

Abstract
Metnase (also known as SETMAR) is a SET and transposase fusion protein in humans and plays a positive role in double-strand break (DSB) repair. While the SET domain possesses histone lysine methyltransferase activity, the transposase domain is responsible for 5′-terminal inverted repeat (TIR)-specific binding, DNA looping, and DNA cleavage activities. We recently demonstrated that human homolog of Pso4 (hPso4) is a Metnase binding partner that mediates Metnase binding to non-TIR DNA such as DNA damage sites. Here we show that Metnase functions as a dimer in its TIR binding. While both Metnase and hPso4 can independently interact with TIR DNA, Metnase’s DNA binding activity is not required for formation of the Metnase-hPso4-DNA complex. A further stoichiometric analysis indicated that only one protein is involved in interaction with dsDNA when Metnase-hPso4 forms a stable complex. Interaction of the Metnase-hPso4 complex with TIR DNA was competitively inhibited by both TIR and non-TIR DNA, suggesting that hPso4 is solely responsible for binding to DNA in the Metnase-hPso4-DNA complex. Together, our study suggests that hPso4, once it forms a complex with Metnase, negatively regulates Metnase’s TIR binding activity, which is perhaps necessary for Metnase localization at non-TIR sites such as DSBs.
INTRODUCTION
Metnase, also known as SETMAR, is a double strand break (DSB) repair factor that has a conserved functional transposase coding sequence spliced into a human gene encoding a SET domain protein [1–6]. The SET domain is responsible for histone lysine methyltransferase activity at histone 3 lysine 4 and lysine 36 [2], while the transposase domain preserves most of the activities of the ancestral Hsmar1 transposase, including 5′-terminal inverted repeat (TIR)-specific DNA binding [1, 3–5], assembly of a paired end complex (PEC) [3], and DNA cleavage activity [3, 4, 7]. Unlike other transposases, however, Metnase-mediated DNA cleavage is not coupled to its sequence-specific DNA binding [3, 4], suggesting that its DNA cleavage action may be involved in other DNA metabolisms such as DNA repair [2, 7].
A recent study showed that Metnase physically interacts with human Pso4 (hPso4) [5], a member of the pre-mRNA splicing complex [8] and a U-box protein with associated E3 ubiquitin ligase activity crucial for its function in pre-mRNA splicing in vivo [9–11]. Human Pso4 contains six successive WD-40 motifs at the C-terminus that is known to form a structural interface for the assembly of multiprotein complexes [12], and has been identified as a component of the nuclear matrix [13]. Pso4 also interacts with terminal deoxynucleotidyl transferase (TdT), a protein that catalyzes addition of nucleotides to the 3-terminus of DNA during V(D)J recombination repair pathways [14]. Yeast studies [15–19] and mammalian experiments with a targeted inhibition of hPso4 expression [14] suggested that hPso4 plays a unique role in DSB repair. It has also been previously linked to DNA repair through a direct physical interaction between Cdc5L and WRN, the protein deficient in Werner Syndrome [20].
Although the transposase domain is essential for Metnase function in DNA repair [2], it is not clear how a protein with a sequence-specific DNA binding activity plays a role in DNA repair. A recent observation showed that hPso4 is a double-stranded DNA binding protein [14] mediating formation of a stable complex with Metnase on non-TIR DNA [5]. In vivo, hPso4 is induced and co-localized with Metnase following IR treatment, and cells treated with hPso4-siRNA failed to show localization of Metnase at DSB sites and Metnase-mediated stimulation of DNA end joining coupled to genomic integration [5], suggesting that hPso4 is necessary to bring Metnase to the DSB sites for its function(s) in DNA repair. In this study, we biochemically analyzed the Metnase-hPso4 interaction with dsDNA, and showed that Metnase dimer forms a 1:1 stoichiometric complex with hPso4 tetramer on dsDNA. Stoichiometric and competition analyses showed that hPso4 is solely responsible for binding to dsDNA in the Metnase-hPso4-DNA complex, suggesting that Pso4 may control Metnase’s TIR-specific DNA binding activity.
MATERIALS AND METHODS
Cells, Enzymes, and Chemicals
Human Embryonic Kidney (HEK 293) cells were grown in Dulbecco’s modified Eagle’s media (DMEM) (GIBCO-BRL) supplemented with 10% fetal calf serum (GIBCO-BRL), penicillin (10 unit/ml, Sigma), and streptomycin (0.1 mg/ml, Sigma). [γ-32P]-ATP (3000 Ci/mmol) was from Perkin-Elmer and Analytical Science (Boston, MA) and Bradford reagents and protein molecular weight markers were purchased from Bio-Rad (Hercules, CA). An anti-Metnase polyclonal antibody was previously described [2] and an anti- Pso4 polyclonal antibody specific for human Pso4 was obtained from Calbiochem (Darmstadt, Germany). Anti-Flag (M2) and -V5 monoclonal antibodies were from Sigma (St. Louis, MO) and Invitrogen (Carlsbad, CA), respectively.
Generation of stable cell lines
Cells stably over-expressing wt-Metnase or wt-hPso4 were generated by transfecting HEK293 cells with a vector harboring FLAG-Metnase, V5-Metnase, or FLAG-hPso4 using FuGENE6 transfection reagent (Roche Molecular Biologicals), followed by selection with G418 (Invitrogen) for 14 days, after which a single colony was isolated and amplified.
Purification of Metnase and hPso4
Purification of both wt-Metnase and hPso4 was previously described [5]. Briefly, Metnase- (or hPso4-) overexpressing human 293 cells (1.6 × 108) were suspended in 20 ml of extraction buffer (TEGDN; 50 mM Tris-HCl pH 7.5, 1 mM EDTA, 10 % glycerol, 5 mM DTT, 1.0 % Nonidet-P40, and mammalian protease inhibitor cocktails containing 0.2M NaCl), and centrifuged (100,000 × g) for 30 min. The supernatant (S100 fraction) was filtered through a Whatman paper and used for immunoaffinity purification. The S100 fraction was incubated at 4 °C for 60 min with anti-FLAG M2 affinity gel (Sigma) that had been pre-equilibrated with TEGDN buffer containing 0.2 M NaCl. The beads were washed three times with TEGDN-2.0 M NaCl buffer prior to elution of the protein with TEGDN-0.2 M NaCl containing FLAG peptide (500 μg/ml). If necessary, the eluant was diluted with 10 volumes of TEGDN buffer, and loaded onto a heparin-Sepharose 6 Fast Flow column (Amersham Biosciences) pre-equilibrated with TEGDN buffer. After washing the column, Metnase (or hPso4) was fractionated using a linear gradient (0 to 2.0 M NaCl) of TEGDN buffer. The eluted protein was dialyzed against TEGDN buffer containing 50 mM NaCl and stored at −80°C.
Preparation of DNA substrates
DNA substrates were obtained from the Integrated DNA Technologies (Coralville, IA). The 32-mer dsDNA containing either the 19-mer core sequence necessary for Metnase binding (TIR32) or 5 nts mutation at the core (MAR3M) was previously described [4].
Electrophoretic mobility shift assay (EMSA) for protein-DNA interaction
Duplex DNA was labeled with [γ-32P]-ATP (3,000 Ci/mmol) and T4 polynucleotide kinase (Roche Molecular Biochemical) according to the manufacture’s instructions. Indicated amount of purified Metnase and/or hPso4 was incubated with 200 or 400 fmol of 5′-32P-labeled DNA at room temperature for 15 min in a reaction mixture containing 50 mM Tris-HCl (pH 7.5), 5 mM DTT, bovine serum albumin (0.2 μg/μl), 5% (w/v) glycerol, and 50 mM NaCl. The protein(s)-DNA complex was analyzed on either 5% polyacrylamide gel (acrylamide: bis-acrylamide = 43.2: 0.8) in 0.5X TBE or 1% vertical agarose gel in 1X TBE. The gels were dried and exposed to x-ray films (Kodak). For quantification, the bands of interest were excised from the gels and measured for radioactivity using a Beckman Scintillation Counter LS 6500. Alternatively, the individual DNA was quantified using the NIH image program (version 1.62).
Glycerol gradient sedimentation
Purified Metnase or hPso4 was sedimented through a linear 10–35 % (vol/vol) glycerol gradient at 45,500 rpm (196,276 × g) for 26 hrs at 4°C. Fractions (175 ul/each) were collected from the bottom, and the aliquots were run on 10% SDS-PAGE for silver staining and western blot analysis and 5% PAGE for dsDNA binding activity.
RESULTS
Metnase functions as a dimer in its binding to TIR DNA
Metnase is a TIR-specific DNA binding protein [1, 3–5] that promotes a paired-end complex (PEC) of two TIR DNAs [3], suggesting that it may function as a dimer. To figure out this possibility, we first examined whether Metnase molecules interact with each other in vivo. A vector harboring FLAG- or V5-Metnase was transfected into HEK-293 cells stably expressing V5- or FLAG-Metnase, respectively, and cell extracts were examined for interaction between Metnase molecules. FLAG- and V5-Metnase were co-immunoprecipitated when a reciprocal pull-down of Metnase was carried out using an anti-V5 or -FLAG antibody (Fig. 1), indicating that Metnase molecules interact with each other in vivo. To see whether Metnase actually exists as a dimer, we examined for native molecular mass of Metnase by a glycerol gradient centrifugation. For this, purified wt-Metnase was mixed with protein markers [urease (260 kDa, 11.8S), alcohol dehydrogenase (A.D., 150 kDa, 7.4S), bovine serum albumin (BSA, 67 kDa, 4.4S), and carbonic anhydrase (C.A., 29 kDa, 2.8S)] and sedimented on a 10–35 % glycerol gradient centrifugation. The 78 kDa of Flag-Metnase possessing the TIR binding activity was eluted at the position of 150 kDa alcohol dehydrogenase (Fig. 2), indicating that Metnase functions as a dimer in binding to TIR DNA.
Figure 1. Interaction of Metnase molecules in vivo.
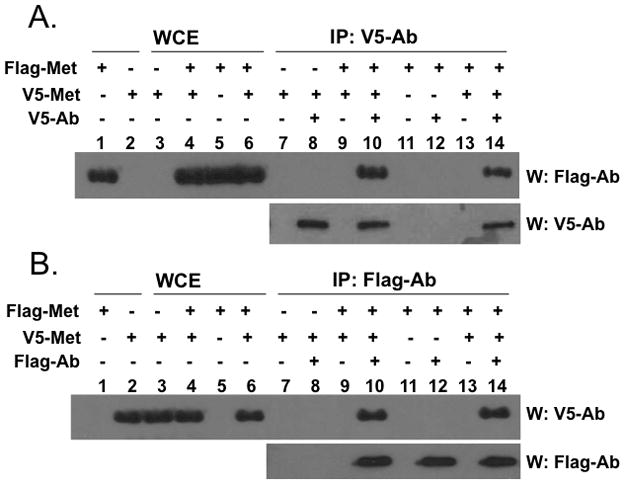
HEK-293 cells stably expressing V5-Metnase or Flag-Metnase were transfected with a vector harboring Flag-Metnase or V5-Metnase, respectively. Forty-eight hrs later, cell extracts were prepared and incubated with either an anti-V5 (panel A) or -Flag (panel B) monoclonal antibody for co-immunoprecipitation of V5- and Flag-Metnase. Following immunoprecipitation, samples were run on 10% SDS-PAGE and immunoblotted using either anti-Flag or -V5 antibody as indicated on the figure. In lanes 1–6, either purified Flag- or V5-Metnase (lanes 1 & 2) or whole cell extracts (WCE, lanes 3–6) were included as loading controls.
Figure 2. Metnase functions as a dimer in its binding to TIR DNA.
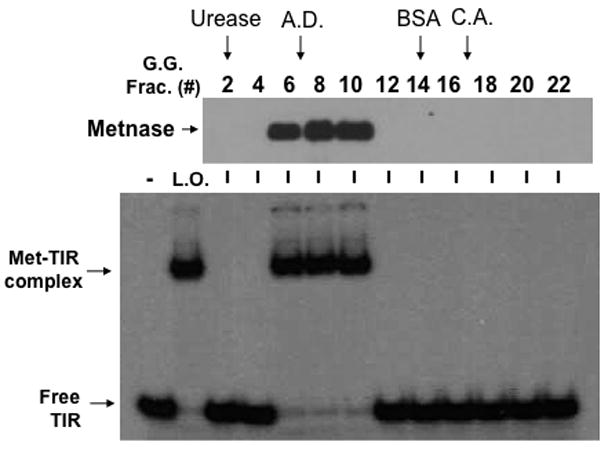
Glycerol gradient sedimentation of wt-Metnase. An aliquot of the immunoaffinity-purified wt-Metnase was diluted with an equal volume of buffer B (50 mM Tris-HCl, pH 7.5, 5 mM DTT, 200 mM NaCl, 0.02 % (w/v) NP40, and 0.5 mM EDTA, pH 8.0) containing protein markers [urease (260 kDa, 11.8S), alcohol dehydrogenase (A.D.; 150 kDa, 7.4S), bovine serum albumin (BSA; 67 kDa, 4.4S), and carbonic anhydrase (C.A.; 29 kDa, 2.8S)] and layered onto a 4.2-ml linear 10–35% (vol/vol) glycerol gradient. Gradients were centrifuged at 45,500 rpm for 26 hr in a Beckman SW55 rotor at 4°C. Fractions were collected from the bottom and analyzed for wt-Metnase (western blot, top panel) and TIR-specific DNA binding activity (bottom panel). Arrows marked indicate positions of protein markers.
Metnase’s DNA binding activity is not required for its interaction with DNA once it forms a stable complex with hPso4
Metnase forms a stable complex with its binding partner, human Pso4 (hPso4) on dsDNA, which is essential for Metnase localization at DNA double-strand break (DSB) in vivo [5]. Metnase possesses TIR-specific DNA binding activity [1, 3–5], whereas hPso4 binds to dsDNA with no sequence preference [14]. Given that both Metnase and hPso4 bind to TIR DNA [4, 5], each protein in the Metnase-hPso4 complex may interact with DNA separately. Alternatively, only one protein in the Metnase-hPso4 complex is responsible for binding to TIR. To figure this out, we compared TIR binding of wt-Metnase with a mutant (R432A) lacking TIR binding activity [4] in the presence of hPso4. In a stoichiometric analysis, addition of increasing amounts of hPso4 tetramer [11] proportionally increased formation of the Metnase-hPso4-TIR complex in the presence of fixed amount of Metnase dimer (Fig. 3B). A similar result was observed when non-TIR DNA (MAR3M) was used instead of TIR (Fig. 3C), suggesting that Metnase dimer forms a 1:1 stoichiometric complex with hPso4 on dsDNA. A mutant (R432A) lacking TIR binding activity [4] also effectively formed a stable complex with hPso4 on TIR DNA (Fig. 3D), indicating that Metnase’s DNA binding activity is not required for formation of a stable Metnase-hPso4-DNA complex.
Figure 3. Formation of a stable Metnase-hPso4-DNA complex does not require Metnase’s DNA binding activity.
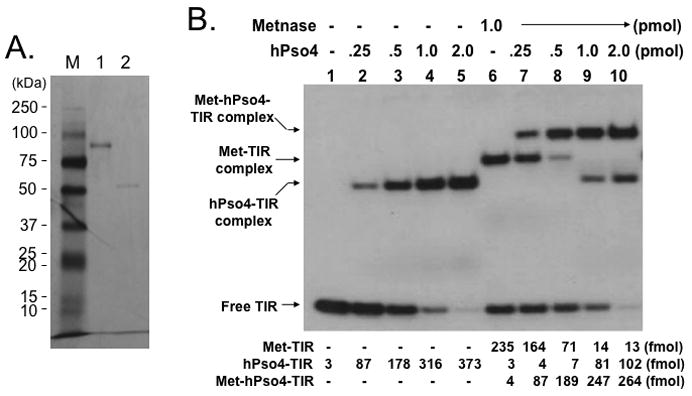
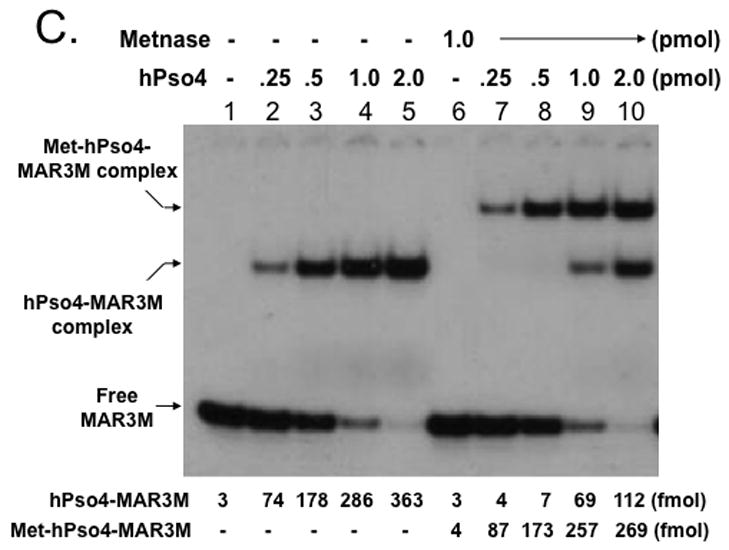
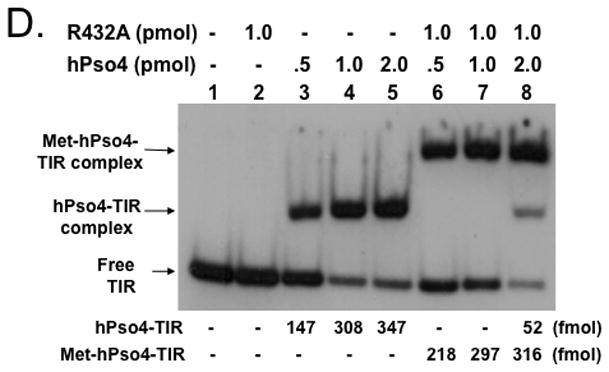
A. SDS-PAGE of immunoaffinity purified Flag-Metnase (lane 1) and Flag-hPso4 (lane 2) used in this study. Lane M represents protein markers. B & C. Formation of a stable Metnase-hPso4 complex with TIR and non-TIR DNA. Reaction mixtures (20 ul) containing fixed amount of Metnase (0 or 1.0 pmol) were incubated with varying amounts of hPso4 (0.25, 0.5, 1.0, and 2.0 pmol) for 15 min prior to addition of 400 fmol of 5′-32P-labeled TIR DNA (panel B) or non-TIR DNA (MAR3M, panel C). Following 15 min incubation at 25°C, the protein-DNA complexes were analyzed by 5% native PAGE in the presence of 1X TBE. For quantification, individual bands were excised from dried gel and measured for radioactivity. D. A Metnase mutant (R432A) lacking DNA binding activity form a stable complex with hPso4 and dsDNA is independent of Metnase’s DNA binding activity. Reaction mixtures (20 ul) containing indicated amounts of R432A lacking TIR-specific DNA binding activity and/or hPso4 were mixed with 200 fmol of 5′-32P-labeled TIR and incubated for 15 min at 25°C, and analyzed by 5% native PAGE.
Only one protein is responsible for binding to TIR DNA when Metnase and hPso4 form a stable complex with DNA
Since Metnase’s DNA binding activity is not necessary for formation of the Metnase-hPso4-DNA complex (Fig. 3D), we reasoned that hPso4 is responsible for binding to dsDNA in the Metnase-hPso4-DNA complex. To test this, we carried out a stoichiometric analysis in which the interaction of Metnase, hPso4, and the Metnase-hPso4 with TIR DNA were quantitatively measured. Although both Metnase and hPso4 possess TIR binding activity, the amount of the Metnase-hPso4-TIR complex formed in a gel mobility shift assay was identical to the amount of Metnase-TIR or hPso4-TIR complex (Fig. 4A & 4B). Similarly, there was no difference between single protein (2 pmol of either Metnase or hPso4) and the mixture (2 pmol of Metnase + 2 pmol of hPso4) in forming the protein-TIR complex when varying amounts of 32P-labeled TIR were added to the reaction mixtures (Fig. 4C). We next examined interaction of Metnase, hPso4, and the Metnase-hPso4 complex with non-TIR DNA (MAR3M). The amount of MAR3M bound to hPso4 was almost identical to those bound to the Metnase-hPso4 complex (Fig. 4D), suggesting that hPso4 is equally effective as the Metnase-hPso4 complex in binding to non-TIR DNA. Together, our observation strongly suggests that only one protein is responsible for interacting with TIR when the Metnase-hPso4 form a stable complex. Given that Metnase’s DNA binding activity is not necessary for formation of the Metnase-hPso4-DNA complex (Fig. 3D), our observation supports a notion that hPso4 is responsible for binding to DNA in the Metnase-hPso4-DNA complex.
Figure 4. Metnase forms a 1:1 stoichiometric complex with hPso4 on dsDNA.
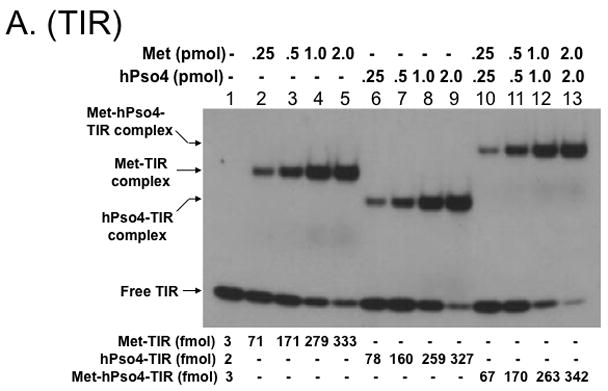
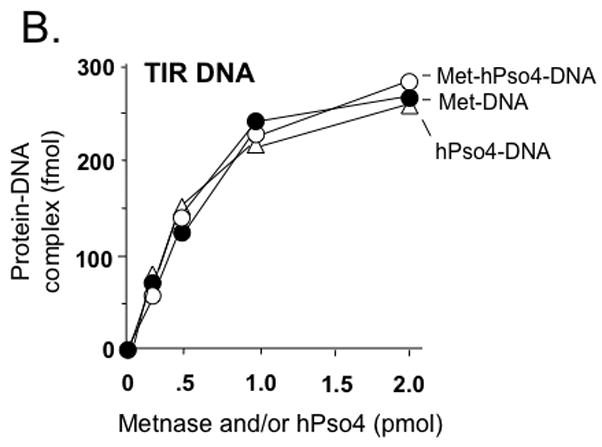
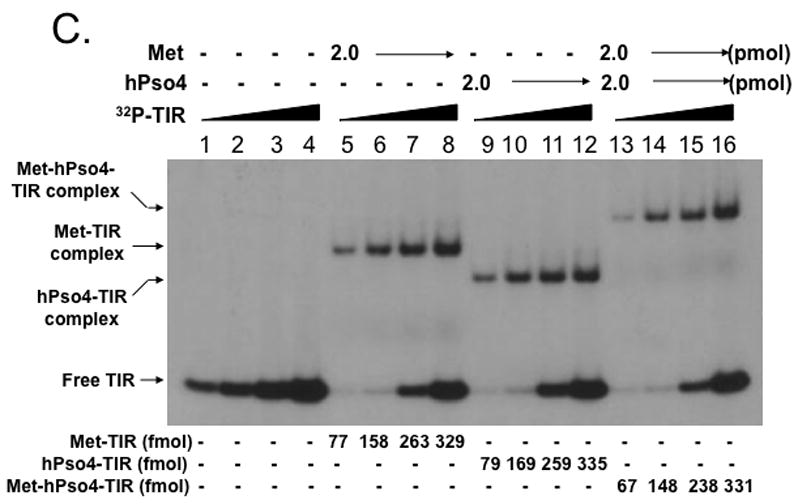
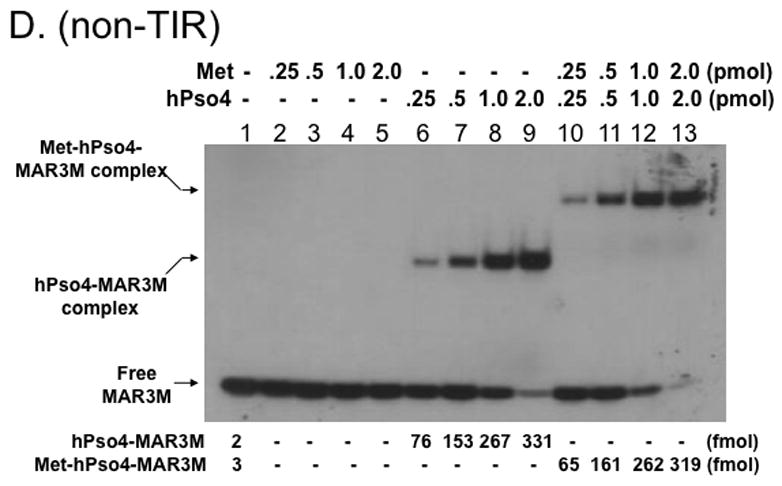
A. Stoichiometric analysis of Metnase and/or hPso4 interaction with TIR DNA. Reaction mixtures (20 ul) containing indicated amounts of Metnase and/or hPso4 were incubated with 5′-32P-labeled DNA (400 fmol) for 15 min prior to 5% native PAGE analysis in the presence of 1X TBE. For quantification, individual bands were excised from dried gel and measured for radioactivity. B. Amounts of the Metnase-TIR (closed circle), hPso4-TIR (open triangle), and Metnase-hPso4-TIR complexes (open circle) from Fig. 4A were plotted. C. Relative TIR-binding activity of Metnase, hPso4, and the Metnase-hPso4 complex. Metnase, hPso4, or the Metnase-hPso4 complex (2 pmol each) was incubated with varying amounts (0.1, 0.2, 0.4, and 0.8 pmol) of 32P-TIR DNA at 25°C for 15 min prior to 5% native PAGE analysis. Individual protein-DNA complexes (marked on the left side of the figure) were quantified using the NIH image program (version 1.62). D. Stoichiometric analysis of Metnase and/or hPso4 interaction with non-TIR DNA (MAR3M). Reaction mixtures (20 ul) were the same as those described in Figure 4A, except for the use of 5′-32P-labeled non-TIR DNA (MAR3M, 400 fmol). Individual protein-DNA complexes were marked on the left side.
hPso4 is solely responsible for binding to dsDNA when it forms a stable complex with Metnase
To further understand interaction of the Metnase-hPso4 complex with dsDNA, we carried out a competition experiment in which the interaction of Metnase and/or hPso4 with 32P-TIR DNA was examined in the presence of non-labeled competitor DNA. Formation of the Metnase-32P-TIR complex was significantly inhibited in the presence of TIR DNA (Fig. 5, lanes 2–6), whereas the hPso4-32P-TIR complex was competitively inhibited in the presence of either TIR or non-TIR DNA (Fig. 5, lanes 7–11). Similar to the hPso4-TIR complex, formation of the Metnase-hPso4-TIR complex was competitively inhibited by both TIR and non-TIR DNA (Fig. 5, lanes 12–16), suggesting that hPso4 is solely responsible for binding to DNA in the Metnase-hPso4-DNA complex.
Figure 5. Interaction of the Metnase-hPso4 complex with TIR DNA is equally inhibited by both TIR and non-TIR DNA.
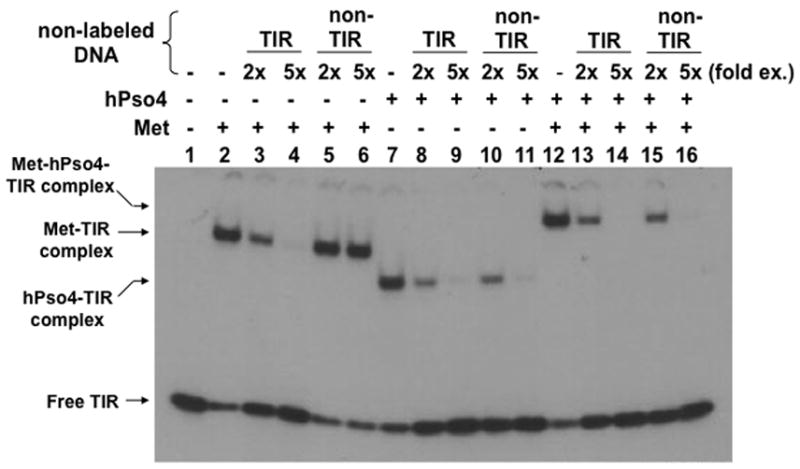
Two pmol of Metnase, hPso4, or the Metnase-hPso4 complex was preincubated for 15 min at 25°C before addition of 200 fmol of 32P-TIR DNA and competitor DNA [400 fmol (2-fold excess) or 1,000 (5-fold excess) of unlabeled TIR or non-TIR (MAR3M) DNA]. Following 15 min incubation, the reaction mixtures were analyzed by 5% native PAGE. Individual protein-DNA complexes were marked on the left side.
DISCUSSION
Metnase is a multi-functional protein with undefined role in mammalian DNA repair and genomic integration of foreign DNA [2, 5, 6]. We recently identified that hPso4 is a Metnase binding partner that mediates the interaction of Metnase with non-TIR DNA such as DSB sites necessary for Metnase’s function in DSB repair [5]. In this study, we carried out a biochemical analysis of the Metnase, hPso4, and the Metnase-hPso4 complex for their interaction with dsDNA, and showed that i) Metnase functions as a dimer in its interaction with TIR DNA, ii) Metnase dimer forms a 1:1 stoichiometric complex with hPso4 (tetramer) on dsDNA, and iii) hPso4, once forming a stable Metnase-hPso4 complex, is solely responsible for binding to DNA. These findings suggest that hPso4, once forming a complex with free Metnase, negatively regulates Metnase-TIR interaction, which may be necessary for Metnase’s localization at non-TIR sites such as DSBs following IR.
Transposases recognize both ends of transposon to excise the element from one site and insert it at another, and it is likely that the ends are brought together and form a synaptic complex comprising two transposase molecules and the two ends of the corresponding element [3, 22–24]. A recent in vitro study with long and short transposon ends demonstrated that Metnase (SETMAR) formed a paired-end complex with two transposon ends [3]. In this study, we provided in vivo and in vitro evidence that Metnase (SETMAR) stably functions as a dimer in its binding to duplex DNA (Fig. 2). Considering that human chromosomes possesses thousands of TIR and TIR-like sequences that Metnase can interact with [1, 3–5, 25], Metnase dimer may simultaneously interact with two distant TIR sites and generate DNA looping and/or unique DNA structure within the nucleus.
Upon DNA damage, hPso4 was induced [14] and formed a stable complex with Metnase [5]. Although the physiologic role of the Metnase-hPso4 interaction is not fully understood, cells lacking hPso4 failed to show Metnase localization at the DSB sites [5], suggesting that hPso4 plays a crucial role in the recruitment of Metnase to the DSB sites. The stoichiometric analysis described in this study has several interesting implications for the architecture of the Metnase-hPso4 complex on dsDNA. First, Metnase dimer forms a 1:1 stoichiometric complex with hPso4 on dsDNA [11] (Fig. 4). Although hPso4 is a direct binding partner of Metnase, Metnase also pulled down the human homolog of Spf27, a member of the Prp19 core complex involved in pre-mRNA splicing [5]. Given that Pso4 is a part of the pre-mRNA splicing complex consisting of Pso4, Cdc5L, Plrg1, and Spf27 [8], the Metnase-hPso4 complex may also be a part of the bigger complex including other members of the pre-mRNA splicing complex in vivo. Secondly, although both Metnase and hPso4 independently interact with TIR DNA, hPso4 is solely responsible for binding to dsDNA once the two proteins form a stable complex. This claim is based on the findings that i) the Metnase-hPso4 complex interacted with same stoichiometric amount of non-TIR DNA as the TIR DNA (Fig. 3), ii) the Metnase-hPso4 complex interacted with same number of TIR molecules as Metnase or hPso4 alone did (Fig. 4), and iii) formation of the Metnase-32P-TIR complex was significantly inhibited by excess of TIR but not by non-TIR, whereas the Metnase-hPso4-TIR complexes were equally inhibited by both TIR and non-TIR DNA (Fig. 5).
It is not clear how Metnase’ TIR binding activity is not functioning when it forms a complex with hPso4, although both Metnase and hPso4 possess very similar binding affinity to TIR DNA [4, 5]. It is possible that hPso4, once forming a complex with Metnase, may directly interfere with Metnase’s DNA binding domain within the transposase [4]. Further structural study would be necessary to clarify this intriguing issue. Considering that hPso4 is induced following IR treatment in vivo [5, 14, 26], formation of a stable Metnase-hPso4 complex likely occurs in response to DNA damage. The Pso4 also undergoes structural alterations in response to DNA damage [26]. The Metnase-hPso4 complex, once formed, likely goes to non-TIR sites such as DSB sites [5], since hPso4 is solely responsible for binding to DNA in forming the Metnase-hPso4-DNA complex.
Acknowledgments
This research was supported by grants from NIH (CA92111 & CA140422), NIH predoctoral training grants (T32 DK0075-20 to BDB), the Walther Cancer Institute, and the IU Simon Cancer Center.
ABBREVIATIONS
- DSB
double strand break
- DTT
dithiothreitol
- DMEM
Dulbecco’s Modified Eagle Medium
- HLMT
histone lysine methyltransferase
- HTH
helix-turn-helix
- IR
ionizing radiation
- MBS
Metnase Binding Site(s)
- PAGE
polyacrylamide gel electrophoresis
- PEC
paired end complex
- SET
Su(var)3–9, Enhancer-of-zeste, Trithorax
- TIR
5′-terminal inverted repeat
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cordaux R, Udit S, Batzer MA, Feschotte C. Proc Natl Acad Sci U S A. 2006;103:8101–8106. doi: 10.1073/pnas.0601161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee SH, Oshige M, Durant ST, Rasila KK, Williamson EA, Ramsey H, Kwan L, Nickoloff JA, Hromas R. Proc Natl Acad Sci U S A. 2005;102:18075–18080. doi: 10.1073/pnas.0503676102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu D, Bischerour J, Siddique A, Buisine N, Bigot Y, Chalmers R. Mol Cell Biol. 2007;27:1125–1132. doi: 10.1128/MCB.01899-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roman Y, Oshige M, Lee YJ, Goodwin K, Georgiadis MM, Hromas RA, Lee SH. Biochemistry. 2007;46:11369–11376. doi: 10.1021/bi7005477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beck BD, Park SJ, Lee YJ, Roman Y, Hromas RA, Lee SH. J Biol Chem. 2008;283:9023–9030. doi: 10.1074/jbc.M800150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williamson EA, Farrington J, Martinez L, Ness S, O’Rourke J, Lee SH, Nickoloff J, Hromas R. Biochimie. 2008 doi: 10.1016/j.biochi.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miskey C, Papp B, Mates L, Sinzelle L, Keller H, Izsvak Z, Ivics Z. Mol Cell Biol. 2007;27:4589–4600. doi: 10.1128/MCB.02027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ajuh P, Kuster B, Panov K, Zomerdijk JC, Mann M, Lamond AI. Embo J. 2000;19:6569–6581. doi: 10.1093/emboj/19.23.6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohi MD, Vander Kooi CW, Rosenberg JA, Chazin WJ, Gould KL. Nat Struct Biol. 2003;10:250–255. doi: 10.1038/nsb906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loscher M, Fortschegger K, Ritter G, Wostry M, Voglauer R, Schmid JA, Watters S, Rivett AJ, Ajuh P, Lamond AI, Katinger H, Grillari J. Biochem J. 2005;388:593–603. doi: 10.1042/BJ20041517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vander Kooi CW, Ohi MD, Rosenberg JA, Oldham ML, Newcomer ME, Gould KL, Chazin WJ. Biochemistry. 2006;45:121–130. doi: 10.1021/bi051787e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gotzmann J, Gerner C, Meissner M, Holzmann K, Grimm R, Mikulits W, Sauermann G. Exp Cell Res. 2000;261:166–179. doi: 10.1006/excr.2000.5025. [DOI] [PubMed] [Google Scholar]
- 13.Gerner C, Sauermann G. J Cell Biochem. 1999;72:470–482. [PubMed] [Google Scholar]
- 14.Mahajan KN, Mitchell BS. Proc Natl Acad Sci U S A. 2003;100:10746–10751. doi: 10.1073/pnas.1631060100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brendel M, Bonatto D, Strauss M, Revers LF, Pungartnik C, Saffi J, Henriques JA. Mutat Res. 2003;544:179–193. doi: 10.1016/j.mrrev.2003.06.018. [DOI] [PubMed] [Google Scholar]
- 16.Brendel M, Henriques JA. Mutat Res. 2001;489:79–96. doi: 10.1016/s1383-5742(01)00066-7. [DOI] [PubMed] [Google Scholar]
- 17.da Silva KV, de Morais Junior MA, Henriques JA. Curr Genet. 1995;27:207–212. doi: 10.1007/BF00326150. [DOI] [PubMed] [Google Scholar]
- 18.Henriques JA, Vicente EJ, Leandro da Silva KV, Schenberg AC. Mutat Res. 1989;218:111–124. doi: 10.1016/0921-8777(89)90017-7. [DOI] [PubMed] [Google Scholar]
- 19.Meira LB, Fonseca MB, Averbeck D, Schenberg AC, Henriques JA. Mol Gen Genet. 1992;235:311–316. doi: 10.1007/BF00279375. [DOI] [PubMed] [Google Scholar]
- 20.Zhang N, Kaur R, Lu X, Shen X, Li L, Legerski RJ. J Biol Chem. 2005;280:40559–40567. doi: 10.1074/jbc.M508453200. [DOI] [PubMed] [Google Scholar]
- 21.Boulikas T. Crit Rev Eukaryot Gene Expr. 1993;3:193–227. [PubMed] [Google Scholar]
- 22.Adams CD, Schnurr B, Skoko D, Marko JF, Reznikoff WS. Mol Microbiol. 2006;62:1558–1568. doi: 10.1111/j.1365-2958.2006.05471.x. [DOI] [PubMed] [Google Scholar]
- 23.Richardson JM, Dawson A, O’Hagan N, Taylor P, Finnegan DJ, Walkinshaw MD. Embo J. 2006;25:1324–1334. doi: 10.1038/sj.emboj.7601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crellin P, Sewitz S, Chalmers R. Mol Cell. 2004;13:537–547. doi: 10.1016/s1097-2765(04)00052-8. [DOI] [PubMed] [Google Scholar]
- 25.Jordan IK. Proc Natl Acad Sci U S A. 2006;103:7941–7942. doi: 10.1073/pnas.0602656103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu X, Legerski RJ. Biochem Biophys Res Commun. 2007;354:968–974. doi: 10.1016/j.bbrc.2007.01.097. [DOI] [PMC free article] [PubMed] [Google Scholar]