

Abstract
Recent reports indicate that the activating transcription factor 5 (ATF5) is required for the survival of cancer cells but not for non-cancer cells. However, the mechanisms by which ATF5 regulates genes and promotes cell survival are not clear. Using a cyclic amplification and selection of targets (CASTing) approach, we identified a novel ATF5 consensus DNA binding sequence. We show in C6 glioma and MCF-7 breast cancer cells that ATF5 occupies this sequence and that ATF5 activates reporter gene expression driven by this site. Conversely, reporter activity is diminished when ATF5 activity is blocked or when ATF5 expression is down-regulated by serum withdrawal. We further show that the early growth response factor 1 (Egr-1), whose promoter contains two adjacent ATF5 consensus binding sites at a conserved promoter position in rat, mouse and human, is targeted and regulated by ATF5 in C6 and MCF-7 cells. These data provide new insight on the mechanisms by which ATF5 promotes gene regulation and cancer-specific cell survival.
Keywords: ATF5, DNA regulatory element, Egr-1, cancer-specific cell survival
Introduction
ATF5 is a member of the activating transcription factor/cAMP response element-binding (ATF/CREB) protein family that represents a large group of basic leucine zipper (bZip) transcription factors with diverse physiological functions (1). Recent studies have shown that ATF5 is a regulator of cell survival that is specific to cancer cells (2, 3). ATF5 is expressed in a number of gliomas (2) and breast cancers (3) and is down-regulated in a number of cancer cell lines following growth factor deprivation, which leads to apoptosis (4). Exogenous expression of ATF5 suppresses apoptosis in HeLa cells induced by serum withdrawal and in FL5.12 cells, an interleukin 3 (IL-3)-dependent cell line, from IL-3 deprivation (4). Conversely, dominant/negative (d/n) ATF5 induces apoptosis of HeLa, FL5.12, and a number of glioma and breast cancer cell lines cultured in the presence of growth factors (2-4). Expression of d/n ATF5 also causes death of brain tumor cells in vivo (2). Death induced by interference with ATF5 expression or activity seems to be limited to cancer cells, however. Similar interference with ATF5 function in non-neoplastic breast cells or in normal cells outside of brain tumors, such as mature neurons and glial cells, did not affect their survival (2, 3, 5, 6). Confirming that interference with ATF5 function by d/n ATF5, rather than an indirect, non-specific effect, leads to death of cancer cells, a small interfering RNA (siRNA) against ATF5 also caused death of glioma cells (2). At present, little is known about how ATF5 functions to promote cancer-specific cell survival.
Studies on protein-protein interactions suggest that a given bZip protein forms stable dimers with a small number of selective partners within the group, using the interfaces formed by the bZip motifs of the two interacting proteins (7-11). ATF5 is believed to be capable of forming either homo-dimers by itself or hetero-dimers with certain members of the ATF/CREB family of bZip proteins (10). As an indication of some potential interplay between ATF5 and CREB, an electrophoretic mobility shift analysis (EMSA) showed that ATF5 binds to a cAMP response element (CRE) consensus sequence (12). Moreover, ATF5 represses cAMP-mediated activation of a CRE reporter in JEG3 cells (13) and inhibits activation of a CRE reporter in PC12 cells in response to nerve growth factor (NGF) treatment (5). These reports suggest that, at least under some circumstances, ATF5 binds CRE and acts as a CRE repressor.
Previous research also suggests that ATF5 may rely on different DNA binding properties for gene regulation. First, while the recombinant ATF5, likely in the form of an ATF5 homo-dimer, binds to CRE (12), an ATF5 hetero-dimer, which ATF5 is predicted of capable, if not preferable, of forming in a living cell (7-11), is more likely to recognize other DNA regulatory sequences that is different from CRE. Second, transcription factors in living cells usually form complexes with their natural protein partner(s) and often posttranslationally modified so that they recognize DNA sequences that are significantly different from those recognized by the corresponding recombinant protein (14-16). Therefore, it is likely that endogenous ATF5 may bind to regulatory DNA elements that are different from CRE in certain cellular contexts.
Here, we describe the identification of a novel ATF5 consensus DNA binding sequence and the activation by ATF5 of a reporter driven by this sequence in C6 glioma and MCF-7 cancer cells. We further show that the promoter of the early growth response factor 1 (Egr-1) gene contains two adjacent ATF5 consensus binding sites that are targeted and regulated by ATF5. Consequently, Egr-1, which is a known regulator of cell growth and survival, is subject to ATF5-dependent regulation in C6 and MCF-7 cells. Thus, conditions such as serum deprivation that lead to down-regulation of ATF5 also result in loss of Egr-1 expression.
Materials and Methods
Cell culture, transfection and stable cell lines
C6 rat glioma cells were grown in Dulbecco's modified Eagle medium (DMEM) with 10% newborn calf serum, 100 μg/ml streptomycin, and 100 IU/ml penicillin. For serum withdrawal, cells were washed with phosphate buffered saline (PBS; 140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 1.8 mM KH2PO4, pH7.4.) and maintained in serum-free DMEM. Cell transfection was carried out using FuGENE 6 reagent (Roche) according to the manufacturer's instructions. Stable cell lines were selected and maintained in growth medium containing 800 μg/ml of G418 (Clontech).
Plasmids
To create a mammalian vector for expressing a Flag-HA-double-tagged ATF5, we fused the rat ATF5 with the Flag-HA-tags in pCIN4 vector (17). Full length rat ATF5 was made by PCR from pCMS-EGFP-Flag-ATF5 (5) with 5′ (TTCTAGACCGGTTAACGCTAGCATGTCACTCCTGGCGACCCTGG) and 3′ (GGATCCGAATTCGCGGCCGCTAGGCACTGCGGGTCCTCTGG) primers, respectively. The PCR fragment was subsequently cloned in pCR2.1TOPO, released by XbaI and BamHI double digestion, and subcloned into the NheI and BamHI sites in pCIN4. To make pGL3-ATF5CON (ATF5CON), double-stranded oligonuclotides encoding the ATF5 consensus binding site (ATF5CON) was formed by annealing both complementary oligonucleotides ATF5u (CTAGCCACCTCTTCCTTAACA) and ATF5d (GATCTGTTAAGGAAGAGGTGG). The double-stranded DNA was directly cloned into NheI and BglII of the pGL3-promoter vector located 5′ to the SV40 promoter. Egr-1 promoter (2.0 kb) was cloned by PCR using rat brain genomic DNA as template with primers GGGGTACCCCCCGATCTTCCTTCTTCTG (Egr-1Pu) and CCGCTCGAGCCCCGAATCGGCCTCTAT (Egr-1Pd). Mutant Egr-1 promoter (1.8 kb) with the ATF5CON sites deletion was produced using PCR primers GGGGTACCGGTTGCTTCGGAGATAGGG (mutEgr-1Pu) and CCGCTCGAGCCCCGAATCGGCCTCTAT (Egr-1Pd). The WT and Mut promoters were cloned, respectively, into pGL-3 basic vector at the KpnI-XhoI sites.
Isolation of native ATF5-containing protein complexes
C6-vector control and C6-Flag-HA-ATF5 cells grown at about 80% confluence were washed 2 times with cold PBS and lysed with cold cell lysis buffer (20mM Tris-Cl, 10mM KCl, 400mM NaCl, pH7.3) containing 0.8% Triton X-100 and 1× protease inhibitor (Roche). Lysates were diluted 4 fold with cold dilution buffer (20mM Tris-Cl, 10mM KCl, pH7.3) containing 1× protease inhibitor, and were clarified by centrifugation at 15,000 rpm for 30 min at 4°C. Cell extract from C6-Flag-HA-ATF5 cells were subjected to affinity chromatography on M2 (Flag antibody)-agarose beads and the bound materials were eluted with 3×Flag peptide (Sigma). The elution was subsequently mixed with HA-affinity agarose beads (Sigma). The bound immunocomplexes were collected after washing four times with buffer (20mM Tris-Cl, 10mM KCl, 100mM NaCl, pH7.3) containing 0.2% Triton X-100. ATF5-containing protein complexes were released after incubating the immunocomplexes with HA peptide (Roche).
Immunoblotting
Immunoblotting was carried out as previously described (18). The primary antibodies were a mouse monoclonal anti-Flag (1:1000) (Stratagene), a rat anti-HA (1:1000) (Roche), and a goat anti-ATF5 antibody (1:250) (Abcam). Corresponding horseradish peroxidase-conjugated secondary antibodies were from Jackson ImmunoResearch. Signal was visualized using the Western LightningTM Chemiluminescent reagent (PerkinElmer).
In vitro translation
In vitro translation was carried out using the TNT Quick Coupled Transcription/Translation Systems (Promega) following the manufacturer's instructions. S35-methionine (Amersham) was used as the labeling reagent.
CASTing for the ATF5 consensus DNA-binding site
CASTing (cyclic amplification and selection of targets) was performed essentially as previously described (14). A target oligonucleotide, CAGCTGAGCGATCCTGCACTAG-N20-GACAGCAGGTGGAATTCCACGTG, which contains flanking PCR primer sites and a central core of 20 degenerate bases was made. 5.0 μg of this target oligonucleotide was converted to double-stranded DNA, named N20, by performing a primer extension reaction with Taq DNA polymerase, using an excess of 3′ primer (CACGTGGAATTCCACCTGCTGTC). The first cycle of CASTing used 5 μg of N20 and 4 μl of ATF5-containing protein complexes purified from C6-Flag-HA-ATF5 cell in a total volume of 20 μl of buffer (100mM NaCl, 20 mM HEPES [pH7.5], 1.5mM MgCl2, 10mM dithiothreitol, protease inhibitor cocktail, 0.1% Trition X-100, 20% glycerol). Following 20-min incubation at room temperature, 10 μl of M2 agarose beads were added, and the mixture was incubated at room temperature on a low-speed shaking platform for 1 h. The beads/ATF5/DNA complexes were retrieved by centrifugation at low speed and were washed four times with HB100 buffer solution (20 mM Tris-Cl, pH7.3, 10 mM KCl, 100 mM NaCl). The beads that contained bound DNA were added to 100 μl of PCR reaction mixture containing final concentration of 0.5 μM of the 5′ (CAGCTGAGCGATCCTGCACTAG) and the 3′ (CACGTGGAATTCCACCTGCTGTC) primers which flank the degenerate oligonucleotides, 200 μM each of deoxyribonucleotide, and 2.5 unit of Taq DNA polymerase in Taq reaction buffer (Promega). DNA was amplified by 20 cycles of PCR (94°C, 30s; 55°C, 30s; 72°C, 30s). The PCR product was subject to a second round of IP-PCR enrichment where 10 μg of sonicated salmon sperm DNA was added as a nonspecific competitor during the incubation of the ATF5 protein complexes with the PCR products. The IP-PCR process was repeated for 4 more rounds. The resulting PCR products were cloned into pCR2.1TOPO (Invitrogen).
EMSA
Nuclear and cytoplasmic extracts were isolated using NE-PER Nuclear and Cytoplasmic Extract Reagent (PIERCE) according to the manufacturer's instructions. Biotin-labeled DNA probes were made with Biotin 3′ End DNA Labeling Kit (PIERCE). Electrophoretic mobility shift assay was performed with LightShift Chemiluminescent EMSA Kit (PIERCE) according to the manufacturer's instructions. EMSA reaction was performed as described previously (19). In brief, approximately 20 fmole of biotin end-labeled double-stranded oligonucleotides were incubated with 1 μl of cell extract (∼2.0 μg of protein) and 1.5 μg of poly (dI·dC) in a total of 20 μl of reaction buffer. After a 20-min incubation at room temperature, the products were separated in nondenaturing polyacrylamide gels (4%) containing 0.5× Tris-borate-EDTA. Materials on the gel were transferred to a nylon membrane. The biotin end-labeled DNA is detected using the Streptavidin-Horseadish Peroxidase Conjugate and visualized with the Chemiluminescent Substrate. Supershifts were carried out by incubating 1 μl of primary antibody with cell extracts in binding buffer for 20 min at room temperature prior to addition of labeled oligonucleotide. Competitive binding analysis was preformed by incubating unlabeled oligonucleotide in excess to that of the probe with cell extracts in binding buffer for 20 min prior to addition of labeled oligonucleotide.
Luciferase assay
This was done as previously (19) with Renilla as an internal control. Cells were harvested 48 h after transfection or 24 h after serum withdrawal, and were lysed with buffer provided in the Promega Luciferase System (Promega). Luciferase and Renilla activities were determined using a TD20/20 Luminometer (Turner Designs). Relative Luciferase activities were obtained by normalizing the luciferase activity against Renilla activity. Data are presented as mean +/- SEM (n=3).
RT-PCR
RT-PCR was carried out using Go-Taq DNA polymerase (Promega) according to manufacturer's instruction. Cycling parameters were as following: 95°C 2 min; 35 cycles of 95°C 30 sec, 57°C 45 sec, and 72°C 45 sec; 72°C 5 min.
Survival assay
Cell survival was determined by trypan blue (0.1% in PBS) staining. Percent of unstained cells are considered viable cells. TUNEL assay (%Apoptotic) was carried out using a TUNEL Kit (Promega) following the manufacturer's instructions. Data are presented as mean +/- SEM (n=3).
ChIP assay
Chromatin immunoprecipitation (ChIP) assay was performed as previously (18) using a kit purchased from Upstate. A 194 bp Egr-1 coding region that can be amplified in PCR with primers CAGGAGTGATGAACGCAAGA and AGCCCGGAGAGGAGTAAGAG was used as a control. Egr-1 promoter region that contains the ATF5CON element (206 bp) was detected in PCR using primers TCTGACGACCCTGATCTTCC and GACGCAAGCAGTGAATGAAA.
Results
Isolation of ATF5 protein complexes from a C6 glioma cell line stably expressing a double-tagged ATF5
To facilitate isolation of native ATF5 complexes that can be used to bind and select DNA binding sequences for ATF5, we first established and characterized a C6 glioma cell line that stably expresses a Flag- and HA- double-tagged ATF5. Rat ATF5 (5) was cloned into the pCIN4 vector (17). The resultant construct, pCIN4-Flag-HA-ATF5, was verified for its correct expression of the Flag- and HA- tagged ATF5 by Western immunoblotting analysis on transient transfected C6 glioma cells using antibodies against Flag, HA, and ATF5, respectively (data not shown). The expression of the Flag-HA-ATF5, which has a calculated molecular weight of 35 kd, was further verified in an in vitro translation assay, using empty vector pCIN4 as a control (Fig 1A). The pCIN4-Flag-HA-ATF5 construct or the empty pCIN4 vector was next transfected into C6 cells and clones of stably transfected cell lines were selected in growth medium containing G418 (0.8 μg/ml). Western immunoblotting analyses, using an anti-HA antibody, on whole cell extracts of the stable cell lines indicated that most of them expressed Flag-HA-ATF5 (Fig 1B and data not shown).
Fig 1. Isolation of native ATF5-containing protein complexes from rat C6 glioma cells.
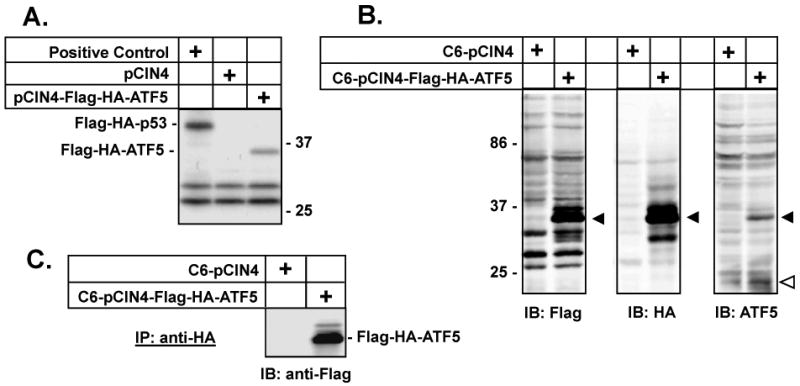
A) Verification of Flag-HA-ATF5 expression from the pCIN4-Flag-HA-ATF5 construct. In vitro translation (Promega) analysis of the pCIN4-Flag-HA-ATF5 showed that a 35 kd protein, consistent with the predicted size of double-tagged Flag-HA-ATF5, was produced as expected. pCIN4-Flag-HA-p53 (Positive Control) and pCIN4 (negative control) were used. Molecular markers (kd) are labeled on the right side. B) Selection of a C6 cell line that stably expresses Flag-HA-ATF5. pCIN4-Flag-HA-ATF5 or pCIN4 were transfected into C6 glioma cells and stable transfected cell lines were selected in growth medium containing G418 (0.8 mg/ml). Cell extracts were prepared from C6-pCIN4 or C6-pCIN4-Flag-HA-ATF5 cells and an equal amount (50 μg) of cell extracts from indicated cells was loaded for each lane and resolved by 10% SDS-PAGE. Immunoblotting (IB) analysis was carried out using indicated antibodies. Solid triangles indicate expression of Flag-HA-ATF5 (35 kd); open triangle indicates endogenously expressed ATF5 that is 22 kd (5). The difference in size (about 13 kd) between the ectopically expressed Flag-HA-ATF5 and the endogenous ATF5 is contributed by the Flag-HA-tag as well as by the apparent preference of the second potential in-frame Kozak start site that is used to produce the endogenous ATF5 in every type of cell tested so far (2, 5). The two translation start sites are separated by 100 amino acids, or about 11 kd. Molecular weights (kd) are labeled on the left side. C) Analysis of ATF5-containing complexes isolated by a dual-tag/double-immunoprecipitation (IP) strategy. Whole cell extracts from C6-pCIN4 and C6-pCIN4-Flag-HA-ATF5 cells were subjected to affinity chromatography on M2 (Flag antibody) agarose beads and an HA-affinity column. The bound proteins were eluted and examined for presence of Flag-HA-ATF5 bait, as described in Materials and Methods. A fraction of this eluate was re-IPed with anti-HA beads and subjected to 10% SDS-PAGE, followed with IB using an anti-Flag antibody. The Flag-HA-ATF5 in the elute gave a prominent band at 35 kd on the Western immunoblot.
Angelastro et al. (5) have shown that endogenous ATF5 in PC12 pheochromocytoma cells is a 22 kd protein, apparently generated from a preferential use of the second of the two Kozak start sites. To determine the size of the endogenous ATF5 expressed in C6 cells and to characterize the ATF5 antibody (Abcam) that we used, we performed an immunoprecipitation (IP)-depletion as well as an IP assay and compared the processed materials with the crude cell extract in their reactivity with the ATF5 antibody by Western immunoblotting analysis. Our data showed that a 22 kd protein in C6 cells was recognized by the ATF5 antibody, that this protein could be precipitated from the whole cell extract with anti-ATF5 beads, and that the abundance of this protein in the whole cell extract could be specifically reduced by incubation with anti-ATF5 beads (Fig S1). These data confirmed that the anti-ATF5 antibody that we used correctly recognized the endogenous ATF5 in C6 cells that has an apparent molecular weight of 22 kd.
We selected one clone (C6-Flag-HA-ATF5) that expressed the ectopic Flag-HA-ATF5 protein at a level that was similar to endogenous ATF5 (Fig 1B). Because of the near-physiological level of expression, the ectopically expressed Flag-HA-ATF5 is less likely to produce non-physiological interactions that might otherwise occur in cells with grossly overexpressed ATF5. Thus, the composition and stoichiometry of the tagged protein complexes in the selected C6-Flag-HA-ATF5 clone are likely to reflect those of native ATF5 complexes.
To isolate native protein complexes containing ATF5, whole cell extract from the stable cell line was sequentially subjected to affinity chromatography on M2 (Flag antibody) agarose beads and an HA-affinity column. A mock control was simultaneously carried out using extract from C6-pCIN4 cells. As expected, Flag-HA-ATF5 was identified in the elution from the C6-pCIN4-Flag-HA-ATF5 cells but not from the control C6-pCIN4 cells (Fig 1C). Confirming that ATF5-associated proteins were pulled down using this dual-tag/double-IP strategy, several protein bands were visualized when the ATF5 containing immunoprecipitate (but not that from control C6-pCIN4 cells) was fractionated by SDS-PAGE and stained with Coomassie Blue (data not shown). In addition, because the 22 kD endogenous ATF5 was not among the major proteins co-IPed with the tagged ATF5 (data not shown), we concluded that ATF5 heterodimer(s) rather than homodimer predominate(s) in C6 cells.
Selection of an ATF5 DNA Binding Site
Previous research has shown that purified ATF5 can bind a CRE consensus sequence in an EMSA analysis (12). In addition, ATF5 expression represses cAMP-mediated activation of a CRE reporter in JEG3 cells (13) and inhibits activation of a CRE reporter in PC12 cells in response to NGF treatment (5). There are several reasons however, that indicate that ATF5 may rely on additional DNA binding properties for other functions. First, as a member of a subfamily of the ATF/CREB family that also includes ATF4, ATF5 is predicted to form either homo- and hetero-dimers (7-11). Such ATF5 complexes may prefer DNA binding sequences other than CRE. Second, studies on the DNA-binding specificity of certain proteins, such as the nuclear phosphoprotein p53 and muscle regulatory factor myogenin, indicate that transcription factors directly isolated from cells usually form complexes with their natural protein partner(s) and often recognize DNA sequences that are significantly different from those that interact with the corresponding recombinant proteins (14, 15). In addition, posttranslational modifications such as phosphorylation, which may occur in certain cells, can play a significant role in modulating a transcription factor's transactivation potential and preference for DNA binding sequences (16). Therefore, such studies raised the possibility that ATF5 may bind to regulatory DNA elements that are different from CRE as previously known.
We carried out our study of DNA binding sites for ATF5 protein complexes in the rat C6 glioma cell, where ATF5 function is required for survival (6). We used a modified reiterative selection procedure – cyclic amplification and selection of targets (CASTing) – as described by Funk et al. (15). A set of 65-bp double-stranded DNAs that contain flanking PCR primer sites that bracket a central core of 20 degenerate bases, (designated N20) was prepared (Fig 2A; also see Materials and Methods). The N20 set was incubated with ATF5 complexes isolated from C6-pCIN4-Flag-HA-ATF5 cells and the ATF5-containing protein-DNA complexes were immunoprecipitated using an anti-HA antibody (Roche). The DNA was then purified from the immunoprecipitated material and amplified by PCR using primers that were complementary to sequences flanking the 20 degenerate bases in N20. The PCR product was subjected to a second round of protein-binding/IP/PCR enrichment and this process was repeated for 4 more rounds. The resulting PCR product was cloned into pCRII and sequenced. 29 of these sequences were aligned to obtain the maximal overlapping sequence and a conserved 11-bp CT-rich region, with the middle 9-nucleotides highly conserved, was identified (Fig 2B). 15 of the 29 sequences conform exactly at the 9-bp core region within the 11-bp consensus sequence (CYTCTYCCTTW); another 13 have only one mismatch. Outside of the 11-bp conserved region, which is boxed in Fig 2B, are sequences that bear no similarity among the clones, suggesting that each clone was an independent isolation event.
Fig 2. Identification of the ATF5 consensus DNA binding sequence.

A) Sequence of the degenerate oligonucleotide used for CASTing. N20 stands for the 20 degenerate nucleotides. 5′- and 3′-primers corresponding to the sequences flanking the N20 were used in PCR reactions to amplify the selected DNA that is bound to ATF5 complexes and immunoprecipitated between each cycle. B) Alignment of 29 sequences that were selected following six rounds of protein-DNA binding/immunoprecipitation/PCR amplification procedures (see Materials and Methods). The sequences of all 29 nucleotides that originated from “N20” and that are flanked by the primers are shown. The sequences are aligned to overlap the conserved 11 bp region that are in boldface and boxed. The lack of similarity outside the 11 bp consensus region indicated that each clone was an independent isolation event. “N”s represent undetermined nucleotides outside of the consensus region. C) ATF5 consensus DNA binding sequence. Upper Panel: the number of times each nucleotide was found in positions relative to the consensus sequence [C(C/T)TCT(T/C)CCTTA] are shown. Lower Panel: percentage conservation is calculated for each of the positions for the consensus. The high degree of conservation is particularly striking for the 9 nucleotides in the middle, omitting the first C (C1) and the last A (A11). Nucleotides T3, C4, and T9 are 100% conserved.
A statistical presentation of the conservation at each of the 11 bases within the ATF5 DNA binding region is shown in Fig 2C. While the two ending nucleotides, C1 and A11 (the number 1 and 11 positions in the conserved 11 bp sequence), are about 60% conserved, the other positions are at least 86% conserved. Three nucleotides, T3, C4, and T9, respectively, are invariable among all the 29 clones.
Specific DNA Binding by ATF5 protein complexes to the ATF5 DNA consensus sequence
To demonstrate specific binding of ATF5 to the identified ATF5 binding sequences, we synthesized a double-stranded oligonucleotide corresponding to the consensus sequence (CCTCTTCCTTA) and tested its ability in EMSA assays to interact with native ATF5-containing complexes isolated from C6 cells. As shown in Fig 3A, a single shifted band was observed when nuclear extract from C6-pCIN4 cells was incubated with the ATF5 consensus DNA binding site (ATF5CON) (compare lane 1 with lane 3). While this binding of the endogenous ATF5 was not affected by addition of antibody against HA (lane 4), it was abolished by ATF5 antibody (lane 5). Nuclear extract from C6-pCIN4-Flag-HA-ATF5 cells produced a shifted band that was more prominent (lane 7), suggesting that over-expressed Flag-HA-ATF5 interacted with ATF5CON. This shifted band was reduced partially by addition of anti-HA monoclonal antibody (lane 8), which is consistent with the interpretation that HA antibody, which can interact with ATF5 complexes in C6-pCIN4-Flag-HA-ATF5 cells (Fig 1), interfered with Flag-HA-ATF5 binding on ATF5CON. Addition of antibodies against ATF5 completely abolished ATF5 binding (lane 9). As controls, neither HA nor ATF5 antibody alone, interacted with the biotin-labeled ATF5CON in the absence of the extracts (lanes 2 and 6). These results show that ATF5 complexes in C6 glioma cells interact with the 11-bp ATF5 DNA binding consensus sequence.
FIG. 3. Specific binding of ATF5 to the consensus DNA binding sequence.
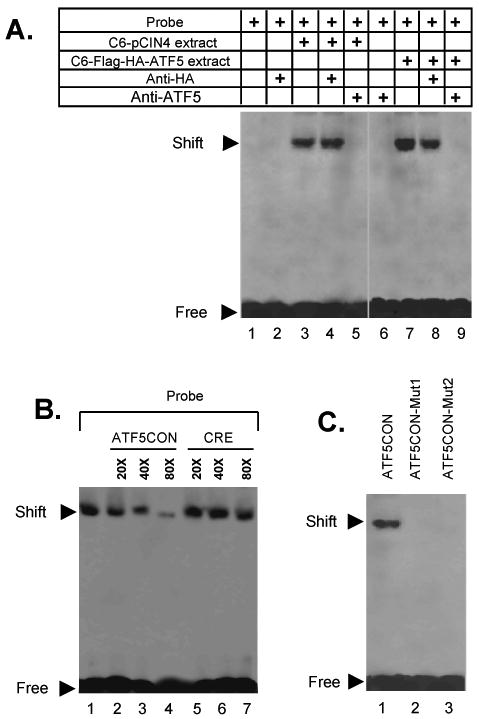
A) EMSA assays on the ATF5 consensus DNA binding sequence and extracts of C6 cells stably transfected with pCIN4 or pCIN4-Flag-HA-ATF5. While probe alone (biotin-labeled ATF5 consensus DNA binding sequence; lane 1) or probe incubated with anti-HA (lane 2) or anti-ATF5 (lane 6) anti-bodies did not show any binding, a specific ATF5 consensus DNA-binding activity was seen in extracts of C6-pCIN4 (lane 3) and of C6-pCIN4-Flag-HA-ATF5 (lane 7) cells. The stronger signal of the shifted band in lane 7 indicated additional binding by over-expressed ATF5. Addition of an anti-HA antibody did not affect binding of endogenous ATF5 to the probe (comparing lanes 3 and 4). In contrast, addition of the anti-HA antibody reduced ATF5 binding activity in the C6-pCIN4-Flag-HA-ATF5 cells (comparing lanes 7 and 8). The ATF5 binding activity that was insensitive to anti-HA antibody was at a similar level as that in the C6-pCIN4 extract (comparing lanes 3 and 8), suggesting that the anti-HA antibody specifically abolished binding of the ATF5 consensus sequence by the over-expressed Flag-HA-ATF5. Anti-ATF5 antibody completely abolished binding in C6-pCIN4-Flag-HA-ATF5 cells (lane 5). The image was generated by aligning together two representative gels. B) Comparison of competition for ATF5 binding by ATF5CON and CRE. EMSAs were carried out as in A) using ATF5 consensus DNA binding sequence as probe and extract from C6-pCIN4-Flag-HA-ATF5 cells, except that excess of non-labeled ATF5CON (CCTCTTCCTTA) or CRE (CGTGACGTCATAG) were added to the reactions prior the addition of cell extracts. C) Mutation of the ATF5CON abolishes ATF5 binding. EMSA was performed as in A) with indicated probes. ATF5CON-Mut1 (CCGATTCCTTA; lane 2) and ATF5CON-Mut2 (CCTCTTCGGTA; lane 3) contained mutations that are located at the invariably conserved T3C4 and T9 residues in the ATF5CON (Fig 2), respectively.
We next carried out EMSA assays to show specific interactions between ATF5 in C6 cells and ATF5CON. As shown in Fig 3B, binding of ATF5 to ATF5CON (probe) was reduced by increasing amounts of unlabeled ATF5CON. In contrast, an excess of unlabeled CRE did not affect ATF5 binding to ATF5CON. We also generated two ATF5CON mutants, ATF5CON-Mut1 and ATF5CON-Mut2, which carried mutations at the invariable T3/C4 and T9 nucleotides, respectively (Fig 2B), and assessed their ability to interact with ATF5 in C6 cells. As shown in Fig 3C, the mutations in ATF5CON-Mut1 and ATF5CON-Mut2 abolished their interaction with ATF5. In addition, an excess of either of the mutant probes interfered with the interaction between ATF5 protein complexes and labeled ATF5COM in a competition assay (data not shown). These results demonstrate that the ATF5 in C6 glioma cells interacts specifically with the newly identified ATF5 DNA binding consensus sequence (ATF5CON). Conspicuously, this interaction is greater than that between ATF5 and CRE, which was previously shown to interact with purified recombinant ATF5 (12).
Activation of the ATF5 consensus DNA binding site by ATF5 in C6 glioma and MCF-7 breast cancer cells
To examine whether the ATF5 consensus DNA binding site functions in vivo, and to determine whether ATF5 could act as a transcriptional regulator for it, we constructed ATF5CON-luc, an ATF5-responsive luciferase reporter in which the ATF5 consensus sequence was inserted in front of the SV40 promoter-luciferase gene in the pGL3-Promoter Vector (Promega). This ATF5CON-luc reporter or the empty control vector (pGL3) was transfected into C6 cells with renilla (for normalization as an internal control) and with or without a full-length ATF5 expression construct. While ATF5 did not have any effect on the basal luciferase activity of the control vector that does not contain the ATF5 binding site, ATF5 activated the ATF5 reporter by 3.5-fold in C6 cells (Fig 4A) and by about 5-fold in MCF-7 cells (Fig 4B). A Western immunoblot analysis of the cell extracts isolated from transfected C6 cells showed that comparative levels of ATF5 were expressed in cells co-transfected with ATF5 and pGL3 control or with ATF5 and pGL3-ATF5CON (Fig 4A insert).
FIG. 4. Specific activation of ATF5CON by ATF5 in C6 glioma and MCF-7 breast cancer cells.
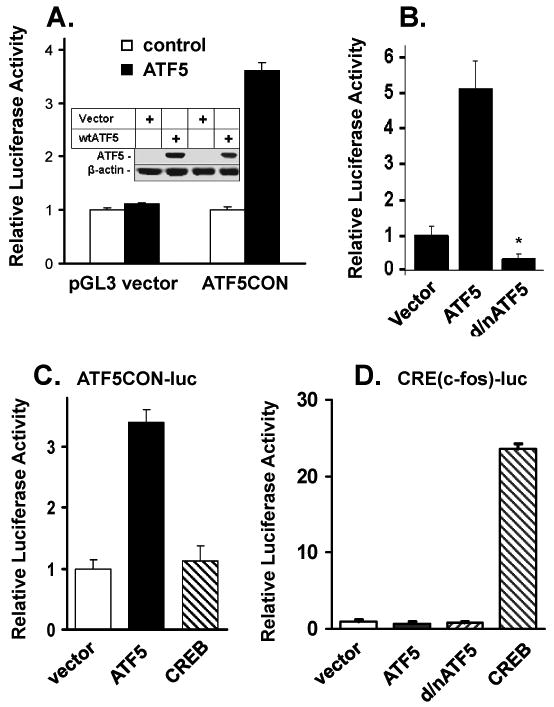
A) Expression of ATF5 stimulates a luciferase reporter containing the ATF5 consensus DNA binding site (ATF5CON). C6 cells were transfected with pGL3 vector (control) or pGL3-ATF5CON (ATF5CON) luciferase reporter with a construct (300ng) empty (control) or expressing ATF5 (ATF5). Renilla was used as an internal control for each transfection. 48 h after transfection, cells were harvested for determination of luciferase and renilla activities. Results are reported in relative light units and normalized with Renilla activity. Data are means +/- SEM (n=3). Insert: Western immunoblotting analysis on cell extracts made from C6 cells transfected as in A). Immunoblotting was carried out using anti-ATF5 (upper panel) or anti-β-actin (lower panel) as indicated. B) Regulation of ATF5CON in MCF-7 cells. Transfection of indicated constructs and luciferase analyses were performed as in A) except MCF-7 cells were used. Data are means +/- SEM (n=3). * Significance comparing with vector p<0.01. C) Specific activation of ATF5CON-luc by ATF5. ATF5CON-luc reporter, Renilla, and indicated mammalian expression vector expressing ATF5 and CREB or vector control were transfected into C6 cells. Determination of luciferase activities was carried out as in A). Data are means +/- SEM (n=3). D) Transfection and luciferase activity assays were carried out as in C) except a CRE(c-fos)-luciferase reporter and indicated expression constructs were used. Data are means +/- SEM (n=3).
We next examined whether and how the ATF5 reporter responded to different levels of ATF5 transfected into C6 cells. We found that ATF5CON-luc activity was steadily increased in response to increasing amounts of ATF5 DNA up to 300 ng per transfection; only a marginal increase was observed when more than 300 ng of ATF5 DNA was used (Fig S2). We concluded that ATF5CON responded to ATF5 expression in a dose-dependent fashion up to 300 ng and we therefore used this amount of ATF5 DNA for all subsequent reporter assays unless otherwise specified.
To determine whether ATF5CON-luc activity is responsive to interfering with endogenous ATF5 in C6 cells, we co-transfected the ATF5 reporter gene into C6 cells with and without a d/nATF5 (2, 3, 5, 6). Expression of this d/nATF5 was previously shown to interfere with the actions of ATF5 in neuroprogenitor cells (5, 6) and to induce death of several types of cancer cells that include C6 glioma and breast cancer cells (2, 3), presumably due to its ability to interfere with ATF5 binding to cognate DNA sequences. Expression of d/nATF5 depressed the ATF5CON activity by more than 50% in C6 glioma cell (data not show) and more than 60% in MCF-7 breast cancer cell (Fig 4B). These data demonstrate that ATF5 in cancer cells is an activator of the ATF5 consensus DNA binding element and that this ATF5 element could play important role in regulating genes that are critical for cancer cell survival.
Selectivity of the activation of CRE and ATF5CON by ATF5
It was previously reported that recombinant ATF5 (presumably ATF5 homo-dimer) could bind to the CRE consensus sequence in an EMSA analysis (12) and that ATF5 expression repressed CRE reporter activity in JEG3 cells (13) and in PC12 cells (5). In light of our findings that ATF5CON competes for ATF5 binding complexes isolated from C6 cells more efficiently than the CRE (Fig 3B), we sought to compare the capacity of ATF5 to activate ATF5CON and CRE in C6 cells. As shown in Fig 4C, in contrast to the robust stimulation of ATF5CON by ATF5, expression of VP16-CREB (20) did not affect the activity of ATF5CON. These data are consistent with the preferential interaction between cellular ATF5 and ATF5CON shown in the EMSA assay (Fig 3B). To examine the selectivity of the ATF5CON and CRE binding sites further, we next compared ATF5 and CREB in regulating a CRE-containing c-fos promoter-luciferase reporter (CRE) activity in C6 cells. Expression of neither ATF5 nor d/nATF5 had measurable effect on this CRE reporter, whereas expression of VP16-CREB up-regulated the CRE reporter dramatically (Fig 4D). We also obtained similar findings using a reporter construct that contains the CRE element of the Bcl-2 promoter (21) (data not shown). These experiments demonstrated that ATF5 effectively activates ATF5CON in living cells and appears to be substantially more effective in activating ATF5CON than CRE.
ATF5CON is downregulated in dying cancer cells induced by serum withdrawal and the downregulation is reversed when survival-promoting ATF5 is expressed
Growth factor deprivation has been shown to lead to down-regulation of ATF5 and the consequent cell death can be blocked by over-expression of ATF5 in HeLa and FL5.12 cells (4). To assess how ATF5CON responds to serum withdrawal in cancer cells, we compared the ATF5CON-driven reporter activity in C6 and MCF-7 cells with or without serum withdrawal. We first showed that serum withdrawal led to a precipitous drop in survival (Fig 5A) and to a dramatic increase in apoptotic cell population (Fig 5B) of C6 cells; both transiently transfected (data not shown) and stably transfected (Fig 5A,B) ATF5 repressed cell death induced by serum withdrawal. We next co-transfected ATF5CON-luc with an ATF5-expression vector or control vector and subjected the cells to serum withdrawal. Serum withdrawal led to a 50% reduction in ATF5CON activity in C6 cells (Fig 5C) and to about 65% reduction in MCF-7 cells (Fig 5D). Serum withdrawal-induced down-regulation of ATF5CON was reversed when co-transfected with an ATF5 construct (Fig 5C). A Western analysis on the C6 cell extracts isolated from these transfected cells showed that the endogenous ATF5 was expressed in cells growing in serum and was lost in those subjected to serum withdrawal, while the ectopic expressed ATF5 remained in cells subject to serum withdrawal (Fig S3). These data are consistent with the conclusion that ATF5CON activity decreases in response to the loss of ATF5 in C6 and MCF-7 induced by serum withdrawal, leading to cell death. Expression of ATF5 reverses the loss of ATF5CON activity induced by serum withdrawal and results in cell survival.
FIG. 5. ATF5 expression and ATF5CON-driven activity are down-regulated in C6 and MCF-7 cells subjected to serum withdrawal and expression of ATF5 reverses serum withdrawal-provoked down-regulation of ATF5CON activity and cell death.

A and B) Serum withdrawal promoted apoptotic death of C6 cells. C6 cells (C6) or a C6 cell line stably expressing Flag-HA-ATF5 (C6-Flag-HA-ATF5; described in Fig 1) were seeded in 24-well plates at a density of 1.5×105 per well and fed with growth medium containing 10% FBS. The medium was replaced with serum-free medium the next day (Day 0) and the cells were subsequently incubated for 3 more days. Cell viability was assessed by Trypan blue staining on each day after serum withdrawal (A) while the ratio of apoptotic cells were determined by TUNEL assay (B). The numbers of surviving cells before serum withdrawal were set at 100% in A). Data are means +/- SEM (n=3). C) Downregulation of ATF5CON activity by serum withdrawal was reversed by expression of ATF5 in C6 cell. C6 cells were transfected with Control vector (pLeGFP) or ATF5 (pLeGFP-Flag-ATF5) together with the ATF5CON-luc reporter and Renilla. 24 h later, cells were either re-fed with growth medium (+serum) or subjected to serum withdrawal (-serum). Cells were harvested 24 h later and luciferase activity was determined as in Fig 4A. Data are means +/- SEM (n=3). D) Serum withdrawal led to down-regulation of ATF5CON activity in MCF-7 cells. Experiment was carried out as in C) except MCF-7 cells were used. Data are means +/- SEM (n=3).
Identification of potential ATF5-dependent genes whose promoters contain the ATF5 consensus DNA binding site
We hypothesized that ATF5-responsive genes can be identified by virtue of their promoters containing regulatory sequences homologous to the ATF5 consensus DNA binding site. As an initial assessment, we searched the rat genome for genes whose promoter contains sequences that match the ATF5CON sequence (CYTCTYCCTTW) completely, using an online Transcription Element Search System (TESS) provided on the University of Penn website - http://www.cbil.upenn.edu/cgi-bin/tess/tess. We set the promoter region from 2000 bp upstream (-2000 bp) to the transcription initiation start site (+1). This search produced 234 potential ATF5-dependent transcription targets. Significantly, many of these genes are known regulators for cell growth and cell survival, among which are Egr1, Eif2b1, GSK3β, RASSF1, Rbp1, Smad4, and Skp1a (Supplemental Table 1). This information seems consistent with ATF5's reported roles in cell cycle regulation (13), maintenance of stem/progenitor cell and neuronal and glia differentiation (5, 6, 22), and cancer specific cell survival (2, 3).
Egr-1, whose promoter contains ATF5 sites, is subject to ATF5-dependent regulation in cancer cells
To see if Egr-1, one of the genes with an ATF5CON site in its putative promoter, is regulated by ATF5, we first examined the expression profile of Egr-1 in response to expression of ATF5. As shown in Fig 6A, the expression level of ATF5 mRNA was elevated in C6-Flag-HA-ATF5 cells compared to that in parental C6 cells and in C6-pCIN4 cells (compare lane 2 with lanes 1 and 3 in the upper panel). Accordingly, the level of Egr-1 mRNA was much higher in C6-Flag-HA-ATF5 cells than in parental C6 cell and in C6-pCIN4 cells (Fig 6A, compare lane 2 with lanes 1 and 3 in the middle panel). The concomitant regulation of ATF5 and Egr-1 is consistent with the possibility that ATF5 directly transactivates Egr-1. As serum withdrawal led to down-regulation of endogenous ATF5 mRNA (4) and protein (Fig S3), we next determined whether Egr-1 would be similarly down-regulated. As shown in Fig 6A, serum withdrawal depleted endogenous ATF5 in C6 and C6-pCIN4 cells (compare lanes 1 with 4, and 3 with 6, in the upper panel). In parallel, we observed that the expression of Egr-1 was also diminished in C6 and C6-pCIN4 cells subjected to serum withdrawal (Fig 6A, compare lanes 1 with 4, and 3 with 6, middle panel). Moreover, ectopically expressed ATF5 in C6-Flag-HA-ATF5 cells (compare lane 5 with lanes 4 and 6, in the upper panel) prevented the loss of endogenous Egr-1 that otherwise would be resulted from serum withdrawal (compare lane 5 with lanes 4 and 6, in the middle panel). A similar corresponding relationship was observed in expression of ATF5 and Egr-1 in MCF-7 cells (compare lane 7 with lane 8; and data not shown). These data support the hypothesis that ATF5 binds to ATF5CON sites on the Egr-1 promoter and stimulates Egr-1 transcription.
FIG. 6. ATF5 transactivates Egr-1 via the ATF5CON sites in the Egr-1 promoter.
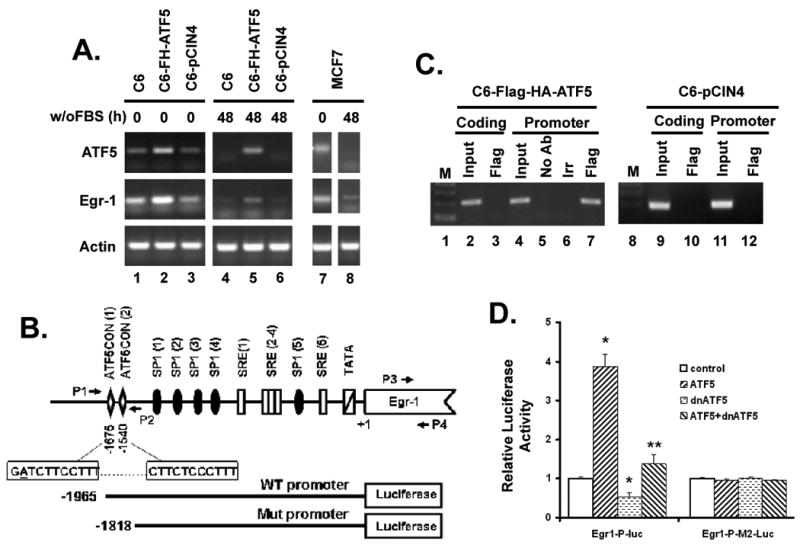
A) Concomitant regulation of ATF5 and Egr-1 in C6 and MCF-7 cells. Total RNA was harvested from C6, C6-pCIN4-Flag-HA-ATF5 (C6-FH-ATF5), and C6-pCIN4 (C6-pCIN4) cells, and from MCF-7 cells grown either in 10% or 0% FBS for 48 hours. mRNA was reversely transcribed into cDNA using oligo(dT)12∼18. Relative levels of ATF5 and Egr-1 transcripts were determined by semi-quantitative RT-PCR using primers specific to their respective mRNAs. β-actin (Actin) was used as loading control. B) Schematic illustration of the rat Egr-1 promoter region and the wild type (WT) and mutant (Mut) Egr-1 promoter luciferase reporters. The rat Egr-1 regulatory region contains many different functional elements, including two ATF5CON sites beginning at nucleotide -1675 and -1540, respectively. P1 and P2 represent the primers flanking the ATF5CON site, which were used to amplify the 201 bp fragment of Egr-1 promoter region from -1689 to -1489; P3 and P4 represent the primers that were used to amplify a 194 bp fragment in the coding region of Egr-1 from +1137 to +1330. The sequence of the two putative ATF5 binding sites was given in the two boxes aligned with the -1675 and -1540 regions. A single tolerable mismatch (A) in the first homologous region is underlined. The wild type (WT) and mutant (Mut) Egr-1 promoter luciferase reporters are aligned at the bottom. C) ATF5 is specifically associated with the endogenous Egr-1 promoter region. ChIP assays were carried out using a kit from Upstate following manufacturer's instruction. Chromatin was prepared using C6-pCIN4-Flag-HA-ATF5 (lanes 2-7) and C6-pCIN4 (lanes 9-12) cells. The amount of chromatin used for “Input” in lanes 2, 4, 9 and 11 was 10% of that used for IP experiments. Primers 1 and 2 were used for detection of precipitated promoter (Promoter, lanes 4-7 and 11-12). Primers 3 and 4 were used to detect Egr-1 coding region (Coding, lanes 2-3 and 9-10) where no sequence resembling the ATF5CON is located, which was used as control. The Flag antibody was expected to pull down the ectopically expressed Flag-HA-ATF5 in C6-pCIN4-Flag-HA-ATF5 cells while an irrelevant X-press (Irr) antibody and a mock experiment (No Ab) were used as controls. The same Flag antibody did not pull down the Egr-1 promoter in C6-pCIN4 cells (lanes 10 and 12). D) Egr-1 promoter is activated by ATF5 and repressed by d/nATF5. Luciferase reporter assay was carried out as in Fig 4A except wild type (WT) and mutant (Mut) Egr-1 promoter-luciferase reporters and indicated ATF5 constructs were used. Data are means +/- SEM (n=3). Significance comparing with control: * p<0.01; ** p<0.05.
To confirm that ATF5 indeed binds to ATF5CON sites on the endogenous Egr-1 promoter, we performed ChIP analysis using the C6-pCIN4 and C6-Flag-HA-ATF5 cell lines. We first examined the rat Egr-1 promoter sequence and found that it contains two ATF5CON sites (Fig 6B). One is located at -1540 with respect to the transcription start site (+1) and it was identified during the initial search (Supplemental Table 1) because it matches perfectly with the 9-Nucleotide core of the ATF5CON. The other site is at -1675 and it contains a single tolerable mismatch (the underlined A in Fig 6B) compared to the consensus sequence. Significantly, the two adjacent ATF5CON sites and the surrounding areas are highly conserved between rat and mouse; the sequences of the ATF5CON sites and their locations relative to the transcription start site are also maintained in the human EGR-1 gene. In Fig 6C, we showed that, in contrast to the chromatin corresponding to the Egr-1 coding sequence, which does not contain sequences homologous to ATF5CON and could not be recovered in a parallel ChIP experiment (lane 3), chromatin of the Egr-1 promoter region that contains the ATF5CON sites was specifically pulled down by an antibody against Flag that recognizes Flag-HA-ATF5 in the C6-Flag-HA-ATF5 cell extracts (lane 7). Neither mock experiments with no antibody (No Ab; lane 5) or an irrelevant control antibody (Irr; lane 6) nor with Flag antibody on chromatin from C6-pCIN4 cells (lanes 10 and 12) precipitated DNA materials. A ChIP analysis on the Egr-1 promoter in MCF-7 cells, using a pair of primers corresponding to the human Egr-1 promoter region, showed a similar result (data not shown). These data indicate that ATF5 in cancer cells binds specifically to the regulatory sequences in the Egr-1 promoter that are homologous to ATF5CON.
To show that ATF5 directly regulates Egr-1 promoter via the ATF5CON sites, we created pGL-3 luciferase reporter constructs that contain either the wild type (WT) Egr-1 promoter or a mutant (Mut) Egr-1 promoter whose ATF5CON sites were deleted (Bottom of Fig 6B). While cotransfection of neither ATF5 nor d/nATF5 with the Mut Egr-1 promoter reporter affected the activity of the Mut reporter, expression of ATF5 stimulated the WT reporter by about 4-fold and expression of d/nATF5 repressed the WT reporter by 40% (Fig 6D). Taken together, these data showed that Egr-1 is a downstream target of ATF5 in cancer cells and that Egr-1 promoter is regulated directly by ATF5 in an ATF5CON sites-dependent manner.
Discussion
Although evidence has recently been presented that the transcription factor ATF5, an ATF/CREB family member and bZip protein, functions as a cancer-specific cell survival factor, the mechanism by which ATF5 promotes survival of cancer cells and the nature of ATF5-regulated genes are not known. In this study, we identified a novel ATF5 binding DNA regulatory element that positively responds to ATF5 expression in cancer cells. In addition, we showed that Egr-1, a transcription factor that promotes cell growth and cell survival, is a downstream target of ATF5; ATF5 binds to the two ATF5 regulatory elements in the promoter of Egr-1 and stimulates Egr-1 expression. These results shed new light on the mechanism by which ATF5 promotes cancer-specific cell survival.
Reiterative selection procedures such as the one we employed here have been widely used to identify consensus DNA binding sites for transcription factors (15, 23). We first purified native ATF5 protein complexes from a C6 glioma cell line that expresses physiological level of double-tagged Flag-HA-ATF5. Because of the subdued level of expression that makes the selected clone to contain 2× ATF5 proteins, the ectopically expressed Flag-HA-ATF5 is less likely to produce non-physiological interactions that might otherwise occur when ATF5 is grossly overexpressed. Importantly, the physiological level of expression of Flag-HA-ATF5 showed exactly the same pro-survival function in C6 and MCF-7 cells as endogenous ATF5, indicating that Flag-HA-ATF5 can substitute for the endogenous ATF5 functionally. Thus, the composition and stoichiometry of the tagged protein complexes in the selected C6-Flag-HA-ATF5 clone are likely to reflect those of native ATF5 complexes. Using purified native Flag-HA-ATF5 protein complexes, we were able to select the DNA sequences that specifically interact with the ATF5-containing protein complexes. Because the composition of the protein complexes that contain the endogenous ATF5 and the tagged ATF5 in the parental C6 and in the selected C6-Flag-HA-ATF5 clone are not yet determined, the purified native Flag-HA-ATF5 protein complexes may in fact represent a variety of ATF5-containing complexes with different capacity to interact with ATF5 consensus DNA site. It would be interesting to identify the ATF5 complex(es) that is(are) responsible for promoting survival of C6 and MCF-7 cells. The ATF5 consensus DNA binding site, which was complied from 29 independently isolated ATF5-bound sequences, is a highly conserved stretch of 11 nucleotides of pyrimidine-rich sequence CYTCTYCCTTW, where T3, C4, and T9 are invariable. Expression of ATF5 stimulated a reporter that contained this conserved ATF5 consensus DNA binding site, while expression of d/nATF5 or serum withdrawal, which down-regulates ATF5 and induces death of cancer cells, depressed the reporter.
Previous studies have shown that recombinant ATF5 can bind to CRE in an EMSA assay (12) and that ATF5 expression represses a CRE-containing reporter in JEG3 cells (13) and in PC12 cells (5). We therefore compared ATF5CON and CRE for their abilities to interact with ATF5 or CREB and for their capacities to drive reporters in response to expression of ATF5 or CREB in cancer cells. Our data indicate that native ATF5 isolated from C6 glioma cells has a much higher affinity to ATF5CON than to CRE, and that ATF5CON responds much more robustly to ATF5 than to CREB in C6 glioma and MCF-7 breast cancer cells. On the other hand, our data showed that, in contrast to the dramatic stimulation of CRE-bearing c-fos and Bcl-2 promoter-luc reporters by CREB, ATF5 expression in C6 and MCF-7 cells did not repress nor stimulate those CRE-bearing reporters. These data indicate that the ATF5CON is a key DNA regulatory element that binds and responds to ATF5 in cancer cells. Therefore, genes whose promoters contain sequences homologous to ATF5CON might be among the major transcriptional targets regulated by ATF5 and they could be the key molecules that carry out ATF5's pro-survival function in cancer cells.
While ATF5 is expressed in and is required for survival of a variety of cancer cell types (2-4), it is also expressed in many non-transformed cell types although not required for their survival (3). A recent report showed that ATF5 is down-regulated in hepatocellular carcinomas and re-expression of ATF5 in those cells led to cell cycle arrest at the G2-M phase (24). In addition, it appears that ATF5 plays an important role in maintaining the proliferative state of progenitor cells for neurons and glia (5, 6, 22) and is induced in response to a variety of cellular stresses (25-27) and by circadian signals (28). Thus, although we have focused here on the role and actions of ATF5 in cancer-specific cell survival, the widespread expression of ATF5 suggests that its interaction with ATF5CON may make important contributions to gene regulation in other functions in both transformed and non-transformed tissues.
A BLAST search of the rat genome database at the National Center for Biotechnology Information revealed 234 genes (Supplemental Table 1) whose promoters contain sequences that match the ATF5CON. Since only promoters (between -2000 to +1 with respect to transcription start site) with a perfect match were identified, this search may vastly underestimate the total number of ATF5-responsive genes. Nevertheless, the identified genes include significant number of molecules that regulate cell proliferation and cell survival, which are functions overlapping with ATF5's. This information indicates the potential to use the ATF5CON sequence for identification of putative transcription targets of ATF5. A systematic bioinformatics search of potential ATF5-responsive targets based on promoter sequence that is homologous to ATF5CON is underway.
We carried out further analysis on Egr-1, which is one of the potential ATF5-regulated target genes identified and is a member of the immediate early gene response family involved in the regulation of cell growth and survival. We showed that ATF5 binds specifically to the promoter region of Egr-1 gene where two ATF5CON sequences are located and are aligned similarly in rat, mouse and human. Expression of Egr-1 responded to expression of ATF5 in both C6 glioma and MCF-7 breast cancer cells. In addition, we demonstrated that Egr-1 promoter is stimulated by ATF5 and repressed by d/nATF5 while a mutant Egr-1 promoter with deletion of the ATF5CON element is insensitive to ATF5 regulation. These data support the conclusion that Egr-1 is a direct down-stream target of ATF5.
Supplementary Material
Acknowledgments
Supported in part by RSG-08-288-01-GMC from American Cancer Society (DXL).
We thank Dr. Muyang Li for helpful discussion and suggestions.
References
- 1.Hai TW, Liu F, Coukos WJ, Green MR. Transcription factor ATF cDNA clones: an extensive family of leucine zipper proteins able to selectively form DNA-binding heterodimers. Genes & development. 1989;3(12B):2083–90. doi: 10.1101/gad.3.12b.2083. [DOI] [PubMed] [Google Scholar]
- 2.Angelastro JM, Canoll PD, Kuo J, et al. Selective destruction of glioblastoma cells by interference with the activity or expression of ATF5. Oncogene. 2006;25(6):907–16. doi: 10.1038/sj.onc.1209116. [DOI] [PubMed] [Google Scholar]
- 3.Monaco SE, Angelastro JM, Szabolcs M, Greene LA. The transcription factor ATF5 is widely expressed in carcinomas, and interference with its function selectively kills neoplastic, but not nontransformed, breast cell lines. International journal of cancer. 2007;120(9):1883–90. doi: 10.1002/ijc.22469. [DOI] [PubMed] [Google Scholar]
- 4.Persengiev SP, Devireddy LR, Green MR. Inhibition of apoptosis by ATFx: a novel role for a member of the ATF/CREB family of mammalian bZIP transcription factors. Genes & development. 2002;16(14):1806–14. doi: 10.1101/gad.992202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Angelastro JM, Ignatova TN, Kukekov VG, et al. Regulated expression of ATF5 is required for the progression of neural progenitor cells to neurons. J Neurosci. 2003;23(11):4590–600. doi: 10.1523/JNEUROSCI.23-11-04590.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Angelastro JM, Mason JL, Ignatova TN, et al. Downregulation of activating transcription factor 5 is required for differentiation of neural progenitor cells into astrocytes. J Neurosci. 2005;25(15):3889–99. doi: 10.1523/JNEUROSCI.3447-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deppmann CD, Acharya A, Rishi V, et al. Dimerization specificity of all 67 B-ZIP motifs in Arabidopsis thaliana: a comparison to Homo sapiens B-ZIP motifs. Nucleic acids research. 2004;32(11):3435–45. doi: 10.1093/nar/gkh653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fong JH, Keating AE, Singh M. Predicting specificity in bZIP coiled-coil protein interactions. Genome biology. 2004;5(2):R11. doi: 10.1186/gb-2004-5-2-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newman JR, Keating AE. Comprehensive identification of human bZIP interactions with coiled-coil arrays. Science (New York, NY. 2003;300(5628):2097–101. doi: 10.1126/science.1084648. [DOI] [PubMed] [Google Scholar]
- 10.Vinson C, Acharya A, Taparowsky EJ. Deciphering B-ZIP transcription factor interactions in vitro and in vivo. Biochimica et biophysica acta. 2006;1759(1-2):4–12. doi: 10.1016/j.bbaexp.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 11.Vinson C, Myakishev M, Acharya A, Mir AA, Moll JR, Bonovich M. Classification of human B-ZIP proteins based on dimerization properties. Molecular and cellular biology. 2002;22(18):6321–35. doi: 10.1128/MCB.22.18.6321-6335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters CS, Liang X, Li S, et al. ATF-7, a novel bZIP protein, interacts with the PRL-1 protein-tyrosine phosphatase. The Journal of biological chemistry. 2001;276(17):13718–26. doi: 10.1074/jbc.M011562200. [DOI] [PubMed] [Google Scholar]
- 13.Pati D, Meistrich ML, Plon SE. Human Cdc34 and Rad6B ubiquitin-conjugating enzymes target repressors of cyclic AMP-induced transcription for proteolysis. Molecular and cellular biology. 1999;19(7):5001–13. doi: 10.1128/mcb.19.7.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wright WE, Binder M, Funk W. Cyclic amplification and selection of targets (CASTing) for the myogenin consensus binding site. Molecular and cellular biology. 1991;11(8):4104–10. doi: 10.1128/mcb.11.8.4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Funk WD, Pak DT, Karas RH, Wright WE, Shay JW. A transcriptionally active DNA-binding site for human p53 protein complexes. Molecular and cellular biology. 1992;12(6):2866–71. doi: 10.1128/mcb.12.6.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deppmann CD, Thornton TM, Utama FE, Taparowsky EJ. Phosphorylation of BATF regulates DNA binding: a novel mechanism for AP-1 (activator protein-1) regulation. The Biochemical journal. 2003;374(Pt 2):423–31. doi: 10.1042/BJ20030455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen D, Kon N, Li M, Zhang W, Qin J, Gu W. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell. 2005;121(7):1071–83. doi: 10.1016/j.cell.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 18.Liu DX, Nath N, Chellappan SP, Greene LA. Regulation of neuron survival and death by p130 and associated chromatin modifiers. Genes & development. 2005;19(6):719–32. doi: 10.1101/gad.1296405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu DX, Greene LA. Regulation of neuronal survival and death by E2F-dependent gene repression and derepression. Neuron. 2001;32(3):425–38. doi: 10.1016/s0896-6273(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 20.Barco A, Alarcon JM, Kandel ER. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002;108(5):689–703. doi: 10.1016/s0092-8674(02)00657-8. [DOI] [PubMed] [Google Scholar]
- 21.Pugazhenthi S, Nesterova A, Sable C, et al. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. The Journal of biological chemistry. 2000;275(15):10761–6. doi: 10.1074/jbc.275.15.10761. [DOI] [PubMed] [Google Scholar]
- 22.Mason JL, Angelastro JM, Ignatova TN, et al. ATF5 regulates the proliferation and differentiation of oligodendrocytes. Mol Cell Neurosci. 2005;29(3):372–80. doi: 10.1016/j.mcn.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. The Journal of biological chemistry. 2000;275(35):27013–20. doi: 10.1074/jbc.M003322200. [DOI] [PubMed] [Google Scholar]
- 24.Gho JW, Ip WK, Chan KY, Law PT, Lai PB, Wong N. Re-expression of transcription factor ATF5 in hepatocellular carcinoma induces G2-M arrest. Cancer research. 2008;68(16):6743–51. doi: 10.1158/0008-5472.CAN-07-6469. [DOI] [PubMed] [Google Scholar]
- 25.Pascual M, Gomez-Lechon MJ, Castell JV, Jover R. ATF5 is a highly abundant liver-enriched transcription factor that cooperates with constitutive androstane receptor in the transactivation of CYP2B6: implications in hepatic stress responses. Drug metabolism and disposition: the biological fate of chemicals. 2008;36(6):1063–72. doi: 10.1124/dmd.107.019380. [DOI] [PubMed] [Google Scholar]
- 26.Zhou D, Palam LR, Jiang L, Narasimhan J, Staschke KA, Wek RC. Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. The Journal of biological chemistry. 2008;283(11):7064–73. doi: 10.1074/jbc.M708530200. [DOI] [PubMed] [Google Scholar]
- 27.Watatani Y, Kimura N, Shimizu YI, et al. Amino acid limitation induces expression of ATF5 mRNA at the post-transcriptional level. Life sciences. 2007;80(9):879–85. doi: 10.1016/j.lfs.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 28.Lemos DR, Goodspeed L, Tonelli L, Antoch MP, Ojeda SR, Urbanski HF. Evidence for circadian regulation of activating transcription factor 5 but not tyrosine hydroxylase by the chromaffin cell clock. Endocrinology. 2007;148(12):5811–21. doi: 10.1210/en.2007-0610. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.