

Abstract
Lysine 315 of mouse polyamine amine oxidase corresponds to a lysine residue that is conserved in the flavoprotein amine oxidases of the monoamine oxidase structural family. In several structures, this lysine residue forms a hydrogen bond to a water molecule that is hydrogen-bonded to the flavin N(5). Mutation of Lys315 in polyamine oxidase to methionine was previously shown to have no effect on the kinetics of the reductive half-reaction of the enzyme (Henderson Pozzi, M., Gawandi, V., and Fitzpatrick, Paul F. (2009) Biochemistry 48, 1508–1516). In contrast, the mutation does affect steps in the oxidative half-reaction. The kcat value is unaffected by the mutation; this kinetic parameter likely reflects product release. At pH 10, the kcat/Km value for oxygen is 25-fold lower in the mutant enzyme. The kcat/KO2 value is pH-dependent for the wild-type enzyme, decreasing below a pKa of 7.0, while this kinetic parameter for the mutant enzyme is pH-independent. This is consistent with the neutral form of Lys315 being required for more rapid flavin oxidation. The solvent isotope effect on the kcat/KO2 value increases from 1.4 in the wild-type enzyme to 1.9 in the mutant protein, and the solvent inventory changes from linear to bowed. The effects of the mutation can be explained by the lysine orienting the bridging water so that it can accept the proton from the flavin N(5) during flavin oxidation. In the mutant enzyme the lysine amine would be replaced by a water chain.
A large number of flavoproteins catalyze the oxidation of amines, with molecular oxygen as the final electron acceptor [1]. Based on their sequences and the three-dimensional structures of a growing number of flavoprotein amine oxidases, most of these enzymes can be grouped into two distinct structural families. D-Amino acid oxidase is the paradigm member of the structural family that also contains sarcosine oxidase, glycine oxidase, and dimethylglycine oxidase [2–5]. Monoamine oxidase is the most-studied member of the family that also contains polyamine oxidase, the histone lysine specific demethylase LSD1, and the L-amino acid oxidases [6–10]. The general reaction of flavoprotein amine oxidases can be divided into two half-reactions. The reductive half-reaction consists of the transfer of a hydride equivalent from the substrate to the flavin, producing reduced flavin and oxidized amine. This step is typically irreversible; therefore, these enzymes exhibit ping pong patterns in steady-state kinetic analyses [11]. While the mechanism of amine oxidation has been controversial [12–14], mechanistic and structural results are most consistent with direct transfer of a hydride from the amine substrate to the flavin [2, 15–20]. In the less-studied oxidative half-reaction, two electrons are transferred from the reduced flavin to molecular oxygen, forming H2O2 [21, 22].
Polyamine and spermine oxidases are involved in the catabolism of polyamines in a variety of eukaryotic cells. The mammalian polyamine oxidases (PAOs)1 prefer N1-acetylated spermine and spermidine as substrates, oxidizing the substrate on the exo side of the secondary nitrogen (Scheme 1) [23]. Spermine oxidases catalyze a similar reaction, but are more active with non-acetylated spermine and spermidine [24, 25]. In contrast, plant polyamine oxidases catalyze the oxidation on the endo side of the secondary nitrogen [26], a reaction also catalyzed by putrescine oxidases [27]. To date, no structure of a mammalian polyamine or spermine oxidase has been described. While there are structures of polyamine oxidases from maize and yeast, the former is more accurately a spermine oxidase based on its substrate specificity [28] and the latter is reported to catalyze the oxidation of N1-acetylspermine and spermine equally efficiently [29]. All of the published structures of proteins in the monoamine oxidase family show a conserved lysine residue in the active site [6–8, 30, 31]. In several of these structures there is a water molecule in an appropriate location to form hydrogen bonds to the lysine amino group and the FAD N(5). This is illustrated in Figure 1 for maize polyamine oxidase. When this lysine in that enzyme is mutated to methionine, the rate constant for reduction of the flavin by spermidine is reported to be 1400-fold slower than the value for the wild-type enzyme, suggesting that this residue plays a critical role in amine oxidation [32]. Mutation of the corresponding lysine in LSD1, Lys661, to alanine yielded a mutant protein with no detectable activity in a gel-based assay [33], consistent with a critical role in that enzyme also. While the lack of kinetic data for the LSD1 K661A enzyme precludes determination of which steps in the reaction are affected by the mutation, amine oxidation is rate-limiting for that enzyme, at least with peptide substrates [19]. Alignment of sequences of mouse polyamine oxidase with other members of the monoamine oxidase family, including maize PAO and LSD1 (Figure 2), shows that Lys315 in mouse PAO corresponds to the conserved lysine residue. Surprisingly, mutagenesis of this lysine in mouse PAO to methionine has no effect on the rate constant for flavin reduction at the pH optimum or on the pH dependence of the reductive half-reaction [34], in contrast to the results with maize PAO and LSD1. We report here that mutagenesis of Lys315 in mouse PAO to methionine instead affects the oxidative half-reaction of the enzyme.
Scheme 1.

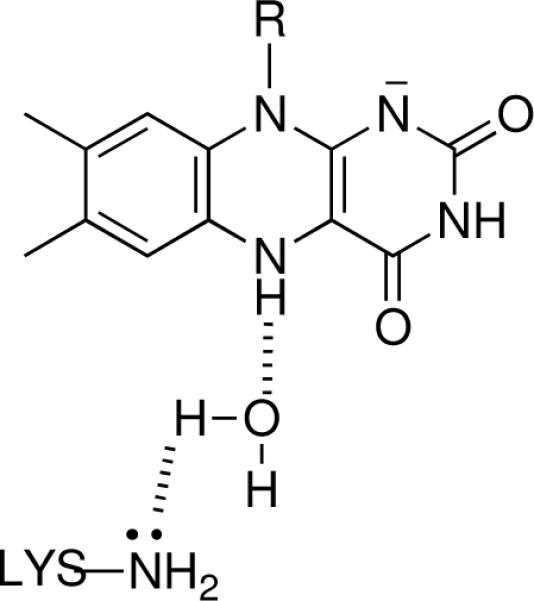
Figure 1.

The water-mediated interaction between the FAD and Lys300 in maize polyamine oxidase. The structure was drawn using pdb file 1h81 [49].
Figure 2.

Multiple sequence alignment for mouse PAO, human MAO-B, human MAO-A, maize PAO, and yeast Fms1. Conserved residues are in bold. The lysine corresponding to Lys315 in mouse PAO is underlined.
EXPERIMENTAL PROCEDURES
Materials
Spermine was purchased from Acros Organics (Geel, Belgium) and N1-acetylspermine was purchased from Fluka (Switzerland). Deuterium oxide was purchased from Cambridge Isotope Laboratories, Inc (Andover, MA). All other materials were of the highest purity commercially available. Wild-type and K315M PAO were expressed and purified as previously described [34].
Assays
Steady-state kinetic assays were performed using a computer-interfaced Hansatech (Hansatech Instruments) oxygen electrode [35]. All assays were initiated by the addition of enzyme. All buffers contained 10% glycerol; 50 mM PIPES, 50 mM Tris-HCl, 50 mM CHES and 50 mM CAPS were used for the pH ranges of 6.6, 7.1–8.6, 9.1–9.6, and 10, respectively. Solvent isotope effects were performed in buffers containing 50 mM CHES (pH 9 or pD 9.4) or 50 mM CAPS (pH 10, or pD 10.4), 10% glycerol. Glycerol buffers with a relative viscosity of 1.3 were prepared as described by Segur and Oberstar [36]. A concentration of 1 mM N1-acetylspermine was used in all assays. Due to the hygroscopic nature of N1-acetylspermine, its concentration was determined enzymatically [20].
Data Analysis
Steady-state kinetic parameters were determined based on fits to the Michaelis-Menten equation using the program KaleidaGraph (Adelbeck Software, Reading, PA). Data for the kcat/KO2-pH profile of wild type PAO were fit to eq 1, which applies for a kinetic parameter that decreases below pK due to the protonation of a single moiety; y is the kcat/KO2 value at the specific pH being measured, c is the pH-independent value of the kcat/KO2, and K is the ionization constant for a residue which must be unprotonated. Eq 2 was used to fit the kcat pH profile for the wild-type enzyme. This describes a kinetic parameter that decreases to a limiting value at low pH; YL and YH are the values of the kinetic parameter at the pH extremes and K is the ionization constant for the transtition [37]. Eqs 3–5 were used to fit the proton inventories for wild type and K315M PAO. Eq 3 describes a proton inventory arising from a single proton. Eq 4 describes a proton inventory when two protons with identical fractionation factors contribute to the isotope effect [38]. Eq 5 describes a proton inventory when a large (≥3) number of protons contribute to the isotope effect [38]. In all three equations, n is the mole fraction of D2O in the buffer, (kcat/KO2)0 and (kcat/KO2)n are the kcat/KO2 values in water and the indicated mole fraction of D2O, respectively, and KIE is the solvent isotope effect.
RESULTS
Steady-state kinetic parameters
Since our previous characterization of K315M PAO showed that the kinetics of amine oxidation were not significantly affected by the mutation, steady-state kinetic parameters were determined at saturating concentrations of N1-acetylspermine (≥50 Km), allowing the kcat and kcat/KO2 values to be determined.2 The analyses were carried out at pH 10, the pH optimum [34], and pH 8, the pH of the peroxisome [39]. The values for the wild-type enzyme and the K315M mutant are shown in Table 1. At pH 8 the kcat/KO2 value for the mutant protein is one-tenth that of the wild-type enzyme, while the effect at pH 10 is slightly larger. The effect on the kcat value is smaller; the high Km for oxygen at pH 10 and the limited solubility of oxygen precluded measuring a reliable kcat value for the mutant protein above pH 8.
Table 1.
Steady-state kinetic parameters for wild type and K315M PAO
| pH 8 | pH 10 | |||||
|---|---|---|---|---|---|---|
| enzyme | kcat/KO2 (mM−1 s−1) | KO2 (mM) | kcat (s−1) | kcat/KO2 (mM−1 s−1) | KO2 (mM) | kcat (s−1) |
| Wild-type | 14 ± 1 | 0.32 ± 0.05 | 4.3 ± 0.2 | 21 ± 3 | 0.99 ± 0.25 | 20.5 ± 2.6 |
| K315M | 1.2 ± 0.1 | 1.0 ± 0.1 | 1.2 ± 0.1 | 0.7 ± 0.1 | 6 ± 6 | 4 ± 4 |
*Conditions: 1 mM N1-acetylspermine, 0.05 mM Tris-HCl (pH 8) or CAPS (pH 10), 20° C
pH profiles
The values in Table 1 show that the kcat/KO2 and kcat values are affected to some extent by pH. Accordingly the effect of pH on these kinetic parameters was determined for the wild-type and mutant enzyme over the pH range of 6.6–10. The kcat/KO2-pH profiles are shown in Figure 3. The kcat/KO2 value for the wild-type enzyme is clearly pH-dependent, with a limiting value of 20 ± 1 mM−1s−1 at high pH and decreasing below a pKa of 7.0 ± 0.1. In contrast, the kcat/KO2 value for the mutant protein is independent of the pH between pH 6.6 and 10, with an average value of 0.8 ± 0.1 mM−1s−1 over that pH range. This is consistent with the mutation resulting in a decrease of 25-fold in the reactivity of the reduced enzyme with oxygen at the pH optimum and with Lys315 being responsible for the pH dependence of the kcat/KO2 value.
Figure 3.

kcat/KO2-pH profile for wild-type (filled circles) and K315M (open circles) PAO with N1-acetylspermine. The line is from a fit of the data to eq 1.
The kcat-pH profile for the wild-type enzyme is shown in Figure 4. There is a decrease in the value of this kinetic parameter at low pH from a limiting value at high pH. The data were fit to eq 2, which assumes the kcat value reaches a limiting value at low pH, with a pKa value of 8.8 ± 0.2 for the transition. Eq 2 gave a better fit than a model which assumes that kcat value goes to zero at low pH. It was not possible to construct a kcat-pH profile for the mutant enzyme due to the high KO2 value above pH 8. The kcat values for the mutant at pH 8 and below are shown in Figure 4 and establish that the kcat value for the mutant protein is close to the wild-type value over that limited pH range.
Figure 4.

kcat-pH profile for wild-type (filled circles) and K315M (open circles) PAO with N1-acetylspermine. The line is from a fit to eq 2 of the wild-type enzyme data.
Solvent Isotope Effects and Proton Inventory
To gain further insight into the role of Lys315 in the reaction of reduced PAO with oxygen, the solvent isotope effects on the kcat/KO2 value for wild-type and K315M PAO were determined. The solvent isotope effect for wild-type PAO at pH 10/pD 10.4, where the kcat/KO2 value is pH-independent, is 1.43 ± 0.05. This value is not very different from the relative viscosity of a D2O solution, 1.3 versus water. To test whether the small isotope effect is simply due to the viscosity of the D2O buffer, the effect of 10% (w/w) glycerol, which similarly increases the viscosity of the solvent by 30%, was determined. This concentration of glycerol resulted in a slight increase in the kcat/KO2 value, for an inverse viscosity effect of 0.93 ± 0.06. Thus, the solvent isotope effect is not due to the viscosity of the D2O solution. The solvent isotope effect on the kcat/KO2 value for the mutant protein was also determined. At pH 10/pD 10.4 the isotope effect is 1.84 ± 0.08. To ensure that data were collected in a pH-insensitive region, the solvent isotope effect was also determined at pH 9/pD 9.4, where the value is 1.98 ± 0.05. These values are within error of each other and give an average solvent isotope effect of 1.91 ± 0.06. These data establish that the mutation has increased the sensitivity of the kcat/KO2 value to D2O.
Solvent isotope effects can be difficult to interpret, in that a large number of protons in the enzyme will exchange with D2O. A proton inventory experiment, in which the solvent isotope effect is determined at various ratios of H2O and D2O, can establish the number of protons in flight in the isotope-sensitive transition state. Accordingly, proton inventories were determined for the wild-type and mutant proteins. The results are shown in Figure 5. For the wild-type enzyme, the data are readily fit by a straight line, consistent with this solvent isotope arising from a single exchangeable proton. Fitting the data to eq 3, which describes a proton inventory in which the isotope effect arises from a single proton in the transition state, gives a solvent isotope effect of 1.39 ± 0.02. In contrast to the result for the wild-type enzyme, the proton inventory for K315M PAO is bowed, consistent with more than one exchangeable proton contributing to the isotope effect. Use of eq 4, which applies for two exchangeable protons, gave only a slightly better fit. Consequently, the data were fit to eq 5, which describes a solvent isotope effect arising from a large number of protons.
Figure 5.

The effect of the mole fraction of D2O, n, on the kcat/KO2 value at pH 10/pD 10.4 for (A) wild-type and (B) K315M PAO. The lines are from fits to eq 3 in A and to eq 5 in B.
DISCUSSION
Previous analyses of the effect of the K315M mutation on the kinetics of PAO focused on the reductive half-reaction, since the data with the maize enzyme and LSD1 implicated steps in amine oxidation as being substantially slower. However, the mutation does not affect the kcat/Km value for spermine at any pH and does not affect the rate constant for flavin reduction by N1-acetylspermine at the pH optimum, although there is a small change in the pH dependence of that kinetic parameter [34]. The present results establish that the mutation instead affects the reaction of the reduced flavin in PAO with oxygen.
While the increase in the KO2 with pH precludes reliable measurement of the kcat value above pH 8, there is clearly no significant effect of the mutation on this kinetic parameter below that pH. It is thus reasonable to conclude that the K315M mutation does not affect the kcat value of the enzyme. For the wild-type enzyme, the kcat value is more than 20-fold less than the value of kred, the rate constant for flavin reduction at saturating concentrations of the amine substrate, over this pH range [34]. Consequently, the kcat value likely reflects the rate constant for product release rather than a chemical step. The rate constant for this step is clearly pH-dependent, increasing ~100-fold when a group with a pKa of about 8.8 is deprotonated. Similar pH-dependent rate constants for product release have been seen for other flavin amine oxidases, where they were attributed to pH-dependent conformational changes that alter the rate constant for product release [40, 41]. An alternative explanation for the pKa seen in the kcat pH profile for PAO is that it reflects deprotonation of a substrate nitrogen in the enzyme active site. The active form of the amine substrate for PAO has one charged nitrogen, with the others neutral, although the enzyme will bind the substrate with the secondary nitrogen that is oxidized in a charged form [34]. Since the protonation of a nonreacting nitrogen in the substrate increases the affinity of the enzyme for the substrate, it is reasonable that deprotonation of that same nitrogen in the product would decrease the affinity of the enzyme for the product, increasing the rate constant for product release and thus the kcat value.
The K315M mutation results in a decrease of the kcat/KO2 value of 25-fold at the pH optimum. For flavin oxidases, including PAO [42], single turnover kinetic analyses show that the oxidation of the reduced enzyme by oxygen occurs as a second order reaction, with no evidence for an intermediate3 [21].
As a result the steady-state kcat/KO2 value equals the second order rate constant for the reaction of the reduced enzyme with oxygen [44]. The pH dependence of the kcat/KO2 value for wild-type and mutant PAO establishes that the protonation state of Lys315 is important for flavin oxidation and specifically that the nitrogen of Lys315 must be uncharged for more rapid flavin oxidation. This is consistent with the nitrogen being a hydrogen bond acceptor, with the water molecule that forms the bridge to the flavin N(5) as the hydrogen bond donor.
The solvent isotope effect on the wild-type enzyme is consistent with the lysine-water-flavin motif (Figure 1) seen in the structures of maize PAO and other members of the monoamine oxidase structural family. Oxidation of the reduced flavin necessarily results in the loss of a proton from the flavin N(5), while the formation of hydrogen peroxide requires a source of protons. The data are consistent with a model in which the role of the lysine is to properly orient the water molecule hydrogen-bonded to the flavin N(5), so that a free lone pair of electrons is available to facilitate the transfer of the proton from the flavin N(5) to oxygen. The linear proton inventory would then result from the movement of the N(5) proton. An alternative explanation for the solvent isotope effect and the role of Lys315 is that the proton is transferred from the flavin to the bridging water as a second proton is transferred from the water to the neutral lysine. This would require another amino acid residue to act as the source of the proton for oxygen. Either model yields the hydrogen bond interactions shown in Scheme 2. The latter model could result in a curved proton inventory if both protons were in flight in the transition state for flavin oxidation. The linear proton inventory seen with the wild-type enzyme would be due to asynchronicity in the extent of transfer of the two protons. While the data are fit better by a model for a single proton, the precision of the data cannot rule out the involvement of two protons with different isotope effects, if the isotope effect arising from one is 1.1 or less.
Scheme 2.
In contrast to the result with the wild-type enzyme, the solvent inventory for the mutant protein is clearly bowed, indicating that multiple protons are in flight in the transition state for flavin oxidation in this case. Our data do not allow us to determine the number of protons involved with any accuracy beyond stating that the best fits occur with three or more protons. This is a general problem with the proton inventory method [38]. The elimination of the amino moiety of Lys315 by the mutation is likely to alter the interaction of the conserved water molecule with the flavin N(5) and result in an additional water molecule in the active to fill the cavity left by the amino group. If the additional water serves as the proton donor to oxygen as the N(5) proton is lost, a curved proton inventory would result. Moreover, transfer of an additional proton between the two water molecules would be required to avoid formation of adjacent hydroxide and hydronium ions. This model thus predicts that there would be at least 3 protons in flight in the key transition state for the mutant protein, consistent with the proton inventory. The alternative model in which the lysine accepts a proton from the bridging water as a second proton is transferred from the flavin is also consistent with the curved proton inventory in the mutant protein. The role of proton acceptor from the bridging water would be be taken by the additional water molecule in the active site. The need to avoid formation of hydroxide suggests that a chain of water molecules would be needed, providing an explanation for the curvature of the proton inventory.4
For several flavoprotein oxidases, rapid flavin oxidation requires the presence of a positively charged active site residue, presumably to neutralize the negative charge developing on oxygen as it accepts electrons from the flavin [22, 40, 45–47]. The role of Lys315 in the PAO reaction is clearly different, since it must be unprotonated for optimal oxygen reactivity. The effect of the K315M mutation on the kcat/Km value of oxygen is much less than has been observed upon mutating the positively charged residue in other flavin oxidases, suggesting that another residue in PAO must play that role.
While the present results provide insight into the role of Lys315 in the reaction of a mammalian PAO and the effects of mutating this residue, they do not provide an obvious rationale for the results with maize PAO and LSD1. The characterization of the K661A mutant of LSD1 was quite qualitative [33], so the explanation may be that a decrease of ~25-fold in the reaction would have resulted in no detectable activity in the assay. However, the characterization of the K300M mutation in the maize enzyme was done by single-turnover kinetic analyses that measured the rate constant for flavin reduction directly [32]. It may be that this lysine has an additional role in the reductive half-reaction of that enzyme. The sequence identity between maize and mouse PAO is only 21%, and they oxidize different sides of the secondary nitrogen in the substrate, so that some differences in the roles of active site residues would not be remarkable. An alternative explanation is that the mutation has a structural effect in the maize enzyme, such that the resting enzyme is in an inactive conformation and the rate constant for reduction reflects the rate constant for a conformational change to an active enzyme. Such a model was recently proposed to explain the effects of mutating a conserved active site histidine in the lactate dehydrogenase flavocytochrome b2 [48].
Acknowledgments
This work was supported in part by grants from the NIH (R01 GM47291) and The Welch Foundation (AQ-1245).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Abbreviations used: PAO, polyamine oxidase.
For the wild-type enzyme, the Km value for N1-acetylspermine is 12 ± 2 μM at pH 8 and 27 ± 6 μM at pH 10, while the values for the mutant enzyme are 8 ± 2 μM at pH 8 and 2 ± 2 μM at pH 10.
The detection of a hydroperoxyflavin intermediate in the oxidative half-reaction of pyranose 2-oxidase is a notable exception [43].
The only structure of a reduced member of the monoamine oxidase family is that of maize PAO. This shows the water bridging the flavin and Lys300, as shown in Figure 1. However, no product is bound in this structure. If the reaction with oxygen involves the reduced enzyme-product complex, as suggested by the low value of kcat compared to kred, the possibility must be considered that the product displaces the bound water. In that case the nitrogen in the newly oxidized carbon-nitrogen bond could fulfill the proposed role of the oxygen in the water molecule.
REFERENCES
- [1].Fitzpatrick PF. Arch. Biochem. Biophys. 2010;493:13–25. doi: 10.1016/j.abb.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mattevi A, Vanoni MA, Todone F, Rizzi M, Teplyakov A, Coda A, Bolognesi M, Curti B. Proc. Natl. Acad. Sci. USA. 1996;93:7496–7501. doi: 10.1073/pnas.93.15.7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Trickey P, Wagner MA, Jorns MS, Mathews FS. Structure. 1999;7:331–345. doi: 10.1016/s0969-2126(99)80043-4. [DOI] [PubMed] [Google Scholar]
- [4].Settembre EC, Dorrestein PC, Park J, Augustine AM, Begley TP, Ealick SE. Biochemistry. 2003;42:2971–2981. doi: 10.1021/bi026916v. [DOI] [PubMed] [Google Scholar]
- [5].Leys D, Basran J, Scrutton NS. EMBO J. 2003;22:4038–4048. doi: 10.1093/emboj/cdg395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ma J, Yoshimura M, Yamashita E, Nakagawa A, Ito A, Tsukihara T. J. Mol. Biol. 2004;338:103–114. doi: 10.1016/j.jmb.2004.02.032. [DOI] [PubMed] [Google Scholar]
- [7].Binda C, Newton-Vinson P, Hubalek F, Edmondson DE, Mattevi A. Nat. Struct. Biol. 2002;9:22–26. doi: 10.1038/nsb732. [DOI] [PubMed] [Google Scholar]
- [8].Binda C, Coda A, Angelini R, Federico R, Ascenzi P, Mattevi A. Structure. 1999;7:265–276. doi: 10.1016/s0969-2126(99)80037-9. [DOI] [PubMed] [Google Scholar]
- [9].Stavropoulos P, Blobel G, Hoelz A. Nat. Struct. Mol. Biol. 2006;13:626–632. doi: 10.1038/nsmb1113. [DOI] [PubMed] [Google Scholar]
- [10].Pawelek PD, Cheah J, Coulombe R, Macheroux P, Ghisla S, Vrielink A. EMBO J. 2000;19:4204–4215. doi: 10.1093/emboj/19.16.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Palmer G, Massey V. In: Biological oxidation. Singer TP, editor. John Wiley and Sons; New York: 1968. pp. 263–300. [Google Scholar]
- [12].Scrutton NS. Nat. Prod. Rep. 2004;21:722–730. doi: 10.1039/b306788m. [DOI] [PubMed] [Google Scholar]
- [13].Edmondson DE, Binda C, Mattevi A. Arch. Biochem. Biophys. 2007;464:269–276. doi: 10.1016/j.abb.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fitzpatrick PF. Acc. Chem. Res. 2001;34:299–307. doi: 10.1021/ar0000511. [DOI] [PubMed] [Google Scholar]
- [15].Faust A, Niefind K, Hummel W, Schomburg D. J. Mol. Biol. 2007;367:234–248. doi: 10.1016/j.jmb.2006.11.071. [DOI] [PubMed] [Google Scholar]
- [16].Ralph EC, Hirschi JS, Anderson MA, Cleland WW, Singleton DA, Fitzpatrick PF. Biochemistry. 2007;46:7655–7664. doi: 10.1021/bi700482h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ralph EC, Anderson MA, Cleland WW, Fitzpatrick PF. Biochemistry. 2006;45:15844–15852. doi: 10.1021/bi061894o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kurtz KA, Rishavy MA, Cleland WW, Fitzpatrick PF. J. Am. Chem. Soc. 2000;122:12896–12897. [Google Scholar]
- [19].Gaweska H, Henderson Pozzi M, Schmidt DMZ, McCafferty DG, Fitzpatrick PF. Biochemistry. 2009;48:5440–5445. doi: 10.1021/bi900499w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Henderson Pozzi M, Gawandi V, Fitzpatrick PF. Biochemistry. 2009;48:12305–12313. doi: 10.1021/bi901694s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Massey V. J.Biol.Chem. 1994;269:22459–22462. [PubMed] [Google Scholar]
- [22].Mattevi A. Trends Biochem. Sci. 2006;31:276–283. doi: 10.1016/j.tibs.2006.03.003. [DOI] [PubMed] [Google Scholar]
- [23].Wu T, Yankovskaya V, McIntire WS. J. Biol. Chem. 2003;278:20514–20525. doi: 10.1074/jbc.M302149200. [DOI] [PubMed] [Google Scholar]
- [24].Cervelli M, Polticelli F, Federico R, Mariottini P. J. Biol. Chem. 2003;278:5271–5276. doi: 10.1074/jbc.M207888200. [DOI] [PubMed] [Google Scholar]
- [25].Wang Y, Murray-Stewart T, Devereux W, Hacker A, Frydman B, Woster PM, Casero RA., Jr. Biochem. Biophys. Res. Commun. 2003;304:605–611. doi: 10.1016/s0006-291x(03)00636-3. [DOI] [PubMed] [Google Scholar]
- [26].Sebela M, Radova A, Angelini R, Tavladoraki P, Frebort I, Pec P. Plant Sci. 2001;160:197–207. doi: 10.1016/s0168-9452(00)00380-0. [DOI] [PubMed] [Google Scholar]
- [27].van Hellemond EW, Van Dijk M, Heuts DPHM, Janssen DB, Fraaije MW. Applied Microbiology & Biotechnology. 2008;78:455–463. doi: 10.1007/s00253-007-1310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Federico R, Ercolini L, Laurenzi M, Angelini R. Phytochemistry. 1996;43:339–341. [Google Scholar]
- [29].Landry J, Sternglanz R. Biochem. Biophys. Res. Commun. 2003;303:771–776. doi: 10.1016/s0006-291x(03)00416-9. [DOI] [PubMed] [Google Scholar]
- [30].Chen Y, Yang Y, Wang F, Wan K, Yamane K, Zhang Y, Lei M. Proc. Natl. Acad. Sci. USA. 2006;103:13956–13961. doi: 10.1073/pnas.0606381103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Huang Q, Liu Q, Hao Q. J. Mol. Biol. 2005;348:951–959. doi: 10.1016/j.jmb.2005.03.008. [DOI] [PubMed] [Google Scholar]
- [32].Polticelli F, Basran J, Faso C, Cona A, Minervini G, Angelini R, Federico R, Scrutton NS, Tavladoraki P. Biochemistry. 2005;44:16108–16120. doi: 10.1021/bi050983i. [DOI] [PubMed] [Google Scholar]
- [33].Lee MB, Winder C, Cooch H, Shiekhattar R. Nature. 2005;437:432–435. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- [34].Henderson Pozzi M, Gawandi V, Fitzpatrick PF. Biochemistry. 2009;48:1508–1516. doi: 10.1021/bi802227m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Adachi MS, Juarez PR, Fitzpatrick PF. Biochemistry. 2010;49:386–392. doi: 10.1021/bi9017945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Segur JB, Oberstar HE. Ind. Eng. Chem. 1951;43:2117–2120. [Google Scholar]
- [37].Cleland WW. Methods Enzymol. 1979;63:103–138. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- [38].Quinn DM, Sutton LD. In: Enzyme mechanism from isotope effects. Chapter 3. Cook PF, editor. CRC Press; Boca Raton, Florida: 1991. pp. 73–126. [Google Scholar]
- [39].Dansen TB, Wirtz KWA, Wanders RJA, Pap EHW. Nat. Cell Biol. 1999;2:51–53. doi: 10.1038/71375. [DOI] [PubMed] [Google Scholar]
- [40].Denu JM, Fitzpatrick PF. J. Biol. Chem. 1994;269:15054–15059. [PubMed] [Google Scholar]
- [41].Emanuele JJ, Jr., Fitzpatrick PF. Biochemistry. 1995;34:3716–3723. doi: 10.1021/bi00011a029. [DOI] [PubMed] [Google Scholar]
- [42].Royo M, Fitzpatrick PF. Biochemistry. 2005;44:7079–7084. doi: 10.1021/bi050347k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sucharitakul J, Prongjit M, Haltrich D, Chaiyen P. Biochemistry. 2008;47:8485–8490. doi: 10.1021/bi801039d. [DOI] [PubMed] [Google Scholar]
- [44].Fitzpatrick PF, Massey V. J. Biol. Chem. 1982;257:12916–12923. [PubMed] [Google Scholar]
- [45].Roth JP, Klinman JP. Proc. Natl. Acad. Sci. USA. 2003;100:62–67. doi: 10.1073/pnas.252644599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ghanem M, Gadda G. Biochemistry. 2006;45:3437–3447. doi: 10.1021/bi052514m. [DOI] [PubMed] [Google Scholar]
- [47].Jorns MS, Chen Z.-w., Mathews FS. Biochemistry. 2010 [Google Scholar]
- [48].Tsai C-L, Gokulan K, Sobrado P, Sacchettini JC, Fitzpatrick PF. Biochemistry. 2007;46:7844–7851. doi: 10.1021/bi7005543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Binda C, Angelini R, Federico R, Ascenzi P, Mattevi A. Biochemistry. 2001;40:2766–2776. doi: 10.1021/bi002751j. [DOI] [PubMed] [Google Scholar]