

Abstract
In this study we used immunohistochemistry to screen for α-synuclein pathology in the brains of 241 individuals without clinical evidence of neurologic disease, and discovered 36 cases (15%) with incidental Lewy bodies (LBs) and one case, a 96-year-old woman (0.4%), with inclusions similar to those seen in multiple system atrophy (MSA), a nonfamilial neurodegenerative disorder characterized by parkinsonism, cerebellar ataxia and autonomic dysfunction and α-synuclein immunoreactive glial cytoplasmic inclusions (GCI). In a routine hospital autopsy series of 125 brains, we detected GCI in a neurologically normal 82-year-old man (0.8%). Both cases showed widespread GCI in the central nervous system, as well as a few neuronal cytoplasmic inclusions, but no neuronal loss or gliosis in vulnerable brain regions, including the substantia nigra, putamen, inferior olive and pontine base. Applying a recently proposed grading scale for MSA, the two cases showed pathology far below that detected in patients with clinically overt MSA, suggesting the possibility that these two individuals had preclinical MSA. The prevalence of clinically overt MSA is estimated to be about 4 per 100,000 persons (0.004%), which is far less than the frequency of GCI in this series (0.4%–0.8%). Further studies are needed to determine is GCI in neurologically normal elderly represents prodromal MSA or a rare non-progressive age-related α-synucleinopathy.
Keywords: α-synuclein, clinicopathologic, glial cytoplasmic inclusion, multiple system atrophy, preclinical
Introduction
The Lewy body is a neuronal inclusion composed of α-synuclein and the histopathologic hallmark of both Parkinson disease (PD) and dementia with Lewy bodies (DLB), which are among the most common neurodegenerative disorders of aging [17]. Lewy bodies are more common in autopsy series of neurologically normal individuals than is the frequency of PD in population-based studies [7]. For example, Lewy bodies have been detected in 10–12% of neurologically normal individuals [8], while PD affects 0.3–2% of people over 60 years of age [7]. The presence of Lewy bodies in neurologically normal individuals has been termed coincidental or incidental Lewy body disease (iLBD) and it remains uncertain if this represents prodromal PD or DLB [6].
Multiple system atrophy (MSA) is another α-synucleinopathy characterized by variable degrees of parkinsonism, cerebellar ataxia and autonomic dysfunction. MSA has neuronal loss and gliosis in the substantia nigra, posterior putamen, inferior olive, pontine nuclei and cerebellum with pathognomonic α-synuclein immunoreactive glial cytoplasmic inclusions (GCI) [21, 25]. It is unknown to what extent GCI are present in clinically normal individuals.
We used immunohistochemistry to study the frequency of α-synuclein pathology in 241 neurologically normal individuals and discovered one case with widespread GCI similar to those found in MSA. In a separate autopsy cohort of 125 cases without neurodegenerative pathology, we discovered another case with GCI in an individual without clinical evidence of neurologic disease. To our knowledge, there is only one previously reported case of MSA in an individual without clinical neurological findings [18]. The purpose of this report is to review clinical and pathological features of these two cases.
Material and Methods
Subjects
We screened brains of 241 individuals without evidence of neurologic disease on antemortem clinical evaluations with α-synuclein immunohistochemistry [6, 15]. The cases had an average age at death of 79 years (range 60 to 103 years) and included 137 women and 108 men. The tissue samples were obtained from the Tissue Registry of the Mayo Clinic, which is linked to the Mayo Medical Records System. Autopsies were performed between 1988 and 2004. All subjects had medical record documentation of several medical contacts during the last five years of life. Subjects were excluded if they had evidence of Parkinsonism, tremor, dementia or other neurodegenerative diseases. Other exclusion criteria were presence of psychiatric disorder sufficient to require hospitalization or medical treatment, epilepsy, primary intracerebral events as a cause of death, brain tumor (except incidental meningioma), systemic disorders likely to cause chronic brain damage or mental retardation.
In neuropathologic evaluations of 139 cases from a community hospital autopsy service, we discovered GCI in a patient without clinical evidence of neurologic disease. Longitudinal clinical documentation was not available in this individual. This sample included 139 cases collected over a 10-year period. The average age of this cohort was 59 years (range 18 to 60 years) and included 49 women and 90 men. Most of the cases (125 cases; 89%) in this series did not have neurodegenerative disease (8 Alzheimer disease, 3 Lewy body disease, 1 frontotemporal lobar degeneration with ubiquitin inclusions, 1 progressive supranuclear palsy and 1 Creutzfeldt Jakob disease, in addition to the 1 MSA case) and the Braak neurofibrillary tangle stage was low (median 0: range 0 to VI).
Pathological evaluation
For histological examination, formalin-fixed and paraffin-embedded sections from cortical and subcortical regions were stained with hematoxylin and eosin (H&E) and thioflavin-S fluorescent microscopy. To assess demyelination, a Luxol fast blue/periodic acid Schiff (LFB-PAS) stain was used.
In the systematic survey of 241 cases from the Mayo Clinic Tissue Registry sections of frontal and temporal cortex, cingulate gyrus, hippocampus, amygdala, basal ganglia at the level of nucleus basalis of Meynert, pons, medulla and spinal cord were screened for α-synuclein pathology with immunohistochemistry. In the autopsy series of 125 cases, α-synuclein immunohistochemistry was used only when indicated by pathologic findings on histologic studies with H&E (e.g., neuronal loss in substantia nigra or locus ceruleus) or thioflavin-S fluorescent microscopy, which shows weak fluorescent staining of Lewy bodies and GCI (unpublished observations). The hospital autopsy series, thus, cannot be considered a systematic survey.
When GCI were detected, additional sections, including motor cortex, basal ganglia at the level of nucleus accumbens and cerebellum were also immunostained for α-synuclein. The deparaffinized and rehydrated sections were pretreated with 95% formic acid for 30 minutes and steamed in distilled water for 30 minutes, and then immunostained with a previously characterized polyclonal antibody to α-synuclein (NACP; 1:3,000) [11], using 3, 3′-diaminobenzidine as the chromogen. After immunostaining, the sections were counterstained with hematoxylin.
Semi-quantitative evaluation
A recently proposed grading scheme for MSA [14] was applied to both cases. This evaluation included semiquantitative assessment of atrophy, neuronal loss and astrogliosis. Given the absence of atrophy, neuronal loss and astrogliosis, a modified rating scale was used for the number of α-synuclein-positive GCI: absent (0), rare (1), few (2), moderate (3) and large numbers (4). Grading striatonigral degeneration included evaluation of the anterior/medial and dorsal/lateral putamen, anterior and posterior caudate nucleus, medial and lateral globus pallidus as well as mediobasal and dorsolateral substantia nigra. Grading olivopontocerebellar degeneration included evaluation of substantia nigra, pontine nuclei, inferior olive and cerebellum.
Results
While screening for α-synuclein pathology in the brains of 241 individuals without clinical evidence of neurologic disease, we found 36 cases with Lewy bodies (LBs) and one case with GCI (case 1). In a separate series of 125 autopsy cases without neurodegenerative pathology, we found GCI in an elderly man without clinical evidence of neurologic disease (case 2).
Case report (case 1)
This elderly 96-year-old obese woman had a long and complicated medical history that included multiple medical problems, but no evidence of significant neurologic disease, although she had presbycusis and bilateral cataracts. Her past medical history was notable for chronic anemia, cardiovascular disease (hypertension, congestive heart failure and cardiac arrhythmia), gastrointestinal problems (esophageal hiatal hernia, gastritis, cholecystectomy, diverticulosis and chronic constipation) and urological problems (hemorrhagic cystitis). She also had osteoarthritis, degenerative joint disease, vertebral compression fractures and osteoporosis. She had urinary incontinence over the last three years of life. She lived independently until 93 years of age, when she was admitted to a nursing home because of severe degenerative joint disease. She never saw a neurologist and her neurologic examination by internists was normal except for mild hyporeflexia. She died of bronchopneumonia and acute bowel ischemia.
Pathological findings (case 1)
The brain was retrieved from the Mayo Clinic Tissue registry and had been previously sectioned. The coronal slices of the cerebrum and the transverse slices of brainstem revealed no significant pathology. In particular, there was no atrophy or discoloration of the basal ganglia, brainstem or cerebellum. There was visible pigmentation in the substantia nigra and locus ceruleus. The cerebral and cerebellar white matter showed no unusual features.
On microscopic examination with H&E stains, the neocortex was unremarkable. With thioflavin-S fluorescent microscopy a few senile plaques, but no neurofibrillary tangles (NFT) were detected. There were rare GCI with α-synuclein immunostaining in the neocortical white matter. The hippocampus had a normal neuronal population and only a few NFT. The Braak neurofibrillary tangle stage [5] was consistent with Stage II. Anti-phospho-tau immunostaining [CP13; 1:1000, Peter Davies, Albert Einstein College of Medicine, Bronx, NY [22]] revealed tau pathology consistent with argyrophilic grain disease [20] confined to the medial temporal lobe (Fig. 1a). The basal nucleus of Meynert had a normal neuronal population and no NFT, but there were a few GCI in the basal forebrain. There was minimal neuronal loss and gliosis in the lateral putamen, but many GCI (Fig. 1b). There was no obvious iron pigment. A few GCI were present in the caudate nucleus and the globus pallidus (Fig. 1c), but no neuronal cytoplasmic inclusions were detected in the basal ganglia. There were GCI in the internal capsule. The substantia nigra has moderate neuronal loss with extraneuronal neuromelanin, but no NFT or Lewy bodies. In contrast, α-synuclein immunostaining revealed many GCI in the substantia nigra (Fig. 1d). The locus ceruleus and reticular formation had normal neuronal populations. There was no myelin loss on LFB-PAS stains in brainstem fiber tracts. There were GCI and a few neuronal cytoplasmic inclusions in the pontine base and the inferior olivary nucleus (Fig. 1e–g). GCI were also found in the spinal white matter, but there was no neuronal loss or gliosis in the intermediolateral nucleus (Fig. 1h). The distribution of α-synuclein pathology is summarized in Table 1. The case was assigned a grade I of MSA-P and grade 0-I of MSA-C [14].
Fig. 1.

Microscopic findings of case 1. Phospho-tau immunohistochemistry reveals tau pathology consistent with argyrophilic grains in the amygdala (a). Immunostaining for α-synuclein reveals GCI in the putamen (b), globus pallidus (c), substantia nigra (d), pontine base (f) and spinal cord (h), as well as neuronal cytoplasmic inclusions in the inferior olive (g). Higher magnifications (lower left corners) show GCI (f) and neuronal inclusions (g). Luxol-fast blue stains reveals no myelin loss in the pontine basis (e). Bar = 50 μm (a, b, c, f. g. h), 100 μm (d, e).
Table 1.
Neuropathological findings of striatonigral degeneration. Atrophy, neuronal loss, gliosis: absent (0), minimal (1), slight (2), moderate (3), and severe (4). The number of α-synuclein-immunoreactive GCI: absent (0), rare (1-), few (2), moderate (3), large numbers (4).
| Case 1 | Case 2 | |
|---|---|---|
| Putamen, ventromedial | ||
| Atrophy | 0 | 0 |
| Neuronal loss | 0 | 0 |
| Gliosis | 0 | 2 |
| GCI | 2 | 4 |
| Putamen, dorsolateral | ||
| Atrophy | 0 | 1 |
| Neuronal loss | 1 | 2 |
| Gliosis | 1 | 3 |
| GCI | 2–3 | 4 |
| Caudate, anterior | ||
| Atrophy | 0 | 0 |
| Neuronal loss | 0 | 0 |
| Gliosis | 0 | 0 |
| GCI | 1 | 1 |
| Caudate, posterior | ||
| Atrophy | 0 | 0 |
| Neuronal loss | 0 | 0 |
| Gliosis | 0 | 0 |
| GCI | 1 | 1 |
| Globus pallidus, medial | ||
| Atrophy | 0 | 0 |
| Neuronal loss | 0 | 0 |
| Gliosis | 0 | 2 |
| GCI | 2 | 2 |
| Globus pallidus, lateral | ||
| Atrophy | 0 | 0 |
| Neuronal loss | 0 | 0 |
| Gliosis | 0 | 2 |
| GCI | 2 | 2 |
| Substantia nigra, ventrolateral | ||
| Atrophy | 0 | 0 |
| Neuronal loss | 3 | 2 |
| Gliosis | 2 | 0 |
| GCI | 3 | 1 |
| Substantia nigra, dorsolateral | ||
| Atrophy | 0 | 0 |
| Neuronal loss | 3 | 2 |
| Gliosis | 2 | 2 |
| GCI | 2 | 1 |
Case report (case 2)
This 82-year-old man had a past medical history of coronary artery disease, hypertension, hypercholesterolemia, obstructive sleep apnea and Ménière’s disease. He was hospitalized 9 months before death when he presented with a transient ischemic attack characterized by vertigo, slurred speech and fatigable nystagmus. He developed hypoxic encephalopathy 5 months before death, and he died due to multi-organ failure and sepsis. His family history was notable for a sister with Alzheimer’s disease. The neurological examination on admission to the hospital was normal except for mild generalized weakness and mild hyporeflexia. He denied any gastrointestinal or urological problems.
Neuropathologic findings (case 2)
The fixed brain weighs 1280 grams. The macroscopic findings were unremarkable, except for mild frontal atrophy and mild enlargement of the frontal horn of the lateral ventricle of coronal sections (Fig. 2). The basal ganglia, brainstem and cerebellum showed no unusual features. The substantia nigra and locus ceruleus had visible pigmentation.
Fig. 2.

Macroscopic findings of case 2. A coronal section of the forebrain shows no atrophy or discoloration in the putamen (a). Transverse sections of midbrain and pons showed well preserved pigmentation of the substantia nigra and locus ceruleus and no atrophy or white matter discoloration (b).
On microscopic examination, the neocortex was unremarkable on H&E, and there were no SP or NFT with thioflavin-S fluorescent microscopy. There was no amyloid angiopathy, but small vessels in the white matter and basal ganglia had mild adventitial collagenosis. In the cortex, a few GCI were detected in the motor cortex. The hippocampus had normal neuronal populations and only a few SP. No NFT were present in the hippocampus, but there were a few in layer II of the entorhinal cortex. The Braak neurofibrillary tangle stage was consistent with Stage II. The basal nucleus of Meynert had a normal neuronal population and no NFT, but there were GCI in the basal forebrain and hypothalamus. No GCI were present in amygdala, which is histologically unremarkable. Isolated GCI were detected In the olfactory bulb (Fig. 3a), consistent with previous reports [16]. The basal ganglia had mild neuronal loss and gliosis with iron pigment in the lateral putamen. The globus pallidus had mild gliosis and vascular calcification. Throughout the basal ganglia, especially in white matter tracts in the lateral putamen, there were many GCI (Fig. 3b). Some α-synuclein immunoreactive neurites were present in the putamen, but no neuronal cytoplasmic inclusions were detected. There were GCI in the internal capsule. The thalamus was unremarkable, except for a few GCI. The substantia nigra has mild focal neuronal loss with extraneuronal neuromelanin and gliosis, most marked in the ventrolateral region (Fig. 3c). There were no NFT or Lewy bodies, but there were a few GCI (Fig. 3d). There were GCI in the cerebral peduncle. The pontine base had no neuronal loss, gliosis or myelin loss, but there were a few GCI and isolated neuronal cytoplasmic inclusions (Fig. 3e, f). The cerebellar peduncles were unremarkable. There were GCI in the pontine and medullary tegmentum. The locus ceruleus had focal neuronal loss, but no Lewy bodies or NFT. The inferior olive had no neuronal loss or gliosis, but there were a few GCI and neuronal cytoplasmic inclusions (Fig. 3g, h). The cerebellum had no neuronal loss in Purkinje or granular cell layers. The cerebellar white matter was histologically unremarkable, but had a few GCI. The dentate nucleus was well preserved. The distribution of α-synuclein immunoreactive pathology and related pathological findings are summarized in Table 1. This case was assigned grade II of MSA-P and grade 0-I of MSA-C.
Fig. 3.
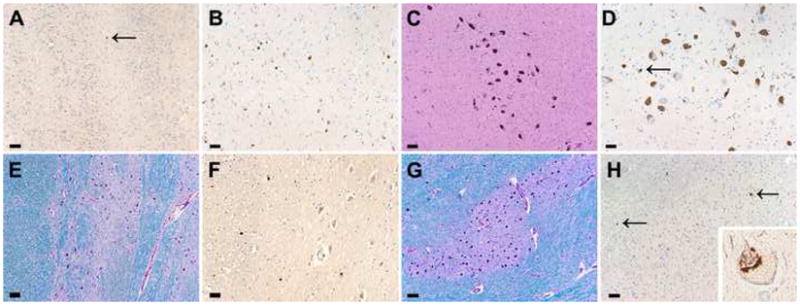
Microscopic findings of case 2. Immunostaining for α-synuclein reveals GCI in the olfactory bulb (a), putamen (b), substantia nigra (d), pontine base (f), inferior olive (h) (arrows). Higher magnification (lower right corner) shows a neuron from the inferior olive with a perinuclear inclusion (h). The neuronal population is well preserved in the substantia nigra (H&E) (c). A Luxol-fast blue stain reveals no myelin loss and well preserved neuronal populations in the pontine base (e) and inferior olive (g). Bar = 25 μm (a, b, d, f, h), 50 μm (c, e, g).
Discussion
This report describes clinical and pathologic findings of two elderly individuals without clinical evidence of neurologic disease who had α-synuclein pathology characterized by widespread GCI and sparse neuronal cytoplasmic inclusions similar to those seen in MSA. One of the two cases was detected in systematic screening for α-synuclein pathology in a series of 241 neurologically normal individuals, for a frequency of 0.4%. In this same series 36 cases had Lewy bodies, for a frequency of 15%. The other case was identified in a hospital autopsy series of 125 cases with no neuropathologic evidence of neurodegenerative disease, for a frequency of 0.8%. The latter series did not have systematic assessment of all cases with α-synuclein immunohistochemistry and the frequency is thus a lower estimate of the frequency of GCI. In a previous study of α-synuclein pathology in 290 cases of autopsy-confirmed progressive supranuclear palsy (PSP), we detected in 31 cases with LBs (11%) and one individual with GCI (0.3%) [22]. Parkkinen and co-workers detected five cases with GCI in α-synuclein immunohistochemical screen of 1800 brains (0.3%) [18]. The frequency of GCI is similar in these series and much higher than the frequency of MSA in population-based studies, which is estimated to be 3 – 4 per 100,00 persons (0.003 – 0.004%) [4, 19]. Interestingly, the frequency of Lewy bodies in autopsy series of neurologically normal individuals (10–15%) is also considerably higher than the frequency of PD in the population, which ranges from 0.3 – 2% for those 60 years of age or older [7]. The discrepancy between the frequency of Lewy bodies and GCI in autopsy series and the frequency of PD and MSA in population-based studies may support the contention that incidental Lewy bodies and GCI are static, age-related neuronal and glial findings that would not necessarily have progressed to overt neurologic disease. Alternatively, they may represent a disease process with such a slow rate of degeneration that they would never be symptomatic in the normal human lifespan.
Of the five cases with GCI reported by Parkkinen and co-workers, only one patient lacked clinical features consistent with MSA. This individual had widespread GCI in the central nervous system, but only mild neuronal loss in the dorsal motor nucleus of vagus and substantia nigra. Wakabayashi and co-workers reported on neuropathologic findings of a patient with clinical features consistent with MSA, but only a 15 month disease duration [23]. Immunostaining for α-synuclein revealed widespread GCI as well as neuronal α-synuclein and neuronal loss in the pontine basis and inferior olive. In addition, GCI and neuronal cytoplasmic inclusions were found in the intermediolateral nucleus of the spinal cord, but no autonomic dysfunction was noted. The authors speculated that autonomic pathology might have become evident if the patient had lived longer.
Results of previous studies suggest that GCI can precede neuronal degeneration [12, 26]. In the two individuals we report, neuronal cytoplasmic inclusions were sparse, restricted to the pontine nuclei and inferior olive and not associated with neuronal loss or gliosis. In contrast, GCI were found in many additional brain areas. In comparing the pathology in our two cases with those of 42 MSA cases reported by Jellinger and co-workers, which served as the basis for a proposed neuropathologic staging scheme for MSA [14], our cases had far less pathology and far less neuronal loss and gliosis. On the other hand, GCI had a similar distribution to those in MSA, which is consistent with the hypothesis that these cases have preclinical MSA.
Increasing clinical attention has been focused on preclinical symptoms in PD with recognition of the wide range of non-motor manifestations of PD, including autonomic dysfunction, olfactory dysfunction and rapid eye movement sleep behavior disorder (RBD) [1, 2, 13]. Less attention has been given to prodromal MSA. We previously reported pathologic findings in iLBD, including decreased nigrostriatal dopaminergic innervation, that suggest that it may be presymptomatic PD rather than merely an incidental age-related change [6]. Boeve and co-workers reported on a patient with idiopathic RBD with brainstem Lewy bodies, but no significant neuronal loss in the substantia nigra, again consistent with iLBD [3]. Only a few studies have reported prodromal symptoms before presentation of overt clinical signs of MSA. In one study RBD was found to antedate more clinical features of MSA by a number of years [13]. In another study Glass and co-workers reported on six individuals in which breathing difficulties were the initial manifestation of MSA [10]. They also described three patients with obstructive sleep apnea for a number of years before clinical diagnosis of probable MSA [10]. One of our two cases (case 2) had a clinical history of obstructive sleep apnea, but it is unclear if this clinical finding represents an early clinical manifestation of MSA. Urologic problems are common in MSA [9]. While one of our two cases (case 1) had urological problems, she had a number of medical reasons that could account for them.
Most cases of MSA begin in the sixth decade of life [24, 25]. Thus, the advanced age of one of our cases (96 years of age) is unusual. If one accepts this case as preclinical MSA, it may be the oldest MSA patient in the literature. It should be reiterated that neither of the individuals in this report had a clinical diagnosis of MSA or any other neurologic disease. Further clinicopathological studies are needed to determine the frequency and clinical significance of GCI in neurologically normal individuals and their possible relationship to prodromal MSA.
Table 2.
Neuropathological findings of olivopontocerebellar system. Atrophy, neuronal loss, gliosis: absent (0), minimal (1), slight (2), moderate (3), and severe (4). The number of α-synuclein-immunoreactive GCI: absent (0), rare (1), few (2), moderate (3), large numbers (4).
| Case 1 | Case 2 | |
|---|---|---|
| Pontine basis | ||
| Atrophy | 0 | 0 |
| Neuronal loss | 0 | 0 |
| Gliosis | 0 | 0 |
| GCI | 2 | 2 |
| Cerebellum | ||
| Atrophy | 0 | 0 |
| Purkinje cell loss | 0 | 0 |
| Hemispheres | 0 | 0 |
| Vermis | 0 | 0 |
| Demyelination | 0 | 0 |
| GCI | 3 | 2 |
| Inferior olive | ||
| Atrophy | 0 | 0 |
| Neuronal loss | 0 | 0 |
| Gliosis | 0 | 0 |
| GCI | 2 | 2 |
| Substantia nigra | ||
| Atrophy/depigm entation | 0 | 0 |
| Neuronal loss | 3 | 2 |
| Gliosis | 2 | 2 |
| GCI | 3 | 1 |
Acknowledgments
Supported by NIH grants P50-AG16574, P50-NS40256 and R01-AG15866, Pacific Alzheimer Research Foundation (PARF) grant C06-01
References
- 1.Abbott RD, Petrovitch H, White LR, Masaki KH, Tanner CM, Curb JD, Grandinetti A, Blanchette PL, Popper JS, Ross GW. Frequency of bowel movements and the future risk of Parkinson’s disease. Neurology. 2001;57:456–462. doi: 10.1212/wnl.57.3.456. [DOI] [PubMed] [Google Scholar]
- 2.Berendse HW, Booij J, Francot CM, Bergmans PL, Hijman R, Stoof JC, Wolters EC. Subclinical dopaminergic dysfunction in asymptomatic Parkinson’s disease patients’ relatives with a decreased sense of smell. Ann Neurol. 2001;50:34–41. doi: 10.1002/ana.1049. [DOI] [PubMed] [Google Scholar]
- 3.Boeve BF, Dickson DW, Olson EJ, Shepard JW, Silber MH, Ferman TJ, Ahlskog JE, Benarroch EE. Insights into REM sleep behavior disorder pathophysiology in brainstem-predominant Lewy body disease. Sleep Med. 2007;8:60–64. doi: 10.1016/j.sleep.2006.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bower JH, Maraganore DM, McDonnell SK, Rocca WA. Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990. Neurology. 1997;49:1284–1288. doi: 10.1212/wnl.49.5.1284. [DOI] [PubMed] [Google Scholar]
- 5.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 6.Dickson DW, Fujishiro H, Delledonne A, Menke J, Ahmed Z, Klos KJ, Josephs KA, Frigerio R, Burnett M, Parisi JE, Ahlskog JE. Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol. 2008;115:437–444. doi: 10.1007/s00401-008-0345-7. [DOI] [PubMed] [Google Scholar]
- 7.Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68:384–385. doi: 10.1212/01.wnl.0000247740.47667.03. [DOI] [PubMed] [Google Scholar]
- 8.Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1988;51:745–752. doi: 10.1136/jnnp.51.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilman S, Low PA, Quinn N, Albanese A, Ben-Shlomo Y, Fowler CJ, Kaufmann H, Klockgether T, Lang AE, Lantos PL, Litvan I, Mathias CJ, Oliver E, Robertson D, Schatz I, Wenning GK. Consensus statement on the diagnosis of multiple system atrophy. J Neurol Sci. 1999;163:94–98. doi: 10.1016/s0022-510x(98)00304-9. [DOI] [PubMed] [Google Scholar]
- 10.Glass GA, Josephs KA, Ahlskog JE. Respiratory insufficiency as the primary presenting symptom of multiple-system atrophy. Arch Neurol. 2006;63:978–981. doi: 10.1001/archneur.63.7.978. [DOI] [PubMed] [Google Scholar]
- 11.Gwinn-Hardy K, Mehta ND, Farrer M, Maraganore D, Muenter M, Yen SH, Hardy J, Dickson DW. Distinctive neuropathology revealed by alpha-synuclein antibodies in hereditary parkinsonism and dementia linked to chromosome 4p. Acta Neuropathol. 2000;99:663–672. doi: 10.1007/s004010051177. [DOI] [PubMed] [Google Scholar]
- 12.Inoue M, Yagishita S, Ryo M, Hasegawa K, Amano N, Matsushita M. The distribution and dynamic density of oligodendroglial cytoplasmic inclusions (GCIs) in multiple system atrophy: a correlation between the density of GCIs and the degree of involvement of striatonigral and olivopontocerebellar systems. Acta Neuropathol. 1997;93:585–591. doi: 10.1007/s004010050655. [DOI] [PubMed] [Google Scholar]
- 13.Iranzo A, Molinuevo JL, Santamaria J, Serradell M, Marti MJ, Valldeoriola F, Tolosa E. Rapid-eye-movement sleep behaviour disorder as an early marker for a neurodegenerative disorder: a descriptive study. Lancet Neurol. 2006;5:572–577. doi: 10.1016/S1474-4422(06)70476-8. [DOI] [PubMed] [Google Scholar]
- 14.Jellinger KA, Seppi K, Wenning GK. Grading of neuropathology in multiple system atrophy: proposal for a novel scale. Mov Disord. 2005;20(Suppl 12):S29–36. doi: 10.1002/mds.20537. [DOI] [PubMed] [Google Scholar]
- 15.Klos KJ, Ahlskog JE, Josephs KA, Apaydin H, Parisi JE, Boeve BF, DeLucia MW, Dickson DW. Alpha-synuclein pathology in the spinal cords of neurologically asymptomatic aged individuals. Neurology. 2006;66:1100–1102. doi: 10.1212/01.wnl.0000204179.88955.fa. [DOI] [PubMed] [Google Scholar]
- 16.Kovacs T, Papp MI, Cairns NJ, Khan MN, Lantos PL. Olfactory bulb in multiple system atrophy. Mov Disord. 2003;18:938–942. doi: 10.1002/mds.10466. [DOI] [PubMed] [Google Scholar]
- 17.McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 18.Parkkinen L, Hartikainen P, Alafuzoff I. Abundant glial alpha-synuclein pathology in a case without overt clinical symptoms. Clin Neuropathol. 2007;26:276–283. doi: 10.5414/npp26276. [DOI] [PubMed] [Google Scholar]
- 19.Schrag A, Ben-Shlomo Y, Quinn NP. Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet. 1999;354:1771–1775. doi: 10.1016/s0140-6736(99)04137-9. [DOI] [PubMed] [Google Scholar]
- 20.Togo T, Cookson N, Dickson DW. Argyrophilic grain disease: neuropathology, frequency in a dementia brain bank and lack of relationship with apolipoprotein E. Brain Pathol. 2002;12:45–52. doi: 10.1111/j.1750-3639.2002.tb00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trojanowski JQ, Revesz T. Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy. Neuropathol Appl Neurobiol. 2007;33:615–620. doi: 10.1111/j.1365-2990.2007.00907.x. [DOI] [PubMed] [Google Scholar]
- 22.Uchikado H, DelleDonne A, Uitti R, Dickson DW. Coexistence of PSP and MSA: a case report and review of the literature. Acta Neuropathol. 2006;111:186–192. doi: 10.1007/s00401-005-0022-z. [DOI] [PubMed] [Google Scholar]
- 23.Wakabayashi K, Mori F, Nishie M, Oyama Y, Kurihara A, Yoshimoto M, Kuroda N. An autopsy case of early (“minimal change”) olivopontocerebellar atrophy (multiple system atrophy-cerebellar) Acta Neuropathol. 2005;110:185–190. doi: 10.1007/s00401-005-1029-1. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe H, Saito Y, Terao S, Ando T, Kachi T, Mukai E, Aiba I, Abe Y, Tamakoshi A, Doyu M, Hirayama M, Sobue G. Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients. Brain. 2002;125:1070–1083. doi: 10.1093/brain/awf117. [DOI] [PubMed] [Google Scholar]
- 25.Wenning GK, Colosimo C, Geser F, Poewe W. Multiple system atrophy. Lancet Neurol. 2004;3:93–103. doi: 10.1016/s1474-4422(03)00662-8. [DOI] [PubMed] [Google Scholar]
- 26.Wenning GK, Quinn N, Magalhaes M, Mathias C, Daniel SE. “Minimal change” multiple system atrophy. Mov Disord. 1994;9:161–166. doi: 10.1002/mds.870090206. [DOI] [PubMed] [Google Scholar]