon undulations suggests primary mechanical failure or rupture at specific points along individual microtubules at the time of injury. Considering that microtubules are the most rigid structural component of the axonal cytoskeleton, broken ends of microtubules may fail to realign and adjacent tortuous segments may not be able to freely slide. This could create a physical blockade that impedes normal axon relaxation to the prestretch length, resulting in undulations. Notably, microtubule stabilization and presumed reduction in flexural rigidity with preinjury application of taxol (31,32,33) failed to mitigate breakage of the microtubules, likely due to the dynamic nature of the deformation.
A potential process that both released the block to relaxation of the axons and triggered subsequent degeneration after injury was found with temporal ultrastructural TEM examination of axon cross-sections. Without treatment, the normal complement of microtubules, seen as ∼24-nm-wide circular profiles, was found to gradually diminish after injury, with almost no profiles found remaining by 3 h. This time course coincides with the relaxation of the undulations and subsequent development of axonal swellings. However, by stabilizing the microtubules with preinjury treatment of taxol, the number of microtubules was maintained over time, with only a slight reduction in their number by 3 h postinjury. Conversely, preinjury treatment with nocodazole (at a nondisruptive, very low dosage of 0.1 μM) greatly accelerated the loss of microtubules, which were almost completely undetectable immediately after injury.
These observations suggest that as the microtubules were essentially dissolved after trauma, their buttressing of the undulated regions along injured axons was released, resulting in relaxation to the straight orientation. While the restoration of normal straight morphology outwardly appeared as a type of recovery, it came at a great cost. With the progressive loss of microtubules, axon transport was essentially derailed, leading to unabated swelling and degeneration.
The present data also demonstrated that not all microtubules were disconnected in one region after trauma. This suggests a process whereby protein transport is interrupted primarily at the individual microtubule level, allowing protein accumulation to occur at multiple sites along injured axons. This same process may also account for the similarly appearing protein accumulation within periodic axonal varicosities along damaged axons that is well characterized after traumatic brain injury (30, 34,35,36,37).
The mechanism triggering gradual microtubule depolymerization after trauma remains unclear. However, it is curious that the traumatic breaks in the microtubules appear similar to the protofilament curling, previously shown to be promoted by hydrolysis of the tubulin-associated GTP, signifying microtubules undergoing active disassembly (27). As such, traumatic microtubule disconnection may trigger a unique type of “catastrophe” or conversion of intrinsic microtubule instability to depolymerization (38, 39). Specifically, breakage points essentially create unstable GDP-tubulin ends that are prone to depolymerization (40), as is classically found in the switch from growth to shrinkage of sprouting axons (41). TEM analysis of randomly selected regions of axons shortly after injury showed that individual microtubules were lost while others remained intact, suggesting that depolymerization was initially limited to regions of mechanical damage and not a generalized effect. Yet, as opposed to axon sprouting (42), no rescue of depolymerization of microtubules was observed after axon trauma resulting in the total loss of microtubules, as has been observed in vivo (14). It is unclear why the microtubules did not ultimately repolymerize following trauma. Future studies are needed to examine whether it is direct damage to the lattice of the stable microtubules that limits their ability to repolymerize or if reassembly of the lost polymer is suppressed due to as yet unidentified effects on microtubule-related proteins.
The observation of microtubule breaking and irreversible depolymerization under a dynamic regime may have broad implications for microtubule and cellular biomechanics. It is interesting that microtubule rupture depends on strain rate but not the strain levels. As microtubules must slide against each other in the lattice during stretching to accommodate the strain, dependence on the strain rate may reflect mechanical sensitivity of the lateral bonding between adjacent polymers and/or microtubule-associated proteins (43).
Collectively, the present results suggest that microtubules are weak links of the axonal cytoskeleton during traumatic axonal injury. Dynamic deformation of axons induces primary mechanical rupture and damage to the microtubules, which in turn triggers delayed elasticity of axons and progressive microtubule disassembly. As a consequence, axonal transport is impaired, leading to protein accumulation and ultimately, axon degeneration. The mechanical induction of this unique microtubule disassembly process distinguishes traumatic axonal injury from other forms of axon injury such as those induced by physiological changes like hypoxia and ischemia.
Supplementary Material
Acknowledgments
The authors thank Tracy Yuen, Catherine von Reyn, and Matthew Weingard from the Center for Brain Injury and Repair, University of Pennsylvania, for technical assistance; Ray Meade from the Biomedical Imaging Core, University of Pennsylvania, for electron microscopic assistance; and Dr. David Meaney from the Department of Bioengineering, University of Pennsylvania, for insightful discussions. This work is supported by NIH grants NS-056202, NS-038104, and NS-048949 (to D.H.S.).
References
- Dennerll T J, Lamoureux P, Buxbaum R E, Heidemann S R. The cytomechanics of axonal elongation and retraction. J Cell Biol. 1989;109:3073–3083. doi: 10.1083/jcb.109.6.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund M, Jorgensen E M, Bastiani M J. Axons break in animals lacking beta-spectrin. J Cell Biol. 2007;176:269–275. doi: 10.1083/jcb.200611117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennarelli T A, Thibault L E, Adams J H, Graham D I, Thompson C J, Marcincin R P. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol. 1982;12:564–574. doi: 10.1002/ana.410120611. [DOI] [PubMed] [Google Scholar]
- Smith D H, Meaney D F. Axonal damage in traumatic brain injury. Neuroscientist. 2000;6:483–495. [Google Scholar]
- Strich S J. Diffuse degeneration of the cerebral white matter in severe dementia following head injury. J Neurol Neurosurg Psychiatr. 1956;19:163–185. doi: 10.1136/jnnp.19.3.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J H, Doyle D, Ford I, Gennarelli T A, Graham D I, McLellan D R. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- Christman C W, Grady M S, Walker S A, Holloway K L, Povlishock J T. Ultrastructural studies of diffuse axonal injury in humans. J Neurotrauma. 1994;11:173–186. doi: 10.1089/neu.1994.11.173. [DOI] [PubMed] [Google Scholar]
- Gentleman S M, Roberts G W, Gennarelli T A, Maxwell W L, Adams J H, Kerr S, Graham D I. Axonal injury: a universal consequence of fatal closed head injury? Acta Neuropathol. 1995;89:537–543. doi: 10.1007/BF00571509. [DOI] [PubMed] [Google Scholar]
- Povlishock J T, Becker D P, Cheng C L, Vaughan G W. Axonal change in minor head injury. J Neuropathol Exp Neurol. 1983;42:225–242. doi: 10.1097/00005072-198305000-00002. [DOI] [PubMed] [Google Scholar]
- Smith D H, Wolf J A, Lusardi T A, Lee V M, Meaney D F. High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. J Neurosci. 1999;19:4263–4269. doi: 10.1523/JNEUROSCI.19-11-04263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povlishock J T. Traumatically induced axonal injury: Pathogenesis and pathobiological implications. Brain Pathol. 1992;2:1–12. [PubMed] [Google Scholar]
- Farkas O, Povlishock J T. Cellular and subcellular change evoked by diffuse traumatic brain injury: A complex web of change extending far beyond focal damage. Prog Brain Res. 2007;161:43–59. doi: 10.1016/S0079-6123(06)61004-2. [DOI] [PubMed] [Google Scholar]
- Adams J H, Graham D I, Gennarelli T A, Maxwell W L. Diffuse axonal injury in non-missile head injury. J Neurol Neurosurg Psychiatr. 1991;54:481–483. doi: 10.1136/jnnp.54.6.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell W L, Graham D I. Loss of axonal microtubules and neurofilaments after stretch-injury to guinea pig optic nerve fibers. J Neurotr. 1997;14:603–614. doi: 10.1089/neu.1997.14.603. [DOI] [PubMed] [Google Scholar]
- Pettus E H, Povlishock J T. Characterization of a distinct set of intra-axonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain Res. 1996;722:1–11. doi: 10.1016/0006-8993(96)00113-8. [DOI] [PubMed] [Google Scholar]
- Jafari S S, Maxwell W L, Neilson M, Graham D I. Axonal cytoskeletal changes after non-disruptive axonal injury. J Neurocytol. 1997;26:207–221. doi: 10.1023/a:1018588114648. [DOI] [PubMed] [Google Scholar]
- Shi R, Pryor J D. Pathological changes of isolated spinal cord axons in response to mechanical stretch. Neuroscience. 2002;110:765–777. doi: 10.1016/s0306-4522(01)00596-6. [DOI] [PubMed] [Google Scholar]
- Gittes F, Mickey B, Nettleton J, Howard J. Flexural rigidity of microtubules and actin filaments measured from thermal fluctuations in shape. J Cell Biol. 1993;120:923–934. doi: 10.1083/jcb.120.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad F J, Pienkowski T P, Baas P W. Regional differences in microtubule dynamics in the axon. J Neurosci. 1993;13:856–866. doi: 10.1523/JNEUROSCI.13-02-00856.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent E W, Gertler F B. Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron. 2003;40:209–227. doi: 10.1016/s0896-6273(03)00633-0. [DOI] [PubMed] [Google Scholar]
- Baas P W, Ahmad F J, Pienkowski T P, Brown A, Black M M. Sites of microtubule stabilization for the axon. J Neurosci. 1993;13:2177–2185. doi: 10.1523/JNEUROSCI.13-05-02177.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M D, Golden A P, Tien J. Molding of three-dimensional microstructures of gels. J Am Chem Soc. 2003;125:12988–12989. doi: 10.1021/ja037677h. [DOI] [PubMed] [Google Scholar]
- Jordan M A, Wilson L. Use of drugs to study role of microtubule assembly dynamics in living cells. Methods Enzymol. 1998;298:252–276. doi: 10.1016/s0076-6879(98)98024-7. [DOI] [PubMed] [Google Scholar]
- Erturk A, Hellal F, Enes J, Bradke F. Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J Neurosci. 2007;27:9169–9180. doi: 10.1523/JNEUROSCI.0612-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf J A, Stys P K, Lusardi T, Meaney D, Smith D H. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci. 2001;21:1923–1930. doi: 10.1523/JNEUROSCI.21-06-01923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata A, Stys P K, Wolf J A, Chen X H, Taylor A G, Meaney D F, Smith D H. Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors. J Neurosci. 2004;24:4605–4613. doi: 10.1523/JNEUROSCI.0515-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard J, Hyman A A. Dynamics and mechanics of the microtubule plus end. Nature. 2003;422:753–758. doi: 10.1038/nature01600. [DOI] [PubMed] [Google Scholar]
- Meaney D F, Smith D H, Shreiber D I, Bain A C, Miller R T, Ross D T, Gennarelli T A. Biomechanical analysis of experimental diffuse axonal injury. J Neurotr. 1995;12:689–694. doi: 10.1089/neu.1995.12.689. [DOI] [PubMed] [Google Scholar]
- Zheng J, Lamoureux P, Santiago V, Dennerll T, Buxbaum R E, Heidemann S R. Tensile regulation of axonal elongation and initiation. J Neurosci. 1991;11:1117–1125. doi: 10.1523/JNEUROSCI.11-04-01117.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X H, Meaney D F, Xu B N, Nonaka M, McIntosh T K, Wolf J A, Saatman K E, Smith D H. Evolution of neurofilament subtype accumulation in axons following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58:588–596. doi: 10.1097/00005072-199906000-00003. [DOI] [PubMed] [Google Scholar]
- Dye R B, Fink S P, Williams R C., Jr Taxol-induced flexibility of microtubules and its reversal by MAP-2 and tau. J Biol Chem. 1993;268:6847–6850. [PubMed] [Google Scholar]
- Mickey B, Howard J. Rigidity of microtubules is increased by stabilizing agents. J Cell Biol. 1995;130:909–917. doi: 10.1083/jcb.130.4.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanBuren V, Cassimeris L, Odde D J. Mechanochemical model of microtubule structure and self-assembly kinetics. Biophys J. 2005;89:2911–2926. doi: 10.1529/biophysj.105.060913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman S M, Nash M J, Sweeting C J, Graham D I, Roberts G W. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett. 1993;160:139–144. doi: 10.1016/0304-3940(93)90398-5. [DOI] [PubMed] [Google Scholar]
- Sherriff F E, Bridges L R, Sivaloganathan S. Early detection of axonal injury after human head trauma using immunocytochemistry for beta-amyloid precursor protein. Acta Neuropathologica. 1994;87:55–62. doi: 10.1007/BF00386254. [DOI] [PubMed] [Google Scholar]
- Gorrie C, Oakes S, Duflou J, Blumbergs P, Waite P M. Axonal injury in children after motor vehicle crashes: extent, distribution, and size of axonal swellings using beta-APP immunohistochemistry. J Neurotr. 2002;19:1171–1182. doi: 10.1089/08977150260337976. [DOI] [PubMed] [Google Scholar]
- Chen X H, Siman R, Iwata A, Meaney D F, Trojanowski J Q, Smith D H. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol. 2004;165:357–371. doi: 10.1016/s0002-9440(10)63303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh J R. Cell biology. Microtubule catastrophe. Nature. 1984;312:196–197. doi: 10.1038/312196b0. [DOI] [PubMed] [Google Scholar]
- Dimitrov A, Quesnoit M, Moutel S, Cantaloube I, Pous C, Perez F. Detection of GTP-tubulin conformation in vivo reveals a role for GTP remnants in microtubule rescues. Science. 2008;322:1353–1356. doi: 10.1126/science.1165401. [DOI] [PubMed] [Google Scholar]
- Drechsel D N, Kirschner M W. The minimum GTP cap required to stabilize microtubules. Curr Biol. 1994;4:1053–1061. doi: 10.1016/s0960-9822(00)00243-8. [DOI] [PubMed] [Google Scholar]
- Tanaka E M, Kirschner M W. Microtubule behavior in the growth cones of living neurons during axon elongation. J Cell Biol. 1991;115:345–363. doi: 10.1083/jcb.115.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuckowree J A, Vickers J C. Cytoskeletal and morphological alterations underlying axonal sprouting after localized transection of cortical neuron axons in vitro. J Neurosci. 2003;23:3715–3725. doi: 10.1523/JNEUROSCI.23-09-03715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers K A, Baas P W. Kinesin-5 regulates the growth of the axon by acting as a brake on its microtubule array. J Cell Biol. 2007;178:1081–1091. doi: 10.1083/jcb.200702074. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.