

Abstract
Currently available synthetic biodegradable elastomers are primarily composed of crosslinked aliphatic polyesters, which suffer from deficiencies including (1) high crosslink densities, which results in exceedingly high stiffness, (2) rapid degradation upon implantation, or (3) limited chemical moieties for chemical modification. Herein, we have developed poly(1,3-diamino-2-hydroxypropane-co-polyol sebacate)s, a new class of synthetic, biodegradable elastomeric poly(ester amide)s composed of crosslinked networks based on an amino alcohol. These crosslinked networks feature tensile Young’s modulus on the order of 1 MPa and reversable elongations up to 92%. These polymers exhibit in vitro and in vivo biocompatibility. These polymers have projected degradation half-lives up to 20 months in vivo.
Keywords: Biodegradable, Elastomer, Tissue engineering
1. Introduction
One critical aspect in realizing therapeutic tissue-engineered systems is the synthesis and fabrication of scaffolds with appropriate chemical, physical, and mechanical properties [1,2]. Typical strategies that guide materials processing and development for tissue engineering applications often focus on biomimicry of the extra-cellular matrix (ECM), a heterogeneous network of proteins that provides support for cell attachment and chemical cues for seeded cells and surrounding tissue. Implants are subjected to dynamic mechanical environments in vivo, which is driving the need for biodegradable elastomers with chemical, physical, and mechanical properties of native ECM for tissue engineering applications. While natural biodegradable elastomers [3-5] are composed of natural proteins and support cell attachment [6], there are difficulties in bulk material processing and potentially dangerous immune responses upon implantation [7]. Numerous synthetic biodegradable elastomers [8-18] have been developed to meet this growing, unmet need. Synthetic biodegradable elastomers are typically composed of crosslinked networks of aliphatic molecules with polyester linkages. These structural elements have led to several deficiencies including either: (1) extremely high crosslinking densities resulting in high modulus materials [15], (2) rapid biodegradation upon implantation [17-19], or (3) limited functional groups that limit the suite of available chemistries for covalent conjugation of biomolecules including proteins and growth factors [8].
Herein we describe the formation and characterization of a new class of synthetic, elastomeric, biodegradable poly-(ester amide) based on the amino alcohol 1,3-diamino-2-hydroxy-propane (DAHP). This new class of biodegradable elastomers, termed poly(1,3-diamino-2-hydroxypropane-co-polyol sebacate)s (APSs), exhibits many advantages of both natural and synthetic materials for tissue engineering applications. We posed five general criteria that led to the selection of DAHP as the appropriate monomer from which to base our material design: (1) the monomer should be non-toxic [20] to enhance biocompatibility and tri-functional in nature to enable the formation of crosslinked networks; (2) the melting point of the monomer should be low (<80 °C) to enable rapid, scalable, thermal batch polymerization processing; (3) the monomer should contain primary amines to participate in hydrogen bonding, and be available for subsequent conjugation reactions; (4) the monomer should be able to form both amide and ester bonds to enable tunable properties including biodegradation half-life; and (5) the monomer should be inexpensive and commonly used in biomedical systems. The polycondensation reaction of an amine-containing monomer is predicted to form primary and secondary amide bonds, which also provide several advantages including a mechanism to control physical properties, mechanical properties, and biological response. DAHP is the most appropriate monomer that satisfies all of these criteria and was therefore used as the multifunctional amine-containing monomer.
We formed biodegradable elastomeric APS polymers through a facile and efficient copolymerization of DAHP with a polyol and a diacid. The polyols and diacid investigated in this study were glycerol, d,l-threitol, and sebacic acid, respectively. Polycondensation of these monomers led to the formation of chemically crosslinked polymers through the formation of ester and amide linkages. These elastomeric polymers are optically transparent, exhibit robust mechanical properties, and demonstrate biocompatibility in vitro and in vivo. The combination of these properties suggests that APS polymers exhibit desirable chemical, physical, and mechanical properties, which are suitable for a variety of biomedical applications.
2. Materials and methods
2.1. Synthesis of poly(1,3-diamino-2-hydroxy-propane-co-polyol sebacate) elastomers
All materials were purchased from Sigma–Aldrich (St. Louis, MO, USA) and used as received unless otherwise specified. The polymer formulation 2DAHP–1G was synthesized in the following manner (Fig. 1). A round-bottom flask was charged with 0.06 mol of 1,3-diamino-2-hydroxy-propane (DAHP), 0.03 mol glycerol (G), and 0.09 mol of sebacic acid (SA) to produce a molar ratio of 2:1:3 of DAP:G:SA, respectively. The polymer formulation 2DAHP–1T was synthesized in a similar manner: a round-bottom flask with 0.06 mol of DAHP, 0.03 mol d,l-threitol (T), and 0.09 mol of sebacic acid (SA) to produce a molar ratio of 2:1:3 of DAHP:T:SA, respectively. The reactants were heated under a nitrogen blanket at 120 °C for 3 h. The pressure was then dropped to approximately 50 mTorr and the contents were allowed to react for 9 h at 120 °C. The product was then stored under a desiccate environment until further use. The product was spread onto glass slides and cured at 170 °C at approximately 50 mTorr for either 24 h or 48 h. Film thicknesses of either 0.5 mm or 1 mm were achieved by applying the reaction product at surface densities of 100 mg/cm2 and 200 mg/cm2, respectively. Films were delaminated in ddH2O at 70 °C for 18 h. Sol was removed by incubating polymer films in 100% ethanol for 24 h followed by washing and incubation with ddH2O. Subsequent characterization techniques were performed on sol-free samples only.
Fig. 1.

Synthesis scheme of APS polymers. The general synthetic scheme of APSs incorporated the following monomers: (1) a multifunctional amine group (A), which was chosen to be 1,3-diamino-2-hydroxypropane in this example, (2) a polyol (P), and (3) a diacid (S). Glycerol and d,l-threitol were chosen as representative polymers while sebacic acid was chosen as the diacid because of its ubiquitous presence in polyesters for biomedical applications. These monomers were melted at 120 °C under a nitrogen blanket followed by a reduction in pressure to induce polymerization. Further polymerization to produce solid slabs was continued at 170 °C at 50 mTorr. In this scheme, R1 represents either a single hydrogen, or bond to either the X-segment or the Y-segment via amide bond. R2 represents either a single hydrogen, or bond to X-segment or Y-segment via ester bond. See Section 2 for details on synthesis.
2.2. Characterization of 1,3-diamino-2-hydroxy-propane-based APS elastomers
The intrinsic viscosity of APS pre-polymers in 1,1,1,3,3,3-hexafluoroiso-propanol (HFIP) was measured using an Ubbelohde viscometer. Briefly, the pre-polymer was dissolved in HFIP in a series of concentrations from approximately 0.1–4 g/dL, filtered, and placed into the viscometer, which was kept at 30 ± 0.05 °C in a constant temperature water bath. At least two replicate measurements were taken for each sample. The intrinsic viscosity was used to determine the molecular weight from the Mark–Houwink equation:
| (1) |
The Mark–Houwink coefficients for APS pre-polymers in HFIP at 30 °C were interpolated from polyamides in HFIP [21] to be Km =5.6 × 10−4 dL/g and a = 0.746. The polymers were assumed to be low-molecular weight oligomers a priori, which would reduce the probability of branching and therefore ensure the validity of the selected Mark–Houwink coefficients. The degree of polymerization was calculated assuming an equal probable of incorporation of the DAHP and the polyol. FT-IR analysis was conducted on both APS pre-polymers and sol-free APS elastomers cured for 48 h. Fragments of pre-polymer and fully cured APS elastomers were mechanically sectioned into small particles and analyzed with an FT-IR microscope (Nikon Instruments, Melville, NY, USA). Spectra were recorded using a Nicolet Magna 550 Series II IR Spectrometer equipped with OMNIC Software using 32 scans across the wave numbers 4000–400 cm−1 at a resolution of 2 cm−1. Sol content was measured by measuring the difference in dry-weight before and after sol removal via ethanol wash (n = 12). The mass densities were measured (n = 12) using a 49.97-mL pycnometer bottle (Humboldt Mfg. Co, Norridge, IL, USA). Atomic composition was measured using a Kratos AXIS Ultra Imaging X-ray Photoelectron Spectrometer with Delay Line Detector. The water-in-air contact angle of polymer films was measured using the sessile-drop method and VCA2000 image analysis software (n = 10). Sol content was determined by measuring dry mass differential before and after incubation with ethanol, which dissolves unincorporated monomers, and using the following relationship where Mwsol is the sample mass including sol, Msolfree is the sample mass without sol:
| (2) |
Water uptake upon hydration was determined in a similar manner by measuring the differential mass of sol-free polymer slabs before and after hydration for 24 h given by the following equation:
| (3) |
Glass transition temperature (Tg) and other potential phase transitions were measured using a Q1000 DSC equipped with Advantage Software v2.5 (TA Instruments, Newcastle, DE, USA) and analyzed with Universal Analysis Software v4.3A (TA Instruments). Cooling and heating rates were 10 °C/min Elastomeric films dedicated for mechanical testing were cut into dog-bone geometries with dimensions of 0.5 mm × 6.5 mm × 30 mm (T × W × L). Tensile testing was performed using an Instron 5542 using a 50 N load cell equipped with Merlin software. Samples (n ≥ 6) were prepared by removing sol and hydrating films in ddH2O for at least 24 h prior to measurement. Polymer slabs were extended at a constant rate of 50 mm/min and were elongated until failure at ambient temperature. Young’s modulus, toughness modulus, ultimate tensile strength and elongation at break were all calculated directly from engineering tensile stress vs. engineering strain curves (Fig. 2). Moles of active network n and molecular weight between crosslinks Mc were calculated from the following equations for an ideal elastomer where Eo is the Young’s modulus, R is the universal gas constant, T is the temperature, and ρ is the mass density:
Fig. 2.

Tensile stress vs. strain curves of APS polymers. Hydrated APSs exhibited elastomeric behavior under tension as indicated by long elongations at break and relatively low Young’s moduli across all tested samples.
| (4) |
2.3. In vitro degradation of polymer films
Cylindrical polymer slabs (n = 4) with dimensions of 0.5 mm × 6 mm (T × D) weighing approximately 20 mg each were incubated at 37 °C in the 3 m sodium acetate buffer solution with a pH of 5.2. Buffer solution was exchanged every 2 d and dry mass loss measurements were made at specified time points. Samples were washed in ddH2O, incubated in ethanol and ddH2O for 24 h each, and finally dried at 70 °C for at least 24 h. Mass loss MLoss was determined by the following formula where Mo is the initial mass and Mf is the final time point:
| (5) |
2.4. Cell attachment and in vitro biocompatibility
Primary human foreskin fibroblasts (HFF, ATCC, Manassas, VA, USA) were cultured in high glucose Dulbecco’s minimal essential medium (DMEM) supplemented with 10% (v/v) fetal bovine serum, 100 μg/mL streptomycin, and 100 U/mL penicillin, at 37 °C and 5% CO2. Cells between passage four and six were harvested for using trypsin 0.025%/EDTA 0.01% and quenched with an equal volume of medium to re-suspend the cells. Polymer films of both 2DAHP–1G and 2DAHP–1T formulations were cured at 170 °C for 48 h for in vitro biocompatibility assessment due to the ability to assess cell morphology using light microscopy (See Section 3). PLGA 6535 (65% l-lactic acid, 35% glycolic acid, High MW, Lakeshore Biomaterials, Birmingham AL, USA) films were used as a control biomaterial and prepared via solvent evaporation of 1% (w/w) solutions in dichloromethane at 100 μL/cm2. Polymer films (n = 4) were seeded with HFFs at 25,000 cells/cm2. Cell metabolism was evaluated by the XTT kit after 24 h according to the manufacturer’s instructions. Cells were incubated for 4 h in medium containing 20% (v/v) XTT solution. Absorption measurements made in triplicate at 450 nm and 690 nm wavelengths using a SpectraMax Plus 384 (Molecular Devices, Sunnyvale, CA, USA) equipped with SOFTmax Pro 4.0. XTT measurements are reported as normalized ratios with respect to TCPS. Phase micrographs of cells were taken at 10× magnification for each sample analyzed. The area and circularity [22] of cell populations were calculated manually using perimeter and area measurements by using Axiovision software (Zeiss, Germany). The circularity C was calculated using the following formula:
| (6) |
where A is the projected area of the cell and P is the perimeter of the cell. Circularity was used as an index of cell spreading. Three distinct cell populations (n = 200 total) were measured to find population means, which were used to determine statistical significance.
2.5. In vivo biocompatibility and tissue response
Polymer slabs of the appropriate APS were synthesized and cut into cylinders with target dimensions of D = 11 mm, T = 1 mm. PLGA slabs were obtained by melt pressing 0.3 g of 6535 PLGA at 172 °C under 5000 MPa into target dimensions of D = 11 mm, T = 1 mm. All polymer slabs were incubated in 100% ethanol to remove sol. Polymer slabs were disinfected for 20 min under UV and incubated in 0.9% saline prior to implantation. Polymer slabs were implanted subcutaneous into 7-wk-old female Sprague–Dawley rates (Charles River Laboratories, Wilmington, MA, USA) by a midline incision and blunt lateral dissection under deep isoflurane-O2 general anesthesia. The animals were cared for in compliance with the regulations of MIT and the NIH [23]. Two implants of each material used were implanted on the upper and lower back of the same animal. Rats were sacrificed at pre-determined time points. Polymer explants were removed from rats at 6 wks, washed gently in Dulbecco’s phosphate buffered solution (DPBS), dried at 70 °C, and weighed to obtain dry mass for degradation calculations. Specimens with intact implants of approximate dimensions of 15 mm × 15 mm were harvested en bloc with surrounding tissues for histology. The samples were fixed in Accustain (Sigma) for 24 h and embedded in paraffin and stained appropriately. Briefly, slides were stained with hematoxylin and eosin (H&E) and Masson’s trichrome stain to determine fibrous capsule thickness. Tissue macrophages were identified by staining sections with primary rabbit anti-rat CD68 (1:200, Abcam, England, UK) followed by goat anti-mouse secondary antibody (Vector Burlingame, CA, USA). Samples were incubated with strepavidin horseradish peroxidase (1:100, Dako, Denmark) and developed with DAB substrate chromogen (Dako). Histology images were recorded with a Zeiss CCD Camera equipped with Openlab v5.5 software.
2.6. Statistics
All measurements of the projected cell area and circularity were based on the sampling over at least n = 100 cells per population with a minimum of three cell populations per data point. XTT metabolism assays were performed in triplicate samples (n = 3) with triplicate readings for each data point. At least four fibrous capsule thickness measurements were made across at least 10 randomly selected images per sample. Histological macrophage scoring was also based on at least 10 randomly selected images. Two-tailed student’s t-tests with unequal variances were performed to determine statistical significance, where appropriate (Microsoft Excel, Redmond, WA, USA). Non-parametric one-way ANOVA tests were also performed where appropriate (GraphPad Prism 4.02, GraphPad Software, San Diego, CA, USA). Dunn’s multiple comparison post-tests were used to determine significance between specific treatments. All tabulated and graphical data are reported as mean ± S.D. Significance levels were set at *p < 0.05, **p < 0.01, and ***p < 0.001.
3. Results
3.1. Chemical, physical, and mechanical characterization of 1,3-diamino-2-hydroxy-propane-based poly(ester amide) elastomers
The APS pre-polymers formed after the initial polymerization step consisted of a transparent viscous fluid at 170 °C. The pre-polymers became opaque and glassy upon cooling down to room temperature. The molecular weight (Mw) of the pre-polymer was measured to be 1278 and 1500 Da for 2DAHP–1G and 2DAHP–1T formulations, respectively (see Table 1). APS pre-polymers were insoluble in common organic solvents such as dicholoromethane, acetone and ethanol. However, both APS pre-polymers were soluble in HFIP, a common solvent used for dissolving polyamides [21]. The presence of esters, carboxylic acids, secondary amides, and primary amines was determined from FT-IR data (see Supplementary Table 1; Supplementary Fig. 1). The polycondensation of 2DAHP–1G and 2DAHP–1T formulations produced solid, elastomeric slabs with similar functional groups as the corresponding pre-polymers (see Supplementary Table 2; Supplementary Fig. 1). One notable exception is the reduction of prevalence of the broad stretch at approximately 3300 cm−1 for the case of 2DAHP–1G cured for 48 h. APS elastomers were optically transparent when dry. Hydrated APS polymer films crosslinked for only 24 h were opaque when hydrated while films cross-linked for 48 h maintained transparency. The FT-IR spectra of APS polymer networks were similar to that of corresponding APS pre-polymers (see Supplementary Fig. 2). The incorporation of amine groups into elastomeric networks is supported by the presence of nitrogen in sol-free slabs as determined by elemental analysis via XPS, a surface technique for elucidating the elemental composition at the surface (see Table 2). The theoretical calculations were made assuming condensation of 6 mol of H2O to form one X-segment and one Y-segment of the polymer backbone. Nitrogen content ranged from 2.24 to 3.97% by mass depending upon the polymer formulation and processing conditions.
Table 1.
Molecular weight of APS pre-polymers as determined by intrinsic viscosity
DOP: degree of polymerization.
Instrinsic viscosity of APS pre-polymers in HFIP at 30 °C.
Weight average molecular weight determined from Mark–Houwink coefficients of polyamides in HFIP [21].
Table 2.
Relative elemental composition of carbon (C, 1s), oxygen (O, 1s), and nitrogen (N, 1s) in APS polymers by mass as measured by XPS
| Formulation | Curing time (h) | C | O | N |
|---|---|---|---|---|
| 2DAHP–1G | 24 | 80.85 | 15.18 | 3.97 |
| 2DAHP–1G | 48 | 85.76 | 10.58 | 3.66 |
| 2DAHP–1G | Theoretical | 66.86 | 25.14 | 8.00 |
| 2DAHP–1T | 24 | 85.48 | 12.28 | 2.24 |
| 2DAHP–1T | 48 | 85.40 | 12.34 | 2.26 |
| 2DAHP–1T | Theoretical | 65.93 | 26.37 | 7.69 |
The sol content of APS networks varied between 16.08 and 3.51% by mass (see Supplementary Table 3). Sol content was reduced by using longer curing times or by increasing the degree of functionality of the polyol by using d,l-threitol. The mass densities of APS networks varied between 1.096 and 1.114 g/cm3 and were dependent upon composition and processing (see Supplementary Table 3). It should be noted that the mass density of the sol-free elastomeric films is significantly lower than the inferred density of the pre-polymer. This is likely due to the removal of low-molecular weight oligomers as well as water due to condensation polymerization. APS networks exhibited water uptake which varied from 14.2 to 19.8% by volume across composition and curing conditions. The water-in-air contact angle of APS films varied between 91.5 and 93.7° and the glass transition temperature varied between 33.7 and 48.0 °C, of which both parameters varied according to composition and curing conditions (see Supplementary Table 4; Supplementary Fig. 2). There were no observable crystallization or melting transitions measured in APS polymers across the measured temperature range (between −80 °C and 250 °C). The mechanical properties of APS networks are summarized in Table 3. The Young’s modulus varied between 1.45 and 4.34 MPa with elongations at break varying between 21 and 92% (Fig. 2). Increasing the maximum elongations could be achieved by reducing the curing time (results not shown).
Table 3.
Mechanical properties of APS polymers
| Formulation | Curing time (h) | Young’s modulus (MPa) | Elongation at break (%) | UTS (MPa) | Toughness (MJ m−3) | n (mol m−3) | Mc (g mol−1) |
|---|---|---|---|---|---|---|---|
| 2DAHP–1G | 24 | 2.45 ± 0.33 | 92 ± 21 | 1.33 ± 0.25 | 0.730 ± 0.31 | 329.24 ± 45.4 | 3345 ± 461 |
| 2DAHP–1G | 48 | 4.34 ± 0.32 | 64 ± 12 | 1.69 ± 0.14 | 0.638 ± 0.18 | 583.51 ± 43.6 | 1879 ± 140 |
| 2DAHP–1T | 24 | 1.45 ± 0.31 | 60 ± 26 | 0.39 ± 0.11 | 0.171 ± 0.18 | 194.90 ± 41.5 | 5718 ± 1218 |
| 2DAHP–1T | 48 | 1.48 ± 0.20 | 21 + 2.2 | 0.24 ± 0.50 | 0.028 ± 0.008 | 199.16 ± 26.4 | 5576 ± 738 |
Despite the fact that the Tg of the tested polymers was above the ambient temperature at which mechanical testing occurred, the films were qualitatively observed to be rubbery. This is likely due to Tg depression upon hydration of the polymer network. Hydration leads to swelling and polymer–water interactions, both of which can lead to increased chain mobility and a corresponding reduction in Tg. The quantitative framework has been previously developed [24] and the effect has been estimated in a variety of polymer network-diluent systems [25] including situations in which the diluent is water [26,27]. The estimated effective Tg of hydrated APS networks was calculated using the following equation:
| (7) |
where xi is the mass fraction, ΔCpi is the incremental change in heat capacity at Tgi, and Tgi is the glass transition temperature for component i. Values for ΔCp and Tg were experimentally determined values that were calculated from DSC thermograms (see Supplementary Fig. 2). Component 1 is defined as the polymer network and component 2 is defined as the diluent, which in this case is water. The effect of crosslinking was assumed to be negligible to obtain a first order approximation. The results are summarized in Supplementary Table 5 where ΔTg1,2 is defined as:
| (8) |
where Tg0 is defined as the glass transition temperature of the polymer network in the dry state (no diluent) and Tg1,2 is the glass transition temperature of the hydrated polymer network (with diluent). The values of Tg were calculated to be reduced significantly as the resulting effective values of Tg ranged from −42.9 to −78.7 °C.
3.2. In vitro degradation
All APS polymers in this study degraded in the presence of sodium acetate buffer. 2DAHP–1G formulations cured for 24 h and 48 h exhibited 97.0 ± 3.5% and 44.3 ± 4.4% dry mass loss, respectively, after 6 wks of in vitro degradation. 2DAHP–1T formulations cured for 24 h and 48 h lost 70.4 ± 2.7% and 42.8 ± 2.3% of dry mass, respectively.
3.3. In vitro biocompatibility
Primary HFFs were chosen to study in vitro cell attachment and spreading because of the importance of these cells in the wound healing response following the implantation of biomaterials [28-30]. HFFs grown on APSs attached and exhibited similar morphology to HFFs grown on standard control biomaterials including PLGA and tissue culture polystyrene (TCPS) (Fig. 3). Cell circularity and projected cell area are two metrics that indirectly measure the ability for cells to adhere and spread upon the biomaterial surface [31,32]. These two parameters were both statistically similar (p > 0.05; p > 0.05, respectively) across all tested materials as determined by one-way ANOVA with Dunn’s multiple comparison tests (Fig. 4). HFFs grown on 2DAHP–1G exhibited a similar level of mitochondrial metabolism at 24 h as compared to HFFs grown on PLGA and TCPS, as measured by XTT cleavage assay. Although metabolism of HFFs grown on 2DAHP–1T was similar when compared to TCPS (p > 0.05), it was reduced when compared to PLGA (*p < 0.05) as measured by one-way ANOVA with Dunn’s multiple comparison tests. Confluent layers of HFFs were obtained after 3 d across all tested biomaterials.
Fig. 3.

Human foreskin fibroblasts cultured on APS polymers. HFFs cultured on APSs appeared to have a similar morphology as other traditional biomaterials including PLGA and tissue culture polystyrene (TCPS). Scale bars in all panels are 200 μm.
Fig. 4.

Characterization of morphology and metabolism of human foreskin fibroblasts cultured on APS films. HFFs exhibited statistically similar circularity (p > 0.05) and projected cell area (p > 0.05), metrics for cell spreading, when compared to PLGA and TCPS as measured by one-way ANOVA. HFFs grown on 2DAHP–1G exhibited similar metabolism as measured by XTT assay when compared to PLGA and TCPS (p > 0.05, p > 0.05) while HFFs grown on 2DAHP–1T exhibited similar metabolism (p > 0.05) when compared to TCPS and reduced metabolism when compared to PLGA (*p < 0.05).
3.4. In vivo biocompatibility and degradation
Subcutaneous (SC) implants of 2DAHP–1G and 2DAHP–1T, both cured for 48 h, exhibited only 4.3 and 5.8% mass loss at 6 wks and 12.9 and 13.0% mass loss at 20 wks, respectively. Fibrous capsules were formed in response to SC implantation of APS polymer slabs. The fibrous capsule thickness in all samples reduced from 1 to 2 wks, which was followed by an increase at 20 and 12 wks for APS and PLGA implants, respectively. This increase was not statistically significant in the case of 2DAHP–1G and PLGA, but was significant in the case of 2DAHP–1T when compared to the fibrous capsule thickness at 1 wk (Fig. 5). Histological sections indicated that the fibrous capsule thickness of both 2DAHP–1G and 2DAHP–1T polymers was significantly less (***p < 0.001) than PLGA at both 1 and 2 wks as determined by two-way ANOVA (see Fig. 7a). These results suggest that APS polymers induce a foreign body response which is milder than PLGA in terms of fibrous capsule formation. The concentration of macrophages was also observed to be lower in 2DAHP–1G and 2DAHP–1T polymers both in the fibrous capsule and in the surrounding tissue (***p < 0.001 and ***p < 0.001, respectively) as measured by the frequency of CD68+ cells at 1 and 2 wks (Fig. 6 and Fig. 7b,c). There was no significant increase in the frequency of CD68+ cells at 20 wks for APS implants (see Supplementary Table 6). The percentage of CD68+ cells at 20 wks was approximately equal to that of 1 wk for the 2DAHP–1G formulation and smaller for the 2DAHP–1T formulation. Neither 2DAHP–1G nor 2DAHP–1T implants became heavily vascularized, recruited macrophages, or allowed fibroblast penetration. PLGA implants remained intact for up to only 12 wks, which prohibited accurate assessment of fibrous capsule and macrophage recruitment beyond this time point.
Fig. 5.

Histology of in vivo foreign body response to APS implants. H&E stain show fibrous capsules are formed at the implantation sites of all polymers. H&E stains show reduced fibrous capsule thickness and for 2DAHP–1G and 2DAHP–1T polymer implants when compared to PLGA (see Fig. 7a). The regions of polymer, fibrous capsule, and muscle are indicated by P, F, and M, respectively, and separated by dashed lines. Long-term histology time points were set at 20 wks for 2DAHP–1G and 2DAHP–1T implants and 12 wks for PLGA implants. Scale bars in all images are 200 μm.
Fig. 7.
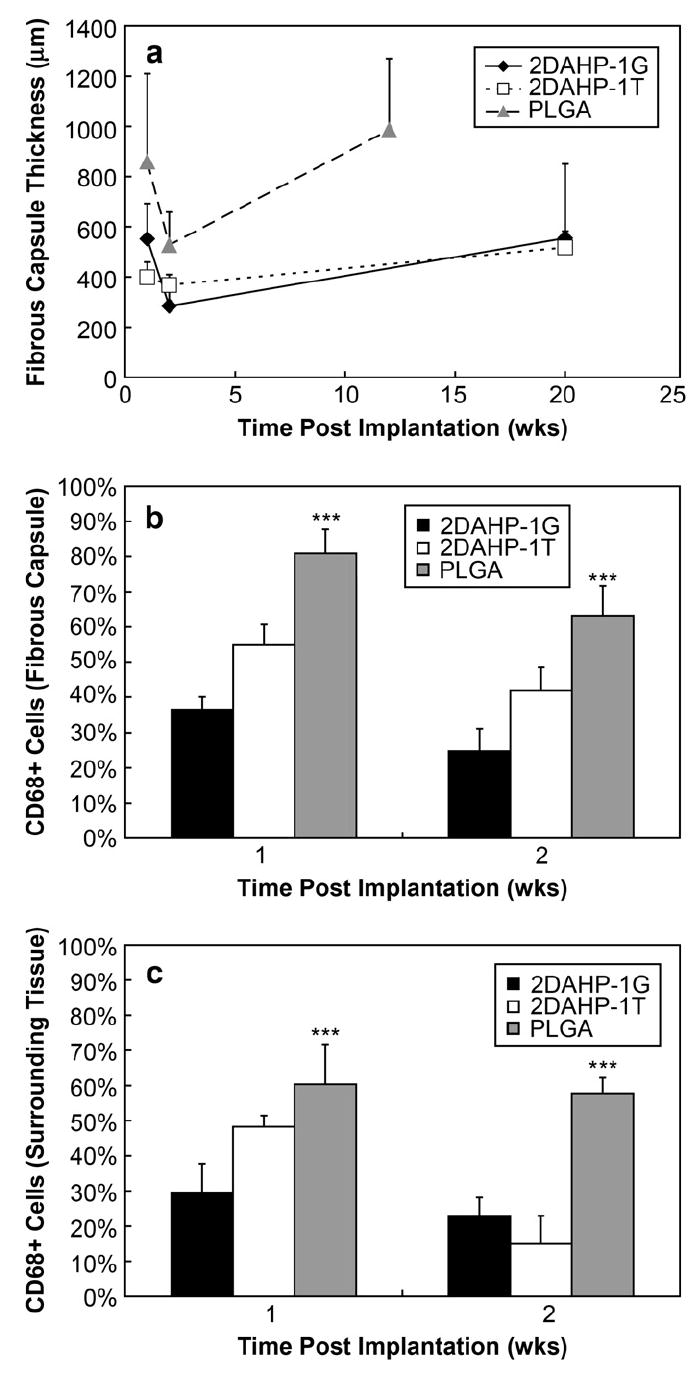
Characterization of foreign body response to APS implants. Fibrous capsule thicknesses were observed to be smaller in 2DAHP–1G and 2DAHP–1T polymers when compared to PLGA at 1 and 2 wks (***p < 0.001). The fibrous capsule thickness for PLGA implants at 12 wks was larger than both 2DAHP–1G and 2DAHP–1T at 20 wks. The presence of macrophages, as determined by CD68+ cells, was reduced within the fibrous capsule and surrounding tissue of 2DAHP–1G and 2DAHP–1T polymers when compared to PLGA at 1 and 2 wks (***p < 0.001).
Fig. 6.

Macrophage recruitment in APS implants. The presence of macrophages, as determined by CD68+ cells, was reduced within the fibrous capsule and surrounding tissue of 2DAHP–1G and 2DAHP–1T polymers when compared to PLGA (see Fig. 7b,c). The regions of polymer, fibrous capsule, and muscle are indicated by P, F, and M, respectively, and separated by dashed lines. Long-term histology time points were set at 20 wks for 2DAHP–1G and 2DAHP–1T implants and 12 wks for PLGA implants. Scale bars in all images are 200 μm.
4. Discussion
The newly synthesized class of compounds, poly(1,3-diamino-2-hydroxypropane-co-polyol sebacate)s (APSs), offers potential advantages of both natural and synthetic elastomeric biomaterials as a material platform for implantable medical devices with a focus on devices that are intended to be semi-permanent including cell-scaffolds for soft-tissue engineering applications. Similar to protein-based elastomers, APSs are slowly degrading polymers that exhibit low moduli, high UTS, and are capable of large deformations. APSs also support cell attachment and are also capable of amine-based chemistries for surface modification. Similar to synthetic biodegradable elastomers, APSs are inexpensive to produce, easy to synthesize, and are adaptable to standard polymer processing capabilities. Previously synthesized biodegradable elastomeric esters have used primarily ester linkages which are susceptible to both esterase activity and hydrolysis [17,18]. The presence of amide bonds leads to reduced susceptibility of degradation via hydrolysis and esterase enzymes, which are excreted by macrophages as a part of the foreign body reaction [30]. This potentially allows for the use of APS polymers in expanded applications where long biomaterial half-lives are required such as scaffolds for cardiovascular grafts and peripheral nerve regeneration conduits [33] where implant half-lives may need to approach 2–6 months or longer.
APS polymers described in this report were synthesized from combining a diacid with a polyol and the tri-functional DAHP. The incorporation of a polyol proved to be important in sufficiently reducing the required thermal processing temperature from approximately 210–170 °C for 2DAHP–1G and 2DAHP–1T formulations. APSs synthesized from only sebacic acid and DAHP were technically challenging to synthesize and subsequently process into films. Although glycerol and d,l-threitol were chosen as representative polyols, other types of polyols could be used including xylitol, sorbitol, and mannitol, which can serve as building blocks for other polymer systems [34]. Similarly, although sebacic acid was chosen since it has been used extensively in other polymeric systems for biomedical applications [35], other types of diacids could be used in the synthesis to achieve a broad range of properties.
The elemental composition of APS polymers in this study generally suggested that there was proportionately less nitrogen and more oxygen present in the elastomeric networks than the theoretical calculations (see Table 2). This suggests that, in general, the polycondensation of sebacic acid with hydroxyl groups occurred more readily as compared to amine groups. The preparation of APS networks under nitrogen and vacuum environments coupled with the lack of nitrate finger-prints in FT-IR spectra suggest that this differential in elemental composition is not due to oxidation of reactants. The relative nitrogen content, which is presumed to exist in the context of primary amines, and secondary amide bonds, was not increased by increasing the curing times. Consider the 2DAHP–1G formulation: the relative nitrogen content is reduced as the curing time is increased. Also, the relative mass fractions of carbon and oxygen are increased and decreased, respectively. This suggests that more sebacic acid molecules are being incorporated into the network, which would reduce the overall relative mass fraction of nitrogen. This result can be inferred from the FT-IR spectrum of the 2DAHP–1G formulation cured for 48 h in which the stretch indicative of hydroxyl and acid groups is significantly reduced (see Supplementary Fig. 1c). The increased incorporation can also be inferred by the effect of curing time on Tg. The Tg increased throughout this variation in material processing. It is known that amide groups exhibit less bond rotation [36], which can lead to more rigid polymer segments, and ultimately increase the Tg [37]. These observations suggest that higher curing time leads to increased amide content despite the relative reduction in overall nitrogen content. The 2DAHP–1T formulation behaved in a similar manner; although the relative nitrogen content is increased only slightly upon increasing the curing time from 24 to 48 h, the Tg rose from 45.4 to 48.0 °C. Although altering the formulation and processing altered the Tg, the observed variation the water-in-air contact angle for both formulations was minimal. This is likely due to the dominating effect of the reagent composition and subsequent surface chemistry of APS films when compared to the effect of material processing conditions.
The calculated Tg depression was extremely pronounced across all polymers and processing conditions. This is likely due to the relatively high mass fraction of water observed in the hydrated polymers as well as the extremely low Tg of water (−139 °C). Hence, all APS polymers in the hydrated state were predicted to be exclusively rubbery, and therefore elastomeric, at physiological conditions. The observed trends in mechanical properties of APS networks are similar to other thermally processed polymeric elastomers [17,18]. Namely, increasing the curing time increases the moles of active network per volume and decreases the molecular weight between crosslinks which in turn leads to increasing the tensile Young’s modulus. This increase in stiffness is often accompanied by a corresponding reduction in elongation at break. While this trend is readily observed in the 2DAHP–1G formulation, the effects of higher crosslinking are not as pronounced in the 2DAHP–1T formulation. This is possibly due to the increase in functionality of the polyol to four hydroxyl groups present in d,l-threitol.
The presence of amine groups on the surface, as determined by XPS and FT-IR, expands the possible suite of chemical modification strategies for functionalization. Amine groups are more nucleophilic than hydroxyl groups and can react with many groups including aldehydes, NHS esters, isocyanates, acyl azides, and anhydrides [38]. The physical properties of APS polymers expand the potential parameter space for biodegradable elastomers. Specifically, the range of attainable values for characteristics such as Tg, contact angle, hydration, elemental composition, chemical modification techniques, and degradation kinetics has been expanded.
PLGA was chosen as a control biomaterial for in vitro and in vivo biocompatibility because of the ubiquitous use of PLGA for biomedical applications. The in vitro response of primary human foreskin fibroblasts on APS polymer films demonstrates the ability for cells to interact favorably with this new class of materials as verified by maintenance of spreading, morphology, and metabolic activity when compared to other commonly used biomaterials. Although APS polymers appear to remain stable in vivo for a much longer time period than the PLGA formulation chosen in this study, PLGA was a suitable control material to assess the acute and chronic tissue responses at 1 and 2 wks, respectively. This is due to the fact that both APS and PLGA implants exhibit very little mass loss over this time scale in vivo [19]. The reduced fibrous capsule thickness and limited macrophage recruitment in response to subcutaneous APS implants suggests a more favorable in vivo response as well, when compared to PLGA implants. The reduced frequency of CD68+ cells found within the fibrous capsules surrounding APS implants at 1 and 2 wk time points suggests a reduced inflammatory response when compared to PLGA implants. Furthermore, the reduction of CD68+ cells in the surrounding tissue also suggests that APS implants do not actively recruit macrophages from the surrounding tissue as readily as PLGA implants. These data suggest that the in vivo stability and favorable immune response are enabling properties of APS polymers, which make them potentially suitable for fabrication of semi-permanent resorbable medical devices for use in long-term implantation applications.
5. Conclusions
APS polymers synthesized using the approach described herein are biomaterials that offer a wide range of potential applications. Favorable in vitro and in vivo cellular responses suggest that this new class of materials could be used as a biomedical device material platform. These materials could be used as a structural material to fabricate resorbable implantable devices [39] such as tissue engineering scaffolds, drug delivery systems, and temporary diagnostic systems.
Supplementary Material
Supplementary materials associated with this article can be found at doi: 10.1016/j.biomaterials.2008.01.029.
{kind=link}
{kind=link}
Acknowledgments
The authors would like to acknowledge the following: Dr. Eliza Vasile from the Center for Cancer Research, Microscopy and Imaging Core Facility at MIT for assistance with imaging, Libby Shaw and Tim McClure at the Center for Materials Science and Engineering at MIT for assistance in XPS and FT-IR, the MIT Institute for Soldier Nanotechnologies (ISN), the MEMS Technology Group at the Draper Laboratory for direct funding for CJB and use of facilities. JPB was financially supported by the J.F.S. Esser Stichting, as well as the Prof. Michaël-Van Vloten Fonds. Funding provided through DL-H-550154; NIH grants R01-DE-013023-06, P41-EB002520-01A1 and R01-HL-076485-01A2. The content of this paper does not necessarily reflect the position or the policy of the government, and no official endorsement should be inferred.
References
- 1.Griffith LG, Naughton G. Tissue engineering – current challenges and expanding opportunities. Science. 2002;295(5557):1009–14. doi: 10.1126/science.1069210. [DOI] [PubMed] [Google Scholar]
- 2.Hollister SJ. Porous scaffold design for tissue engineering. Nat Mater. 2005;4:518–24. doi: 10.1038/nmat1421. [DOI] [PubMed] [Google Scholar]
- 3.Chevallay B, Herbage D. Collagen-based biomaterials as 3D scaffold for cell cultures: applications for tissue engineering and gene therapy. Med Biol Eng Comput. 2000;38(2):211–8. doi: 10.1007/BF02344779. [DOI] [PubMed] [Google Scholar]
- 4.Berglund JD, Nerem RM, Sambanis A. Incorporation of intact elastin scaffolds in tissue-engineered collagen-based vascular grafts. Tissue Eng. 2004;10(9/10):1526–35. doi: 10.1089/ten.2004.10.1526. [DOI] [PubMed] [Google Scholar]
- 5.Altman GH, Diaz F, Jakuba C, Calabro T, Horan RL, Chen J, et al. Silk-based biomaterials. Biomaterials. 2003;24:401–16. doi: 10.1016/s0142-9612(02)00353-8. [DOI] [PubMed] [Google Scholar]
- 6.Sofia S, McCarthy MB, Gronowicz G, Kaplan DL. Functionalized silk-based biomaterials for bone formation. J Biomed Mater Res. 2001;54:139–48. doi: 10.1002/1097-4636(200101)54:1<139::aid-jbm17>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 7.Hsu SH, Whu SW, Hsieh SC, Tsai CL, Chen DC, Tan TS. Evaluation of chitosan–alginate–hyaluronate complexes modified by an RGD-containing protein as tissue-engineering scaffolds for cartilage regeneration. Artif Organs. 2004;28(8):693–703. doi: 10.1111/j.1525-1594.2004.00046.x. [DOI] [PubMed] [Google Scholar]
- 8.Amsden B, Wang S, Wyss U. Synthesis and characterization of thermoset biodegradable elastomers based on star-poly(∈-caprolactone-co-d,l-lactide) Biomacromolecules. 2004;5(4):1399–404. doi: 10.1021/bm034538j. [DOI] [PubMed] [Google Scholar]
- 9.Amsden BG, Tse MY, Turner ND, Knight DK, Pang SC. In vivo degradation behavior of photo-cross-linked star-poly(∈-caprolactone-co-d,l-lactide) Biomacromolecules. 2006;7(1):365–72. doi: 10.1021/bm050731x. [DOI] [PubMed] [Google Scholar]
- 10.Guan J, Sacks MS, Beckman EJ, Wagner WR. Synthesis, characterization, cytocompatibility of elastomeric, biodegradable poly(ester-urethane)ureas based on poly(caprolactone) and putrescine. J Biomed Mater Res. 2002;61(3):493–503. doi: 10.1002/jbm.10204. [DOI] [PubMed] [Google Scholar]
- 11.Guan J, Sacks MS, Beckman EJ, Wagner WR. Biodegradable poly(ether ester urethane)urea elastomers based on poly(ether ester) triblock copolymers and putrescine: synthesis, characterization and cytocompatibility. Biomaterials. 2004;25(1):85–96. doi: 10.1016/s0142-9612(03)00476-9. [DOI] [PubMed] [Google Scholar]
- 12.Lee SH, Kim BS, Kim SH, Choi SW, Jeong SI, Kwon IK, et al. Elastic biodegradable poly(glycolide-co-caprolactone) scaffold for tissue engineering. J Biomed Mater Res. 2003;66A(1):29–37. doi: 10.1002/jbm.a.10497. [DOI] [PubMed] [Google Scholar]
- 13.Nagata M, Machida T, Sakai W, Tsusumi N. Synthesis, characterization, enzymatic degradation of network aliphatic copolyesters. J Polym Sci A. 1999;37:2005–11. [Google Scholar]
- 14.Nijst CLE, Bruggeman JP, Karp JM, Ferreira L, Zumbuehl A, Bettinger CJ, et al. Synthesis and characterization of photocurable elastomers from poly(glycerol-co-sebacate) Biomacromolecules. 2007;8(10):3067–73. doi: 10.1021/bm070423u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poirier Y, Nawrath C, Somerville C. Production of polyhydroxyalkanoates, a family of biodegradable plastics and elastomers, in bacteria and plants. Nat Biotechnol. 1995;13(2):142–50. doi: 10.1038/nbt0295-142. [DOI] [PubMed] [Google Scholar]
- 16.Sodian R, Sperling JS, Martin DP, Egozy A, Stock U, Mayer JE, et al. Technical report: fabrication of a trileaflet heart valve scaffold from a polyhydroxyalkanoate biopolyester for use in tissue engineering. Tissue Eng. 2000;6(2):183–8. doi: 10.1089/107632700320793. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Ameer GA, Sheppard BJ, Langer R. A tough biodegradable elastomer. Nat Biotechnol. 2002 June;20:602–6. doi: 10.1038/nbt0602-602. [DOI] [PubMed] [Google Scholar]
- 18.Yang J, Webb AR, Ameer GA. Novel citric acid-based biodegradable elastomers for tissue engineering. Adv Mater. 2004;16(6):511–5. [Google Scholar]
- 19.Wang Y, Kim YM, Langer R. In vivo degradation characteristics of poly(glycerol-sebacate) J Biomed Mater Res A. 2003;66:192–7. doi: 10.1002/jbm.a.10534. [DOI] [PubMed] [Google Scholar]
- 20.Roy DR, Parthasarathi R, Maiti B, Subramanian V, Chattaraj PK. Electrophilicity as a possible descriptor for toxicity prediction. Bioorg Med Chem. 2005;13(10):3405–12. doi: 10.1016/j.bmc.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 21.Mattiussi A, Gechele GB, Francesconi R. Polyamides in solution. III. Viscometry of linear polycaprolactam. J Polym Sci A2. 1969;7:411–22. [Google Scholar]
- 22.Thurston G, Jaggi B, Palcic B. Measurement of cell motility and morphology with an automated microscope system. Cytometry. 1988;9(5):411–7. doi: 10.1002/cyto.990090502. [DOI] [PubMed] [Google Scholar]
- 23.National Institute Health. Principles of laboratory animal care. 1985 [Google Scholar]
- 24.Couchman PR, Karasz FE. A classical thermodynamic discussion of the effect of composition on glass-transition temperatures. Macromolecules. 1978;11(1):117–9. [Google Scholar]
- 25.Ellis TS, Karasz FE, Brinke GT. The influence of thermal properties on the glass transition temperature in styrene/divinylbenzene network-diluent systems. J Appl Poly Sci. 1983;28:23–32. [Google Scholar]
- 26.Ellis TS, Karasz FE. Interaction of epoxy resins with water: the depression of glass transistion temperature. Polymer. 1984;25:664–9. [Google Scholar]
- 27.Jina X, Ellis TS, Karasz FE. Studies on the water induced plasticization behavior of nylon 4: the influence of crystallinity and radiation crosslinking. Makromol Chem. 1985;186:191–201. [Google Scholar]
- 28.Maloney WJ, Smith RL, Castro F, Schurman DJ. Fibroblast response to metallic debris in vitro. Enzyme induction cell proliferation, toxicity. J Bone Joint Surg Am. 1993 June 1;75(6):835–44. doi: 10.2106/00004623-199306000-00005. [DOI] [PubMed] [Google Scholar]
- 29.Lam KH, Schakenraad JM, Groen H, Esselbrugge H, Dijkstra PJ, Feijen J, et al. The influence of surface morphology and wettability on the inflammatory response against poly(l-lactic acid): a semi-quantitative study with monoclonal antibodies. J Biomed Mater Res. 1995;29(8):929–42. doi: 10.1002/jbm.820290804. [DOI] [PubMed] [Google Scholar]
- 30.Hu WJ, Eaton JW, Ugarova TP, Tang L. Molecular basis of biomaterial-mediated foreign body reactions. Blood. 2001 August 15;98(4):1231–8. doi: 10.1182/blood.v98.4.1231. [DOI] [PubMed] [Google Scholar]
- 31.Ramires PA, Mirenghi L, Romano AR, Palumbo F, Nicolardi G. Plasma-treated PET surfaces improve the biocompatibility of human endothelial cells. J Biomed Mater Res. 2000;51(3):535–9. doi: 10.1002/1097-4636(20000905)51:3<535::aid-jbm31>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 32.Keresztes Z, Rouxhet PG, Remacle C, Dupont-Gillain C. Supramolecular assemblies of adsorbed collagen affect the adhesion of endothelial cells. J Biomed Mater Res. 2006;76A(2):223–33. doi: 10.1002/jbm.a.30472. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez FJ, Gomez N, Perego G, Navarro X. Highly permeable poly-lactide–caprolactone nerve guides enhance peripheral nerve regeneration through long gaps. Biomaterials. 1999;20(16):1489–500. doi: 10.1016/s0142-9612(99)00055-1. [DOI] [PubMed] [Google Scholar]
- 34.Bruggeman JP, Bettinger CJ, Nijst CLE, Kohane D, Langer R. Xylitol-based biodegradable elastomers. Adv Mater. doi: 10.1002/adma.200702377. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langer R, Peppas NA. Advances in biomaterials, drug delivery, bionano-technology. AIChE J. 2003 December;49(12):2990–3006. [Google Scholar]
- 36.Stryer L. Biochemistry. 4. New York, USA: W H Freeman and Co; 1996. [Google Scholar]
- 37.Young RJ, Lovell PA. Introduction to polymers. 2. Nelson Thornes; 1991. [Google Scholar]
- 38.Hermanson GT. Bioconjugate techniques. Boston: Academic Press; 1996. [Google Scholar]
- 39.Daniel K, Kim GY, Vassiliou C, Jalali-Yazdi F, Langer R, Cima MJ. Multi-reservoir device for detecting a soluble cancer biomarker. Lab Chip. 2007;7:1288–93. doi: 10.1039/b705143c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials associated with this article can be found at doi: 10.1016/j.biomaterials.2008.01.029.