

Abstract
The fitness effects of mutations are central to evolution, yet have begun to be characterized in detail only recently. Site-directed mutagenesis is a powerful tool for achieving this goal, which is particularly suited for viruses because of their small genomes. Here, I discuss the evolutionary relevance of mutational fitness effects and critically review previous site-directed mutagenesis studies. The effects of single-nucleotide substitutions are standardized and compared for five RNA or single-stranded DNA viruses infecting bacteria, plants or animals. All viruses examined show very low tolerance to mutation when compared with cellular organisms. Moreover, for non-lethal mutations, the mean fitness reduction caused by single mutations is remarkably constant (0.10–0.13), whereas the fraction of lethals varies only modestly (0.20–0.41). Other summary statistics are provided. These generalizations about the distribution of mutational fitness effects can help us to better understand the evolution of RNA and single-stranded DNA viruses.
Keywords: evolution, mutation, robustness, selection, site-directed mutagenesis, virus
1. Introduction
Evolutionary biologists have long sought for a detailed knowledge of the fitness effects of mutations. Basic questions related to this topic are, for instance, how many spontaneous mutations are selectively neutral, deleterious and beneficial, whether mutations of small effect are more abundant than those of large effect, or whether mutations have similar effects across genetic backgrounds, environments and species. Mutational fitness effects (MFE) can be quantified using the selection coefficient of single mutants, that is, their fitness relative to a common reference genotype, as estimated from progeny sizes, growth rates or related quantities. Therefore, the above questions could be addressed by inferring experimentally the statistical properties of the selection coefficient, such as its mean and variance and, whenever possible, its full statistical distribution. Site-directed mutagenesis offers us a powerful tool for achieving this goal. Moreover, it can be implemented with relative ease in viruses with small genomes, including most RNA viruses and single-stranded DNA (ssDNA) viruses. Here, I review previous contributions and present the data in a standardized manner in order to facilitate the comparison between viruses and evolutionary model testing. First, I outline the relevance of MFE in general. Then, I summarize the basic aspects of the site-directed mutagenesis technique and collect data obtained using this methodology. This leads me to suggest that MFE behave similarly across the viruses studied so far. Finally, I discuss the implications for the evolution of RNA and ssDNA viruses.
The selection coefficient is, together with other parameters such as the mutation rate, the effective population size, the epistasis coefficient or the recombination rate, central to evolutionary theory. Several examples can be given. First, the population genetic dynamics of a given allele depends on the relative strength of random genetic drift and natural selection. As a rule of thumb, if the product of the effective population size and the selection coefficient exceeds one, selection will be the dominant force, whereas drift will be dominant otherwise (Kimura 1983; Ohta 1992). Second, the Haldane-Muller principle establishes that the mean fitness of a population strictly depends on the deleterious mutation rate (Haldane 1937; Muller 1950), but in order to apply this principle from the available information on total mutation rates (Drake et al. 1998), it is necessary to know the fraction of neutral mutations, which implies having some information about the distribution of MFE. Third, according to Muller's ratchet model, fitness declines in finite asexual populations at a rate determined by the selection coefficient and the mutation rate (Muller 1964; Haigh 1978). Fourth, the expected genome mutation frequency at the mutation–selection balance is inversely proportional to the harmonic mean of the selection coefficient (Orr 2000), which could be obtained directly from the distribution of MFE. Finally, the mutation rate that maximizes the long-term adaptation depends directly on the selection coefficient, at least in asexuals (Orr 2000; Johnson & Barton 2002).
The fact that most mutations are neutral or deleterious may lead us to conclude precipitously that the fitness effects of random mutations are not relevant to adaptation. However, this view is incorrect because the fixation probability of a beneficial mutation will depend on the genetic background or the environment. For instance, if a beneficial mutation is linked to other, deleterious mutations, it will go extinct as long as the combined effect is deleterious or, alternatively, the latter may be driven to fixation through genetic hitchhiking (Smith & Haigh 1974). Therefore, knowledge of the distribution of MFE helps us to better predict the fate of beneficial mutations. Also, at least in some situations, beneficial and deleterious mutations may be viewed as the two sides of the same coin. Deleterious mutations can become fixed during population bottlenecks and then revert to the wild-type or be compensated by secondary mutations during a subsequent population expansion. This demographic alternation is typical of pathogens experiencing strong transmission bottlenecks followed by rapid growth. The strength of selection during the growth phase will be determined by the effects of the deleterious mutations fixed by the bottleneck. Similarly, host-shift mutations may be costly in the original host (Ferris et al. 2007), and immune-escape mutations critical for virus survival in one individual may become detrimental for the virus in another individual host (Davenport et al. 2008). Therefore, random mutations, be they neutral or deleterious, are clearly relevant to adaptation.
2. Material and methods
(a). Site-directed mutagenesis
A major problem limiting our ability to study MFE is the bias in mutation sampling owing to selection. In addition to removing this bias, it would be good to know precisely the type and the location of each mutation examined, and to avoid over-representation of large deletions or insertions such as, for instance, single-gene knockouts, which are probably infrequent in nature compared with point mutations. A direct and powerful approach to achieve these goals is site-directed mutagenesis, which allows us to engineer genotypes with single-nucleotide substitutions. Owing to their small genome sizes, RNA viruses (including retroviruses) and ssDNA viruses are excellent experimental systems for applying this technique.
The first step of a site-directed mutagenesis protocol consists of performing a PCR or a non-PCR (linear) amplification of a full-length cDNA clone (RNA viruses) or DNA (ssDNA viruses) using primers that carry the desired mutation (figure 1a). The product can be circularized and used directly to transfect bacteria in the case of phages, whereas for eukaryotic viruses it is generally necessary to clone it in Escherichia coli to obtain a large amount of purified DNA, which is then used for transfection or first transcribed in vitro to synthesize the viral RNA. Tens of single-nucleotide mutants can be generated in this way and assayed for fitness. To minimize the risk of non-desired mutations, the template DNA should be as clonal as possible. This is not a concern in the case of RNA viruses because the template is a cDNA clone but, for DNA viruses, the template DNA should be preferably isolated from a plaque-purified virus to increase its sequence homogeneity. Also, using a high-fidelity DNA polymerase is critical. Typically, the error rate of these enzymes is 10−6 per base per round of copying or lower, as provided by the manufacturers (New England Biolabs, Stratagene and others). Hence, for a linearly amplified DNA and a genome size of 104 bases, the fraction of molecules carrying additional mutations should not exceed 1 per cent, although this fraction might be larger if the PCR method is used.
Figure 1.

Diagram showing different steps of the site-directed mutagenesis protocol for RNA or ssDNA viruses. (a) The viral genome can be amplified using adjacent divergent primers (PCR) or complementary primers (non-PCR, linear amplification). The mutated residue is indicated with a white dot. Cloning and purification, eventually followed by in vitro transcription, are required for eukaryotic viruses, whereas for phages the mutagenesis product can be directly used to transfect bacteria. Sequencing is needed at some point (preferably after virus recovery) to verify the presence of the mutation. (b) Typical lethality tests for phage ΦX174. Left picture: agarose gel electrophoresis of several PCR mutagenesis reactions. The upper band corresponds to the viral genome in linear form. The genome position and nucleotide substitution of each mutant are shown above. The two numbers below each mutation label correspond to the number of plaques obtained in transfection assays for the real mutagenesis (left) and a control reaction using non-mutagenic but otherwise identical primers (right). In this particular test, all mutations expect A402T were confirmed to be lethal. Right picture: typical aspect of E. coli lawns transfected with the real versus control mutagenesis reactions.
To measure the amplification error, it is possible to verify mutation uniqueness by hybridizing the DNA of the mutant and the reference genotype and digesting the heteroduplex with an enzyme that recognizes point mismatches, such as DNA endonuclease S. This method was used to show that more than 95 per cent of the clones obtained by site-directed mutagenesis were actually single-nucleotide mutants in a study with tobacco etch virus (TEV; Carrasco et al. 2007). However, the presence of additional mutations in the engineered clones is not the only source of error. In principle, compensatory mutations may arise early in the fitness assays, leading to underestimation of MFE, but this is unlikely in the short term. More importantly, experimental error in fitness assays adds variance to the observed distribution of MFE. To remove this component, we can carry out experiments in which the whole procedure is repeated using control primers, which do not carry any mutations, instead of the mutagenesis primers. Fitness assays of a set of independent controls will provide baseline values for the average selection coefficient and its variance (Domingo-Calap et al. 2009), or even an entire ‘blank distribution’. Using appropriate statistical tools, it is then possible to infer a distribution of MFE free of experimental error (Peris et al. in preparation).
One advantage of site-directed mutagenesis is that it allows us to determine the fraction of lethal mutations. These mutations typically show up as repeated failures in transfection assays. Appropriate control assays as, for instance, use of the above-mentioned control primers, are needed to ensure that our inability to recover viruses is because of the presence of the engineered mutation and not a consequence of experiment failures (figure 1b).
(b). Fitness: definitions and assays
In order to make quantitative studies on MFE meaningful, the fitness definition used in each particular study has to be stated explicitly and the information for converting units provided. The fitness of a viable mutant in a given environment can be measured by head-to-head assays against the reference virus or by performing growth assays of each virus in parallel. The first approach is more accurate because it relies less on the reproducibility of the assay but requires using a neutral phenotypic or genotypic marker to differentiate the two types of virus. Also, fitness can be measured during the exponential growth phase or throughout the entire course of an infection. The latter is more general but the former has the advantage of focusing on a well-defined fitness component, the exponential growth rate r, which is especially important for fast-growing systems such as viruses.
The selection coefficient, s, is the quantity that allows us to predict changes in genotype frequencies under the action of selection. It can be defined as the relative fitness (Wi/0) minus one, i.e. si = Wi/0 − 1 = Wi/W0 − 1, where Wi and W0 are the absolute fitness values of genotype i and the reference (or wild-type) genotype, respectively. Hence, si = 0 for neutral mutations, si < 0 for deleterious mutations (si = −1 for lethals), and si > 0 for beneficial mutations. In experimental studies, relative fitness has been defined in different ways depending on convenience. For instance, we can use the progeny size per individual per hour divided by that of the reference genotype, Wi/0 = (eri − 1)/(er0 − 1). Here, the −1 term subtracts the parent virus, which means that this is a discrete-time definition (non-overlapping generations). Under a continuous-time model, we would have Wi/0 = e(ri−r0) and si = ri − r0. The two definitions converge when r-values are large (which is not always the case). A problem of the earlier two definitions, however, is that they depend on absolute time units, complicating the comparison between viruses with very different growth rates. To avoid this problem, we can define Wi/0 = ri/r0 and si = ri/r0 − 1 (notice that this is equivalent to scaling s by dividing it by r0 in the continuous-time model). Finally, a biologically meaningful definition of relative fitness is the number of progeny per individual per viral generation relative to the reference virus. If we call K the progeny size of the reference virus, W0 = K − 1 for non-overlapping generations. Since under exponential growth, the population grows as Nt = N0ert, the time required for the reference virus to reach Nt = K starting from one parent is 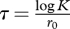 (generation time). After this time, a single parent of genotype i will reach a population size of
(generation time). After this time, a single parent of genotype i will reach a population size of 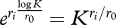 . Hence, the relative fitness per generation is Wi/0 = (Kri/r0 − 1)/(K − 1), and the corresponding selection coefficient is still si = Wi/0 − 1. In the continuous-time model, the equivalent definition is Wi/0 = Kri/r0−1 and
. Hence, the relative fitness per generation is Wi/0 = (Kri/r0 − 1)/(K − 1), and the corresponding selection coefficient is still si = Wi/0 − 1. In the continuous-time model, the equivalent definition is Wi/0 = Kri/r0−1 and 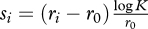 (notice that here the generation time is used to scale s). Defining a viral generation is not straightforward, but the usual convention is to use the cell infection cycle. Then, K is the burst size or viral yield per infected cell.
(notice that here the generation time is used to scale s). Defining a viral generation is not straightforward, but the usual convention is to use the cell infection cycle. Then, K is the burst size or viral yield per infected cell.
3. Results and discussion
(a). Datasets
I review data from published studies in which MFE were characterized by site-directed mutagenesis in vesicular stomatitis virus (VSV; Sanjuán et al. 2004a), TEV (Carrasco et al. 2007) and the bacteriophages ΦX174 and Qβ (Domingo-Calap et al. 2009). Preliminary data of an unpublished study with bacteriophage F1 are also presented (Peris et al. in preparation). An HIV-1 dataset (van Opijnen et al. 2006) is not included because it contains few single-mutants (15) belonging exclusively to a non-protein-coding region (however, this study is an interesting test of genotype–environment interactions). To standardize the results, I use the definitions Wi/0 = ri/r0 and si = ri/r0 − 1, as was also done in the VSV, Qβ, ΦX174 and F1 studies. In the TEV study, relative fitness was defined as Wi/0 = (Ri(t)/Ri(t=0))1/t, where Ri(t) is the relative amount of mutant i and the reference virus at time t. The use of a different definition has to be addressed before establishing any meaningful comparison between the five viruses. As a rough approximation, the latter expression is equivalent to Wi/0 = e(ri −r0) if ri and r0 are defined as average growth rates. Hence, I applied the transformation x → (log x + r0)/r0, which gives us the growth rate ratio for x = e(ri −r0). I used r0 = 3.3 d−1, a value compatible with virus accumulation experimental data (S. F. Elena 2010, personal communication).
For readers interested in using alternative definitions, typical r0 values are 0.8 h−1, 3.6 h−1, 10.0 h−1 and 4.3 h−1 for VSV, Qβ, ΦX174 and F1, respectively. K-values were not determined in the above studies. However, roughly speaking, K ≈ 100–1000 for VSV (Cuevas et al. 2005; Furió et al. 2005) and K ≈ 100 for ΦX174 (Denhardt & Silver 1966; De Paepe & Taddei 2006). For leviviruses, K ≈ 400–4000 (De Paepe & Taddei 2006), whereas K ≈ 3–6 for tobacco mosaic virus (Malpica et al. 2002), although this virus is phylogenetically unrelated to TEV. Finally, measuring K for F1 is problematic because it is not a lytic virus, but taking 1 h as a reasonable generation time, we would have K = er0τ ≈ 100.
Table 1 shows the number of mutations tested in each study, the fraction of lethal mutations and several summary statistics of the selection coefficient for viable mutants. In all cases, mutations consisted of single-nucleotide substitutions located at random sites of the viral genome. Details about each dataset, including the fitness of each individual mutant, can be retrieved from the supplementary information files of the original publications. The procedures used for constructing mutants and assaying their fitness were generally similar but differed in some details. For instance, the PCR method was used for bacteriophages, whereas the non-PCR method was used for TEV and VSV. In all studies, a mutant was considered lethal if it reproducibly failed to transfect the host, and the possibility that this negative result was because of errors during transfection assays was ruled out. Failures in previous steps were also discarded in the case of phages ΦX174, Qβ and F1 by running appropriate control mutagenesis reactions and, in the case of TEV, by checking the uniqueness of each mutation, but similar controls were not carried out in the VSV study. Finally, for TEV and phages, the natural hosts were used, whereas for VSV, fitness assays were carried out in cell cultures. Fitness assays also differed slightly. For the phages, the growth rate of the mutants and the reference virus was measured in parallel and during the exponential growth phase (1–3 h post-inoculation), whereas for TEV and VSV it was measured in a head-to-head competition against the reference virus. For TEV, the proportion of each virus was determined after the end of the exponential growth phase (7 days post-inoculation), whereas for VSV this was done during the exponential growth phase (10 h post-inoculation).
Table 1.
MFE summary statistics from site-directed mutagenesis studies in five different viruses.
| virus | VSV | TEV | Qβ | ΦX174 | F1 |
|---|---|---|---|---|---|
| type | ss(−) RNA | ss(+) RNA | ss(+) RNA | ssDNA | ssDNA |
| host | animals | plants | bacteria | bacteria | bacteria |
| sample size | 48 | 66 | 42 | 45 | 100 |
| fraction of lethal mutations | 0.396 | 0.409 | 0.286 | 0.200 | 0.210 |
| arithmetic meana | −0.132 | −0.112 | −0.103 | −0.126 | −0.107 |
| variancea | 0.036 | 0.041 | 0.018 | 0.047 | 0.037 |
| skewnessa | −1.795 | 0.285 | −1.167 | −1.957 | −1.909 |
| kurtosisa | 3.007 | −0.382 | 0.238 | 4.022 | 3.165 |
aMFE for viable mutations only, measured as the growth rate ratio minus one.
(b). Summary parameters
Comparison of the results obtained with the five viruses studied indicates that the fraction of lethals is always high, ranging from 0.20 to 0.41. This interval is not too wide if we take into account the diversity of this group of viruses, both in terms of genome organization (positive-stranded RNA, negative-stranded RNA or ssDNA) and host use (bacterium, plant or animal). The fraction of lethals is apparently higher in eukaryotic viruses than in phages, and higher in RNA viruses than in ssDNA viruses. However, methodological issues may partially explain this observation. For instance, as indicated above, lethality was less well-demonstrated in the VSV study.
Focusing on viable mutations, the average s is remarkably constant across viruses, ranging from −0.103 to −0.132 (table 1). The variance also shows a relatively narrow range (0.018–0.047). All skewness values are negative and reasonably similar, except for TEV. This negative skewness reflects two very general properties of MFE, namely that mutations are much more often deleterious than beneficial and that mutations of mild effect are more likely than those of large effect (Eyre-Walker & Keightley 2007), as can be seen in figure 2. Departure of TEV from this rule is unlikely to reflect a biologically unique feature of this virus and is more probably owing to large measurement error. Supporting this possibility, mutants with s-values as high as +0.3 were found to be not significantly different from zero (Carrasco et al. 2007). Kurtosis, which measures the ‘peakedness’ of the distribution, is within the range 3–4 for VSV, ΦX174 and F1 but is close to zero for TEV and Qβ. A positive kurtosis value (leptokurtosis) means that the probability density function has a heavier tail and more values near the mean than a Gaussian distribution. The lack of agreement between Qβ and the first three viruses is because of the absence of highly deleterious s-values, i.e. those falling within the range of −0.8 to −0.5. Indeed, adding a single hypothetical datum within this range would make all Qβ parameters shown in table 1 consistent with those of the VSV–ΦX174–F1 group. For instance, if we added the single observation s = −0.7, we would obtain −0.122, 0.029, −1.733 and 3.215 for the Qβ mean, variance, skewness and kurtosis, respectively. Given that data within this range are rare in all cases and that the Qβ dataset is comparatively small, it does not seem unlikely that the actual MFE distribution of Qβ is actually very close to those of VSV, ΦX174 and F1.
Figure 2.

Observed MFE frequency histograms for four different RNA or ssDNA viruses. (a) VSV; (b) TEV; (c) Qβ; (d) ΦX174.
(c). Probability density functions
Different statistical models have been used to more comprehensively characterize MFE. Model fit was done by nonlinear regression between the observed and predicted cumulated probabilities in the VSV, TEV, Qβ, ΦX174 and F1 studies. For VSV, the best-fit was obtained using a compound log-normal plus uniform distribution, whereas among more usual models the log-normal performed best. The Weibull, beta, gamma and exponential distributions performed progressively worse but the squared correlation coefficient (r2) between the expected and observed cumulated probabilities was always higher than 0.95. For TEV, using the original (untransformed) fitness values, it was found that the beta provided the best-fit, followed by the half-normal, the Weibull, the gamma, the normal, the log-normal, the exponential and the Pareto distributions, with r2 > 0.91 in all cases. In the phage study, the only models tested were the exponential, the gamma and the beta and, among these, the exponential provided the best-fit for phage ΦX174, whereas the gamma performed best for phage Qβ, although r2 > 0.96 in all cases.
An important limitation of using cumulative distribution functions for inferring probabilistic models is that the observations are not statistically independent, because the cumulated probability of a given observation depends on the other observations. To avoid this problem, one could fit probability density functions to the observed frequency histogram, but this would entail the problem of choosing the appropriate interval length for constructing the histogram. A better approach consists of estimating the maximum likelihood probability density function given the dataset.
A source of error comes from the fact that a fraction of the total variance is due to the measurement error, as mentioned earlier. This becomes more obvious in the vicinity of s = 0, where some values larger than zero will not correspond to truly beneficial mutants but rather to neutral mutations or slightly deleterious effects measured with error. Since the probability distributions used are typically not defined on either side of zero, these values were removed from the analysis in the original articles, but this clearly introduces a bias. To address this problem, it is necessary to calibrate the measurement error first. Then, the probability density function used to model MFE can be convoluted with the error function and fit to the data by maximum likelihood including observations that fall out of the definition interval of the MFE probability model. This approach has been developed in the bacteriophage F1 study (Peris et al. in preparation). A re-analysis of all data using maximum likelihood and explicitly accounting for experimental error could be attempted in future work. This would help us to test alternative MFE models more rigorously.
(d). Evolutionary implications
Considerable progress has been made recently in characterizing MFE in cellular organisms using different techniques (Eyre-Walker & Keightley 2007). For instance, it has been shown that, in E. coli, more than 90 per cent of single-gene knockout mutations are viable (Baba et al. 2006) and reduce fitness by 3 per cent or less on average (Elena et al. 1998) and that, in Caenorhabditis elegans, the effects of most nucleotide substitutions are nearly undetectable (Davies et al. 1999). It seems clear that most if not all RNA and ssDNA viruses are less robust to mutation than cellular organisms. In a previous survey, the median selection coefficient was found to be approximately 10-fold lower in the latter group (Elena et al. 2006), although the variety of techniques used makes it difficult to establish an accurate comparison. The most likely explanation for the strong MFE found in RNA and ssDNA viruses is that robustness mechanisms, such as alternative metabolic or regulatory pathways, genetic redundancy and modularity are absent from their simple and compact genomes, leading to extensive functional constraints (Holmes 2003; Elena et al. 2006; Belshaw et al. 2008). If genome complexity is a major determinant of mutational robustness, we should expect double-stranded DNA viruses with large genomes to show weaker MFE than RNA and ssDNA viruses. Although site-directed mutagenesis studies have not been undertaken with these viruses, a recent chemical mutagenesis study has revealed a remarkable ability to tolerate deleterious mutations in the bacteriophage T7 (Springman et al. 2009).
The low mutational robustness of RNA viruses and ssDNA viruses can help us to understand better some of their evolutionary properties. First, these viruses respond very rapidly to strong selective pressures such as those imposed by immune pressure or antiviral drugs (Domingo & Holland 1997; Holmes 2009). At least for RNA viruses, this is primarily because of their high per-base mutation rate, which is generally in the order of 10−6 to 10−4 substitutions per round of copying (Drake & Holland 1999). However, MFE are also important and, indeed, they are intricately correlated with mutation rates from an evolutionary standpoint. For instance, the mutation rate that maximizes adaptation depends on the balance between the benefits of generating adaptive mutations and the costs of increasing the mutational load, and these two terms are given by the distribution of MFE. Theory predicts that, neglecting recombination, the optimal rate is determined exclusively by the selection coefficient (Orr 2000; Johnson & Barton 2002), although it is unclear whether mutation rates can be optimized by natural selection (Clune et al. 2008). Conversely, for a given per-base mutation rate, the genome size cannot increase indefinitely because the mutational load would become too high (Eigen et al. 1988). This restriction also imposes a limit indirectly on mutational robustness, because redundancy and other mechanisms of robustness cannot be accommodated easily in very small genomes. The resulting low robustness may in turn favour high mutation rates, as explained above, hence potentially establishing a feedback, which might lead to an evolutionarily stable situation.
There are other reasons why MFE may explain the rapid response to selection shown by RNA viruses. As discussed above, strong selection against deleterious mutations can correlate with strong selection for beneficial mutations during evolutionary reversals. Also, the lack of buffering mechanisms might result in large phenotypic variation in general and strong selection for beneficial mutations (Lenski et al. 2006; Frank 2007; Wagner 2008). Consistent with this view, the fixation of big-benefit mutations has been reported in several phages (Bull et al. 2000; Rokyta et al. 2005, 2008) and lower mutational robustness has been associated with faster adaptation in VSV (Cuevas et al. 2009). Further, greater MFE imply that deleterious mutations are removed more efficiently from populations, favouring the spread of beneficial ones (Orr 2000). However, the relationship between mutational robustness and evolvability is controversial. By reducing MFE, robustness facilitates the accumulation of effectively neutral genetic variants that might become beneficial upon changes in the environment or the genetic background. Hence, over the long term, mutational robustness may foster evolvability (Wagner 2005, 2008; Bloom et al. 2006; Ciliberti et al. 2007; Elena & Sanjuán 2008). Theory predicts that this is a likely scenario if occasional failures in robustness mechanisms occur (Kim 2007). Further, lattice protein models and random mutagenesis experiments suggest that proteins with increased thermostability are more robust to mutation and more likely to evolve new catalytic capabilities (Bloom et al. 2006). Finally, experiments with bacteriophage Φ6 have shown that robust genotypes are more likely to evolve thermotolerance (McBride et al. 2008).
Another seemingly general property of RNA viruses (and probably also of ssDNA viruses) related to MFE is antagonistic epistasis, defined as the tendency of deleterious effects of mutations to diminish as mutations accumulate (Bonhoeffer et al. 2004; Burch & Chao 2004; Sanjuán et al. 2004b; Sanjuán & Elena 2006). Theoretical and experimental work indicates that epistasis and selection coefficients are negatively correlated, such that low robustness is associated with antagonistic epistasis, whereas high robustness is associated with synergistic epistasis (Wilke & Adami 2001; Azevedo et al. 2006; Bershtein et al. 2006; Elena et al. 2006; Sanjuán & Nebot 2008). One explanation for this correlation is the multiple-hit effect, whereby successive mutations damaging the same functional unit have lower and lower deleterious effects (Wilke & Adami 2001; Elena et al. 2006; Sanjuán & Nebot 2008).
In conclusion, the low mutational tolerance of RNA and ssDNA viruses indicates the presence of strong functional constraints. Therefore, a large fraction of the mutations that might a priori be expected to be beneficial when focusing on a given function (for instance an immune-escape mutation) may compromise other viral functions (replication, protein structure, etc.) and thus have a strong fitness cost. These mutations will be selected against unless the fitness benefit is high enough to overcome the cost. As a consequence, the repertoire of adaptive mutations given an environmental challenge might be quite limited, potentially explaining the abundant parallel or convergent evolution observed in RNA and ssDNA viruses at the molecular level (Bull et al. 1997; Wichman et al. 1999; Crill et al. 2000; Cuevas et al. 2002; Rico et al. 2006; Agudelo-Romero et al. 2008). This suggests that, although RNA and ssDNA viruses often respond rapidly to strong selective pressures, their evolutionary plasticity and long-term evolvability might be less spectacular.
Acknowledgements
This work was supported by the Ramón y Cajal research programme from the Spanish MICIIN.
Footnotes
One contribution of 14 to a Theme Issue ‘New experimental and theoretical approaches towards the understanding of the emergence of viral infections’.
References
- Agudelo-Romero P., de la Iglesia F., Elena S. F.2008The pleiotropic cost of host-specialization in Tobacco etch potyvirus. Infect. Genet. Evol. 8, 806–814 (doi:10.1016/j.meegid.2008.07.010) [DOI] [PubMed] [Google Scholar]
- Azevedo R. B., Lohaus R., Srinivasan S., Dang K. K., Burch C. L.2006Sexual reproduction selects for robustness and negative epistasis in artificial gene networks. Nature 440, 87–90 (doi:10.1038/nature04488) [DOI] [PubMed] [Google Scholar]
- Baba T., et al. 2006Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2: 2006.0008 (doi:10.1038/msb4100050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshaw R., Gardner A., Rambaut A., Pybus O. G.2008Pacing a small cage: mutation and RNA viruses. Trends Ecol. Evol. 23, 188–193 (doi:10.1016/j.tree.2007.11.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershtein S., Segal M., Bekerman R., Tokuriki N., Tawfik D. S.2006Robustness-epistasis link shapes the fitness landscape of a randomly drifting protein. Nature 444, 929–932 (doi:10.1038/nature05385) [DOI] [PubMed] [Google Scholar]
- Bloom J. D., Labthavikul S. T., Otey C. R., Arnold F. H.2006Protein stability promotes evolvability. Proc. Natl Acad. Sci. USA 103, 5869–5874 (doi:10.1073/pnas.0510098103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhoeffer S., Chappey C., Parkin N. T., Whitcomb J. M., Petropoulos C. J.2004Evidence for positive epistasis in HIV-1. Science 306, 1547–1550 (doi:10.1126/science.1101786) [DOI] [PubMed] [Google Scholar]
- Bull J. J., Badgett M. R., Wichman H. A., Huelsenbeck J. P., Hillis D. M., Gulati A., Ho C., Molineux I. J.1997Exceptional convergent evolution in a virus. Genetics 147, 1497–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull J. J., Badgett M. R., Wichman H. A.2000Big-benefit mutations in a bacteriophage inhibited with heat. Mol. Biol. Evol. 17, 942–950 [DOI] [PubMed] [Google Scholar]
- Burch C. L., Chao L.2004Epistasis and its relationship to canalization in the RNA virus ϕ6. Genetics 167, 559–567 (doi:10.1534/genetics.103.021196) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco P., de la Iglesia F., Elena S. F.2007Distribution of fitness and virulence effects caused by single-nucleotide substitutions in Tobacco Etch virus. J. Virol. 81, 12 979–12 984 (doi:10.1128/JVI.00524-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciliberti S., Martin O. C., Wagner A.2007Innovation and robustness in complex regulatory gene networks. Proc. Natl Acad. Sci. USA 104, 13 591–13 596 (doi:10.1073/pnas.0705396104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clune J., Misevic D., Ofria C., Lenski R. E., Elena S. F., Sanjuán R.2008Natural selection fails to optimize mutation rates for long-term adaptation on rugged fitness landscapes. PLoS Comput. Biol. 4, e1000187 (doi:10.1371/journal.pcbi.1000187) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crill W. D., Wichman H. A., Bull J. J.2000Evolutionary reversals during viral adaptation to alternating hosts. Genetics 154, 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas J. M., Elena S. F., Moya A.2002Molecular basis of adaptive convergence in experimental populations of RNA viruses. Genetics 162, 533–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas J. M., Moya A., Sanjuán R.2005Following the very initial growth of biological RNA viral clones. J. Gen. Virol. 86, 435–443 (doi:10.1099/vir.0.80359-0) [DOI] [PubMed] [Google Scholar]
- Cuevas J. M., Moya A., Sanjuán R.2009A genetic background with low mutational robustness is associated with increased adaptability to a novel host in an RNA virus. J. Evol. Biol. 22, 2041–2048 (doi:10.1111/j.1420-9101.2009.01817.x) [DOI] [PubMed] [Google Scholar]
- Davenport M. P., Loh L., Petravic J., Kent S. J.2008Rates of HIV immune escape and reversion: implications for vaccination. Trends Microbiol. 16, 561–566 (doi:10.1016/j.tim.2008.09.001) [DOI] [PubMed] [Google Scholar]
- Davies E. K., Peters A. D., Keightley P. D.1999High frequency of cryptic deleterious mutations in Caenorhabditis elegans. Science 285, 1748–1751 (doi:10.1126/science.285.5434.1748) [DOI] [PubMed] [Google Scholar]
- Denhardt D. T., Silver R. B.1966An analysis of the clone size distribution of ΦX174 mutants and recombinants. Virology 30, 10–19 (doi:10.1016/S0042-6822(66)81004-8) [DOI] [PubMed] [Google Scholar]
- De Paepe M., Taddei F.2006Viruses' life history: towards a mechanistic basis of a trade-off between survival and reproduction among phages. PLoS Biol. 4, e193 (doi:10.1371/journal.pbio.0040193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo E., Holland J. J.1997RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 51, 151–178 (doi:10.1146/annurev.micro.51.1.151) [DOI] [PubMed] [Google Scholar]
- Domingo-Calap P., Cuevas J. M., Sanjuán R.2009The fitness effects of random mutations in single-stranded DNA and RNA bacteriophages. PLoS Genet. 5, e1000742 (doi:10.1371/journal.pgen.1000742) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake J. W., Holland J. J.1999Mutation rates among RNA viruses. Proc. Natl Acad. Sci. USA 96, 13 910–13 913 (doi:10.1073/pnas.96.24.13910) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake J. W., Charlesworth B., Charlesworth D., Crow J. F.1998Rates of spontaneous mutation. Genetics 148, 1667–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigen M., McCaskill J., Schuster P.1988Molecular quasi-species. J. Phys. Chem. 92, 6881–6891 (doi:10.1021/j100335a010) [Google Scholar]
- Elena S. F., Sanjuán R.2008The effect of genetic robustness on evolvability in digital organisms. BMC Evol. Biol. 8, 284 (doi:10.1186/1471-2148-8-284) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elena S. F., Ekunwe L., Hajela N., Oden S. A., Lenski R. E.1998Distribution of fitness effects caused by random insertion mutations in Escherichia coli. Genetica 102/103, 349–358 (doi:10.1023/A:1017031008316) [PubMed] [Google Scholar]
- Elena S. F., Carrasco P., Daròs J. A., Sanjuán R.2006Mechanisms of genetic robustness in RNA viruses. EMBO Rep. 7, 168–173 (doi:10.1038/sj.embor.7400636) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A., Keightley P. D.2007The distribution of fitness effects of new mutations. Nat. Rev. Genet. 8, 610–618 (doi:10.1038/nrg2146) [DOI] [PubMed] [Google Scholar]
- Ferris M. T., Joyce P., Burch C. L.2007High frequency of mutations that expand the host range of an RNA virus. Genetics 176, 1013–1022 (doi:10.1534/genetics.106.064634) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S. A.2007Maladaptation and the paradox of robustness in evolution. PLoS ONE 2, e1021 (doi:10.1371/journal.pone.0001021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furió V., Moya A., Sanjuán R.2005The cost of replication fidelity in an RNA virus. Proc. Natl Acad. Sci. USA 102, 10 233–10 237 (doi:10.1073/pnas.0501062102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigh J.1978The accumulation of deleterious genes in a population: Muller's ratchet. Theor. Popul. Biol. 14, 251–267 (doi:10.1016/0040-5809(78)90027-8) [DOI] [PubMed] [Google Scholar]
- Haldane J. B. S.1937The effect of variation on fitness. Am. Nat. 71, 337–349 (doi:10.1086/280722) [Google Scholar]
- Holmes E. C.2003Error thresholds and the constraints to RNA virus evolution. Trends Microbiol. 11, 543–546 (doi:10.1016/j.tim.2003.10.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E. C.2009The evolution and emergence of RNA viruses Oxford, UK: Oxford University Press [Google Scholar]
- Johnson T., Barton N. H.2002The effect of deleterious alleles on adaptation in asexual populations. Genetics 162, 395–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.2007Rate of adaptive peak shifts with partial genetic robustness. Evolution 61, 1847–1856 (doi:10.1111/j.1558-5646.2007.00166.x) [DOI] [PubMed] [Google Scholar]
- Kimura M.1983The neutral theory of molecular evolution Cambridge, UK: Cambridge University Press [Google Scholar]
- Lenski R. E., Barrick J. E., Ofria C.2006Balancing robustness and evolvability. PLoS Biol. 4, e428 (doi:10.1371/journal.pbio.0040428) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malpica J. M., Fraile A., Moreno I., Obies C. I., Drake J. W., García-Arenal F.2002The rate and character of spontaneous mutation in an RNA virus. Genetics 162, 1505–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride R. C., Ogbunugafor C. B., Turner P. E.2008Robustness promotes evolvability of thermotolerance in an RNA virus. BMC Evol. Biol. 8, 231 (doi:10.1186/1471-2148-8-231) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller H. J.1950Our load of mutations. Am. J. Hum. Genet. 2, 111–176 [PMC free article] [PubMed] [Google Scholar]
- Muller H. J.1964The relation of recombination to mutational advance. Mutat. Res. 1, 2–9 (doi:10.1016/0027-5107(64)90047-8) [DOI] [PubMed] [Google Scholar]
- Ohta T.1992The nearly neutral theory of molecular evolution. Annu. Rev. Ecol. Syst. 23, 263–286 (doi:10.1146/annurev.es.23.110192.001403) [Google Scholar]
- Orr H. A.2000The rate of adaptation in asexuals. Genetics 155, 961–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris J. B., Davis P., Cuevas J. M., Nebot M. R., Sanjuán R.In preparation Distribution of fitness effects caused by single-nucleotide substitutions in a DNA virus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico P., Ivars P., Elena S. F., Hernández C.2006Insights into the selective pressures restricting Pelargonium flower break virus genome variability: evidence for host adaptation. J. Virol. 80, 8124–8132 (doi:10.1128/JVI.00603-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokyta D. R., Joyce P., Caudle S. B., Wichman H. A.2005An empirical test of the mutational landscape model of adaptation using a single-stranded DNA virus. Nat. Genet. 37, 441–444 (doi:10.1038/ng1535) [DOI] [PubMed] [Google Scholar]
- Rokyta D. R., Beisel C. J., Joyce P., Ferris M. T., Burch C. L., Wichman H. A.2008Beneficial fitness effects are not exponential for two viruses. J. Mol. Evol. 67, 368–376 (doi:10.1007/s00239-008-9153-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán R., Elena S. F.2006Epistasis correlates to genomic complexity. Proc. Natl Acad. Sci. USA 103, 14 402–14 405 (doi:10.1073/pnas.0604543103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán R., Nebot M. R.2008A network model for the correlation between epistasis and genomic complexity. PLoS ONE 3, e2663 (doi:10.1371/journal.pone.0002663) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán R., Moya A., Elena S. F.2004aThe distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc. Natl Acad. Sci. USA 101, 8396–8401 (doi:10.1073/pnas.0400146101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuán R., Moya A., Elena S. F.2004bThe contribution of epistasis to the architecture of fitness in an RNA virus. Proc. Natl Acad. Sci. USA 101, 15 376–15 379 (doi:10.1073/pnas.0404125101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. M., Haigh J.1974The hitch-hiking effect of a favourable gene. Genet. Res. 23, 23–35 (doi:10.1017/S0016672300014634) [PubMed] [Google Scholar]
- Springman R., Keller T., Molineux I., Bull J. J.2009Evolution at a high imposed mutation rate: adaptation obscures the load in phage T7. Genetics 184, 221–232 (doi:10.1534/genetics.109.108803) [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Opijnen T., Boerlijst M. C., Berkhout B.2006Effects of random mutations in the human immunodeficiency virus type 1 transcriptional promoter on viral fitness in different host cell environments. J. Virol. 80, 6678–6685 (doi:10.1128/JVI.02547-05) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A.2005Robustness and evolvability in living systems NJ, USA: Princeton University Press [Google Scholar]
- Wagner A.2008Robustness and evolvability: a paradox resolved. Proc. R. Soc. B 275, 91–100 (doi:10.1098/rspb.2007.1137) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichman H. A., Badgett M. R., Scott L. A., Boulianne C. M., Bull J. J.1999Different trajectories of parallel evolution during viral adaptation. Science 285, 422–424 (doi:10.1126/science.285.5426.422) [DOI] [PubMed] [Google Scholar]
- Wilke C. O., Adami C.2001Interaction between directional epistasis and average mutational effects. Proc. R. Soc. Lond. B 268, 1469–1474 (doi:10.1098/rspb.2001.1690) [DOI] [PMC free article] [PubMed] [Google Scholar]