

Abstract
Rationale
Mitochondrial dysfunction plays a pivotal role in the development of heart failure. Animal studies suggest that impaired mitochondrial biogenesis due to down-regulation of the PGC-1 transcriptional pathway is integral of mitochondrial dysfunction in heart failure.
Objective
The study sought to define mechanisms underlying the impaired mitochondrial biogenesis and function in human heart failure.
Methods and Results
We collected left ventricular (LV) tissue from end-stage heart failure patients and from non-failing hearts (n = 23, and 19, respectively). The mitochondrial DNA (mtDNA) content was decreased by >40% in the failing hearts, after normalization for a moderate decrease in citrate synthase activity (p<0.05). This was accompanied by reductions in mtDNA-encoded proteins (by 25%–80%) at both mRNA and protein level (p<0.05). The mRNA levels of PGC-1α/β and PRC were unchanged while PGC-1α protein increased by 58% in the failing hearts. Among the PGC-1 co-activating targets, the expression of ERRα and its downstream genes decreased by up to 50% (p<0.05) while PPARα and its downstream gene expression were unchanged in the failing hearts. The formation of D-loop in the mtDNA was normal but D-loop extension, which dictates the replication process of mtDNA, was decreased by 75% in the failing hearts. Furthermore, DNA oxidative damage was increased by 50% in the failing hearts.
Conclusions
Mitochondrial biogenesis is severely impaired as evidenced by reduced mtDNA replication and depletion of mtDNA in the human failing heart. These defects are independent of the down-regulation of the PGC-1 expression suggesting novel mechanisms for mitochondrial dysfunction in heart failure.
Keywords: mtDNA, mitochondrial biogenesis, human heart failure, PGC-1, oxidative damage
Introduction
Mitochondrial dysfunction has been observed in a variety of cardiac diseases, including myocardial ischemia, diabetic cardiomyopathy and heart failure. Accumulating evidence have suggested that mitochondrial dysfunction accounts for impaired myocardial energetics and increased cell death during myocardial injury and the development of heart failure 1, 2. However, the molecular mechanisms responsible for the mitochondrial dysfunction under these pathological conditions are poorly understood making it difficult to develop mitochondria-targeted therapy.
Recent studies have revealed a central role of peroxisome proliferator-activated receptor gamma co-activator 1 (PGC-1) family proteins in mitochondrial biogenesis and function in multiple organs including the heart 3–5. Down-regulation of PGC-1α and its target genes have been observed in a number of rodent models of heart failure raising the intriguing possibility that impaired mitochondrial biogenesis can be a causal mechanism for mitochondrial dysfunction in heart failure 6, 7. However, despite strong evidence suggesting a critical role of PGC-1 mediated mitochondrial biogenesis for normal cardiac function in animal models 6, 8, 9 changes of mitochondrial biogenesis in human heart failure has not been fully characterized, and the regulatory mechanisms not understood. PGC-1 co-activates a number of transcriptional factors involved in mitochondrial biogenesis, e.g. PPARs, ERRs, and NRFs. The respective role of each of these pathways has not been defined in human heart failure.
In this study therefore, we sought to determine whether defective mitochondrial biogenesis was characteristic of human failing heart, and if so, whether PGC-1 mediated transcriptional cascade plays a role. Our results showed that mitochondrial DNA (mtDNA) content as well as the expression of mtDNA-encoded proteins were markedly decreased in the human failing heart. Furthermore, we have identified defects in mtDNA replication and maintenance in the failing heart that were independent of the PGC-1 level but was associated with down-regulation of ERRα and increased oxidative DNA damage.
Materials and Methods
Myocardial samples from non-failing and failing hearts
The failing heart samples (HF, n=23) were obtained from the left ventricular (LV) free wall or apex of the heart during heart transplantation or implantation of a LV assist device. The non-failing heart samples (NF, n=19) were obtained from the LV free wall of donor hearts found unsuitable for transplantation for a variety of reasons e.g. history of blood transfusion in the emergency room, unacceptable age, or history of resuscitation before death etc. All donors had no history, macroscopic, or laboratory signs of cardiac diseases. The local ethics committee approved the study protocol.
RNA Isolation and Real Time PCR
Total RNA was isolated from frozen LV tissue using the RNeasy Kit (Qiagen), and cDNA was synthesized using Omniscript reverse synthase and random hexamers according to manufactures guidelines. Real Time PCR was performed using SYBR green (Bio-Rad). The primers are described in the Online Table I. The Real Time PCR results for the mRNA levels of each gene were normalized to 18S rRNA levels.
Immunoblot analysis
Frozen myocardial tissue from LV was homogenized with the RIPA lysis buffer, and 50 μg of proteins per sample were separated by SDS–polyacrylamide gel electrophoresis. The protein levels for human TFAM (ab47517), TFB2M (ab66014), GAPDH (ab9485), ERRα (ab76228), ERRγ (ab82319), ND6 (ab81212), Flavoprotein (SDHA; MS204), Cyclophilin D (CYPD; MSA04), Cytochrome B (CYTB; sc11436) and ND1 (H00004535-A01) were detected using commercial antibodies from Abcam, Mitosciences, Santa Cruz and Abnova. Several antibodies against PGC-1α were tested using heart tissue from PGC-1α-null or PGC-1α over-expression mice (kindly provided by Dr. Zoltan Arany). Results presented here were obtained using the PGC-1α antibody from Calbiochem (ST1202) that demonstrated appropriate sensitivity and specificity against both mouse and human PGC-1α protein.
Measurements of enzyme activity, mtDNA content, and mtDNA replication
Citrate Synthase activity was measured in tissue homogenate as previously described10. The content of mtDNA was determined by Real Time PCR using primers amplifying the 16S and Cytochrome C Oxidase subunit I (COI) region, and was normalized to that of 18S in nuclear DNA (nDNA). The RNA-DNA hybrid region (7S DNA) that initiated the mitochondrial D-loop formation was measured in the RNA extracts (free of DNA contamination) using Real Time PCR11. To assess the replication of mtDNA, we quantified single stranded DNA generated from the extension of the 7S DNA in the D-loop region, a committed step in mtDNA replication 11, 12. This was achieved by treating the DNA preparation with Mnl I endonuclease that selectively digested double stranded DNA in the presence of Mn2+ at its target sequence: 5′-CCTC(N)7^-3 13 followed by Real Time PCR with primers that spanned 185bp segment in the CYTB region (proximal to D-Loop, and contains two distinct sites of Mnl I). We found that Mnl I cleaved the doubled stranded DNA region of CYTB with >99.9% efficiency under our experimental conditions while leaving a segment of COI region devoid of 5′-CCTC(N)7^-3 sequence totally intact. Thus, the ratio of the PCR products amplifying the CYTB and the COI regions after the Mnl I treatment reflected the fractional amount of mtDNA in replication.
8-hydroxydeoxyguanosin (8-OH-dG) assay
Genomic DNA was isolated from LV tissue using the DNeasy Tissue Kit (Qiagen) in the presence of the free radical spin trap phenyl-tert-butyl nitrone (50mM, Sigma). All assay buffers were purged with N2 to prevent in vitro oxidation of the DNA. Approximately 300ng of genomic DNA was used for 8-OH-dG measurements using a commercially available kit from Cayman according to manufacturer’s instructions.
Statistical analysis
Comparisons among the groups were performed by 1-way ANOVA, followed by Tukey’s post hoc comparisons. Pearson’s correlation coefficients were computed to determine the correlation between mRNA and mtDNA or the mRNA level. All analyses were performed using GraphPad Prism 4.0. All data are expressed as mean ± SEM and a p≤0.05 was considered significant.
Results
General Characteristics
Table 1 summarizes the general characteristics of all patients included in the study. The median age of the NF group was slightly younger compared to the HF although the age range was similar for the NF and HF groups. Approximately 1/3 of the HF patients had ischemic heart diseases. Table 2 shows the clinical characteristics in the ischemic versus non-ischemic heart failure subgroups recorded one week prior to the sample collection. The inotropic treatment was used extensively in both the non-ischemic (100%) and the ischemic heart failure (78%) patients while more ischemic patients received statin treatment. Despite the different etiologies of heart failure, no significant differences in the results presented here were observed between the end-stage non-ischemic and ischemic HF samples. The mRNA levels of heart failure marker genes BNP and SERCA2 were determined in all samples (Figure 1). Consistent with previous reports, significant up-regulation of BNP and down-regulation of SERCA2 were observed in HF (Figure 1A–B).
Table 1.
Clinical Data of the Study Population.
| Non-Failing | Failing | |
|---|---|---|
| n | 19 | 23 |
| Age in years (median; min-max) | 49 (21–70) | 57 (22–72) |
| Gender (M/F) | 12/7 | 16/7 |
| Ischemic | N/A | 9 |
| E.F. (%) | No data | 16 (±5) |
| C.I. (l/min/m2) | No data | 1.8 (±0.5) |
| P.W.P. (mm Hg) | No data | 26 (±8) |
Data are shown as mean ±SEM unless indicated otherwise. E.F.=ejection fraction, C.I.=cardiac index, P.W.P.=pulmonary wedge pressure,
Table 2.
Clinical Characteristics of the Heart Failure Subgroups.
| Non-Ischemic | Ischemic | |
|---|---|---|
| n | 14 | 9 |
| Age in years (median; min-max) | 51 (22–67) | 58 (46–72) |
| Gender (M/F) | 9/5 | 7/2 |
| BMI (kg/m2) | 27(±5) | 28(±4) |
| E.F. (%) | 14 (±3) | 19 (±6) |
| C.I. (l/min/m2) | 1.9 (±0.5) | 1.8 (±0.5) |
| P.W.P. (mm Hg) | 26 (±9) | 25 (±6) |
| Beta Blockers | 6 | 3 |
| Catecholamines | 6 | 4 |
| Phosphodiesterase inhibitors | 8 | 3 |
| Statins | 2 | 8 |
| Digoxin | 6 | 0 |
| ACE Inhibitors | 4 | 3 |
Data are shown as mean ±SEM unless indicated otherwise. BMI=body mass index, E.F.=ejection fraction, C.I.=cardiac index, P.W.P.=pulmonary wedge pressure
Figure 1.
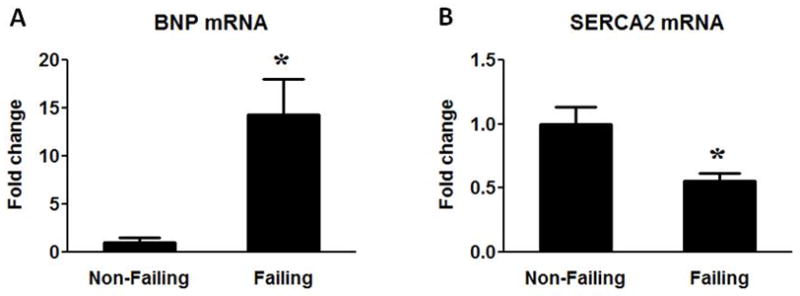
BNP and SERCA2 mRNA expression. Fold change of BNP (A) and SERCA2 (B) mRNA levels in the Failing (n=23) group compared to the Non-Failing controls (n=9). Data are given as the mean of the fold changes ± SEM relative to the Non-Failing hearts (* p<0.05 vs. Non-Failing).
Mitochondrial biogenesis is impaired in heart failure
In order to determine whether human heart failure was associated with changes in the total mitochondria volume mass, we assessed a variety of mitochondrial markers at the levels of enzyme activity, protein expression and mtDNA content. The activity of citrate synthase (CS), a key enzyme in the Krebs cycle and a widely used marker for mitochondrial matrix proteins, was decreased by ~25% in HF samples (Figure 2A). The inner mitochondrial membrane protein CYPD was only reduced by ~10% in HF and the Complex II protein SDHA, encoded by the nuclear DNA, was not different between the groups (Figure 2C and D). In contrast, the Complex I and III proteins ND1, ND6 and CYTB, encoded by the mtDNA, were markedly reduced in HF (Figure 2C and D). Furthermore, we detected a 57% reduction of mtDNA (normalized by nDNA) in the failing samples (Figure 2B). Note that the reduction of the mtDNA copy number was more than 2-fold greater than the decreases in mitochondrial enzyme activity or nDNA encoded mitochondrial proteins. These findings indicated a greater reduction of mtDNA content than that of mitochondria mass in the failing hearts. To rule out the possibility that relative increases of the non-cardiomyocyte population in the failing hearts might have “diluted” the average mitochondria amount per nucleus in the myocardium, mtDNA was also normalized to CS activity. The mtDNA/CS activity remained 43% lower in HF samples compared to NF heart samples (Online Figure I A). Additionally, there was no significant change in the expression of the fibroblast marker DDR2 or in the total nDNA content normalized to the total protein content (Online Figure I B and C). Taken together, these results showed that mtDNA content and mtDNA-encoded proteins were significantly reduced in human failing heart, indicative of impaired mitochondrial biogenesis.
Figure 2.
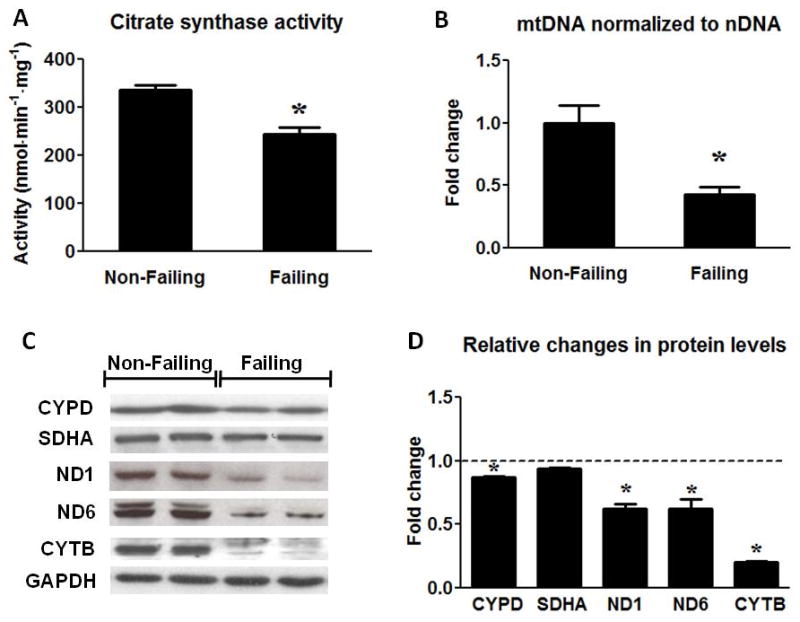
Assessment of mitochondrial protein and mtDNA content. (A) Citrate Synthase activity in the Non-Failing (n=10) and the Failing (n=23) hearts. (B) mtDNA content normalized to nDNA in the Failing (n=23) group compared to the Non-Failing controls (n=9). (C) Representative Western blots for CYPD, SDHA, ND1, ND6, CYTB and GAPDH (two samples per group) and (D) average fold changes of the Failing (n=6) relative to the Non-Failing controls (indicated by the dashed line; n=5). Data are given as the mean of the fold changes ± SEM relative to the Non-Failing hearts (* p<0.05 vs. Non-Failing).
PGC-1 is maintained while ERRα is down-regulated in human failing heart
The evidence of reduced mtDNA and proteins prompted us to assess the PGC-1 transcriptional cascade in the human failing heart. In contrast to the previous observations in animal models, we found that the mRNA levels of the 3 members of the PGC-1 family (PGC-1α, PGC-1β and PRC) were not altered in human failing hearts (Figure 3A). Despite the unremarkable changes in the mRNA expression of the PGC-1 family, the PGC-1α protein was increased by 58% in human HF (Figure 3B). This finding was unlikely due to the differences in the time duration between heart excision and tissue freezing for the two groups, as we observed no change in the mRNA and protein levels of PGC-1α in mouse and human hearts kept in the cold preservation solution for up to 12 hours (Online Figure II).
Figure 3.
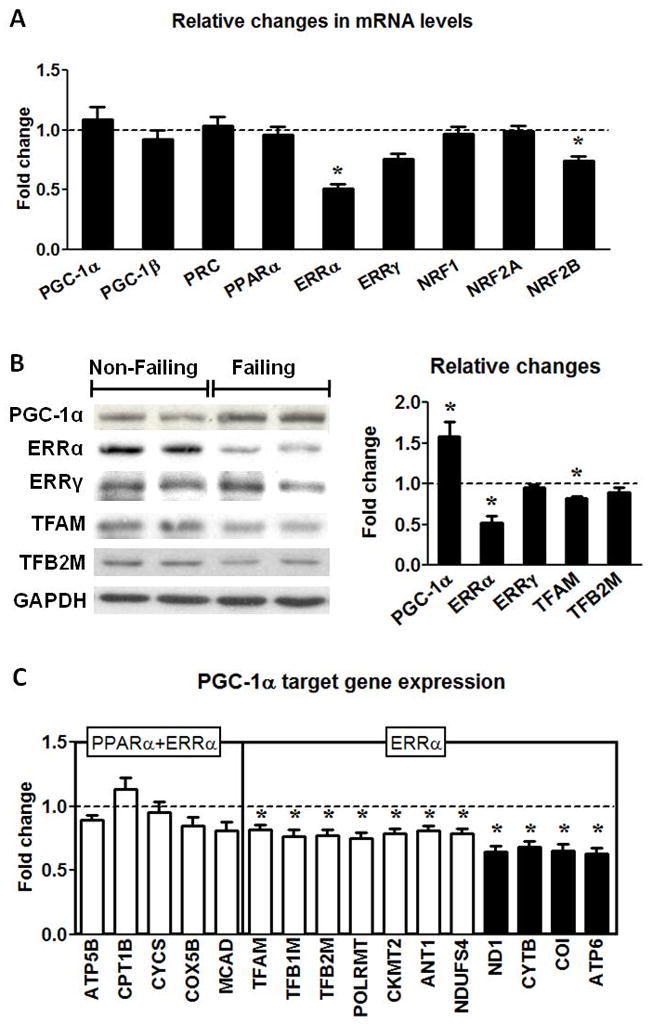
PGC-1 pathway in human failing hearts. (A) Fold changes ± SEM in gene expressions for the PGC-1 pathway in the Failing (n=23) hearts relative to Non-Failing controls (n=9). (B) Protein levels of the transcription regulators for mitochondrial biogenesis. Representative Western blots for PGC-1α, ERRα, ERRγ, TFAM, TFB2M and GAPDH, and average fold changes (mean ± SEM) of the Failing (n=6) hearts relative to the Non-Failing controls (indicated by the dashed line; n=5; * p<0.05). (C) mRNA levels of the PGC-1α downstream gene targets mediated by ERRα or ERRα and PPARα encoded by nDNA (white bars) or mtDNA (black bars) in the Failing hearts (n=23). Data are shown as the mean of the fold changes ± SEM over the Non-Failing hearts (indicated by the dashed line; n=9; * p<0.05 vs. Non-Failing).
Among the transcriptional factors PGC-1 co-activates in mitochondrial biogenesis, there was no change in the expressions of PPARα, NRF1 and NRF2A but there was a significant reduction in the expression of ERRα in the failing hearts at the mRNA as well as at the protein level (Figure 3A and B). ERRγ, another member of the ERR family, decreased modestly at mRNA level (~25%, p=NS) but did not change at protein level. Furthermore, there was also a slight but significant decrease in the mRNA and protein of TFAM (Figure 3B and C) which is an important protein for the mtDNA maintenance. The expression of the partner of TFAM, TFB2M, was reduced at the mRNA but not at the protein level (Figure 3B and C).
To assess the functional significance of the changes in the PGC-1 proteins as well as its co-activating targets we measured the expressions of their downstream genes involved in mitochondrial biogenesis. The expression of genes that were under the control of both PPARα and ERRα did not change significantly but the expression of genes that were predominantly under the control of ERRα alone was significantly reduced in the failing group (Figure 3C). Note that the expression of mtDNA-encoded genes that are indirect targets of ERRα was further reduced. In addition, there was a strong correlation between the mtDNA content and the mRNA levels of mtDNA-encoded proteins (Figure 4). Regression analysis revealed a curvilinear relationship between the mtDNA and COI mRNA (r=0.71) or ATP6 mRNA (r=0.76). These results suggested that the co-activation function of the PGC-1 was sustained in human failing hearts. Although down-regulation of ERRα likely contributed to the decreased transcription of a selection of nuclear-encoded mitochondrial proteins, the depletion of mtDNA played a critical role in the decreased expression of the mtDNA-encoded proteins in the failing hearts.
Figure 4.
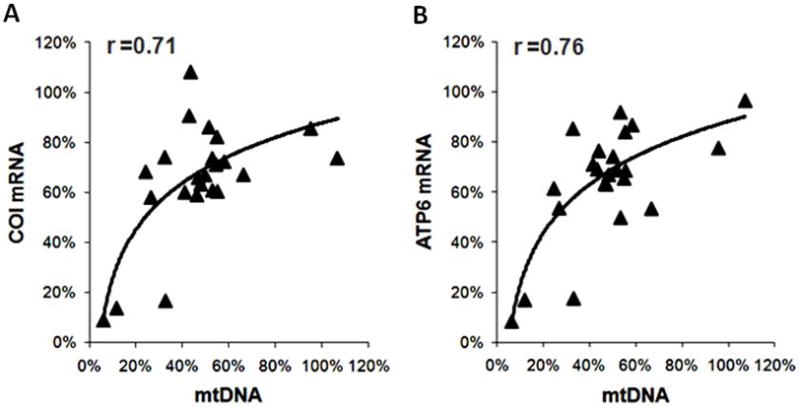
Correlation between the mitochondrial transcripts for COI (A) and ATP6 (B) and mtDNA in the failing hearts. All values are expressed as % of the mean of the Non-Failing controls.
Impaired DNA replication leads to mtDNA depletion
To investigate the mechanism(s) that accounts for reduced mtDNA, we tested whether mtDNA replication was impaired in the failing heart. We examined two major steps in the mtDNA replication: the formation and the extension of the D-loop region. The synthesis of 7S DNA in the D-Loop region was proportional to the abundance of mtDNA in the failing hearts compared to the non-failing controls (Figure 5A). This result suggested that a similar fraction of mtDNA formed D-loop in the failing versus non-failing hearts. In support of this, the expression of the single strand DNA binding protein (mtSSBP1) was unaltered in the failing hearts (Figure 5C). Since the formation of the D-loop was initiated by the synthesis of RNA14, this finding was consistent with our observation that the transcription of the mitochondrial genome was intact. In contrast, the extension of 7S DNA beyond the D-Loop region (indicative of DNA replication in progress) was substantially reduced in the failing hearts. We found that the amount of single strand DNA extended immediately beyond the D-loop was decreased by 75% per mtDNA copy (Figure 5B). We also observed that the expressions of proteins in the DNA replication complex, i.e. polymerase γ (POLG), polymerase γ accessory subunit (POLG2), PEO1 and Topoisomerase I (TOP1MT) were significant down-regulated in the failing hearts (Figure 5C). These results suggested that mechanism(s) involved in the extension of DNA strand rather than the initiation of mtDNA replication was defective in the human failing heart.
Figure 5.
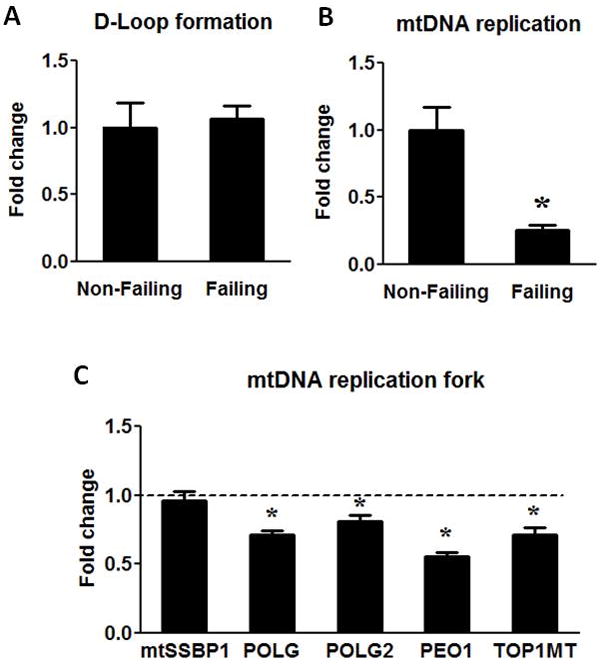
mtDNA replication. (A) The D-Loop formation was assessed by measuring the 7S DNA and normalized to mtDNA amount. (B) mtDNA replication was assessed by measuring the extension of 7S DNA beyond the D-Loop and normalized to mtDNA. (C) mRNA levels of mtSSBP1, POLG, POLG2, PEO1 and TOP1MT. Data are given as the mean of the fold changes ± SEM of the Failing (n=23) group relative to the Non-Failing (n=9) group (* p<0.05 vs. Non-Failing).
Another possible cause of impaired DNA replication and mtDNA depletion in the failing heart is increased DNA damage due to a greater oxidative stress15–17. DNA oxidation by reactive oxygen radicals occurs most readily at guanine residues to form 8-hydroxydeoxyguanosin (8-OH-dG) due to the high oxidation potential of this base. We found that the levels of 8-OH-dG in the DNA were ~50% higher in the failing hearts (Figure 6), suggesting an increased oxidative DNA damage in the failing heart.
Figure 6.
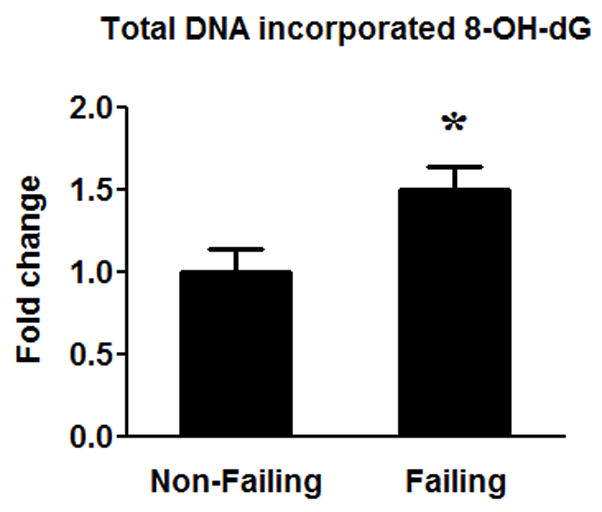
Oxidative DNA damage. The amount of 8-OH-dG in DNA was measured in the failing (n=16) hearts compared to the Non-Failing (n=8) hearts. Data are given as the mean of the fold changes ± SEM over the Non-Failing (* p<0.05).
Discussion
Our results demonstrate that significant decreases in the expression of mtDNA-encoded proteins in human failing hearts are attributable to mtDNA depletion rather than decreased transcriptional activity of the mitochondrial genome. Furthermore, we have identified defects in mtDNA replication and maintenance in the failing heart that are independent of the PGC-1 level but is associated with down-regulation of ERRα and increased oxidative DNA damage. These observations suggest novel mechanisms for impaired mitochondrial biogenesis and provide an important basis for the future development of mitochondria-targeted therapy for heart failure.
Mitochondrial dysfunction and mtDNA depletion in heart failure
Mitochondrial dysfunction has been repeatedly observed in the failing hearts of animal models and patients but its underlying mechanisms are poorly understood. There has been increasing evidence of impaired mitochondrial biogenesis in animal models of heart failure 6, 7, 18, 19. The present study shows that mtDNA content and replication are markedly decreased in human failing hearts, resulting in lower expression of mtDNA-encoded proteins. Previous studies have demonstrated that quantitative and/or qualitative defects of mtDNA lead to mitochondrial dysfunction, cardiomyopathy and heart failure 15, 20–23. Mouse models of mitochondrial gene knockout have not been reported, but more than 50% of disease mutations in mtDNA cause cardiomyopathy suggesting an essential role of mtDNA-encoded proteins in mitochondrial function of the heart 24. Additionally, chemotherapy drugs that inhibit DNA replication, such as doxorubicin or the nucleoside-transcriptase inhibitors (NRTIs), deplete mtDNA and causes cardiac toxicity 20, 21, 25. Conversely, overexpression of human TFAM in mouse hearts maintains high levels of mtDNA post myocardial infarction and significantly delays the development of heart failure 26. Since human TFAM lacks transcriptional activity in mouse mitochondrial genome, this study suggests that the maintenance of mtDNA level plays an independent role in protecting against the heart failure26. Thus, results of the present study add to the growing evidence suggesting that depletion of mtDNA is an important aspect of the biological defects underlying human heart failure and may represent a novel therapeutic target.
We have identified two potential mechanisms for mtDNA depletion in the failing heart. We show that the mtDNA replication is severely impaired at the step of single strand DNA extension. This is associated with a down-regulation in the expressions of all proteins involved in the replication fork. In addition, the oxidative DNA damage is increased more than 50% in the failing heart. Although it has been suggested that oxidative damage of mtDNA occurs at a much greater rate compared to nuclear DNA17, 27 we were unable to distinguish the oxidative damage of mtDNA from that of nDNA in this study. Nevertheless, a recent study has demonstrated that mitochondria-targeted antioxidants protect against mtDNA depletion and left ventricular remodeling post myocardial infarction in mice 28.
PGC-1 mediated mitochondrial biogenesis in heart failure
Recent studies have identified PGC-1 co-activated transcriptional mechanism as a master regulator of mitochondrial biogenesis in a variety of cell types including cardiac myocytes 6, 8, 9, 29. In rodent models of heart failure decreased PGC-1α mRNA levels have been associated with impaired mitochondrial biogenesis and function, suggesting that down-regulation of the PGC-1 pathway may lead to mitochondrial dysfunction and pathogenesis of heart failure 6, 29. Two prior studies examining the PGC-1α levels in human failing hearts have found ~30% decreases in PGC-1α mRNA levels or protein 30, 31. Surprisingly, the present study shows that the expressions of all three members of the PGC-1 family are unaltered in human failing hearts. The discrepancy of the observations between animal models and human diseases can be multi-factorial, including but not limited to, the treatment of patients versus untreated animal models, the stage of diseases, and the uniqueness of human biology. For example, majority of patients with end-stage heart failure are treated with catecholamines or phosphodiesterase inhibitors that are known to stimulate PGC-1α expression via cAMP-mediated mechanisms 5. Differences in the severity of heart failure as well as the lack of age-matched control group could partially contribute to the different findings between the previous and the present human study since the expression of PGC-1α declines with age 32, 33. Importantly, these observations suggest that PGC-1α down-regulation does not uniformly present in human heart failure. Consistent with this notion, a recent analysis of gene expression database of human heart failure also found a 36% increase of PGC-1α expression in the failing hearts 34. These results indicate that impaired mitochondrial biogenesis in the end stage human failing heart can occur in the absence of PGC-1α down-regulation.
Among the transcriptional regulators that PGC-1αco-activates, we have observed a decreased expression of ERRα but not that of PPARα or NRF. The mRNA levels of genes that are regulated by ERRα are down-regulated but not of those that are co-regulated by both ERRα and PPARα. This is consistent with the prior report showing that down-regulation of PGC-1α target genes in human failing hearts is associated with a higher level of PGC-1α but a lower level of ERRα 34. These observations further pinpoint the role of ERRα-mediated transcription in human heart failure although this does not necessarily exclude the possibility that further stimulation of the PGC-1 mechanism may sustain mitochondrial function in the failing hearts. It has been shown in a mouse model that deletion of PGC-1α accelerates the development of heart failure 6. However, given the previous findings that over-expression of PGC-1α in the mouse heart causes cardiomyopathy 35, 36 restoration of the ERRα protein levels may be a more effective strategy targeting mitochondrial biogenesis via the PGC-1 pathway in human heart failure.
In summary, we have found that mitochondrial biogenesis is severely impaired in the human failing heart as evidenced by depletion of mtDNA, attributable to reduced mtDNA replication, and significant loss of mtDNA encoded proteins. These defects are independent of the down-regulation of PGC-1 gene expression but are associated with decreased ERRα expression. Thus, our findings provide a basis for investigating novel mechanisms and therapeutic strategies for mitochondrial dysfunction in human heart failure.
NOVELTY AND SIGNIFICANCE.
What Is Known?
Mitochondrial dysfunction plays an important role in heart failure.
PGC-1α down-regulation has been proposed to underlie impaired mitochondrial biogenesis and function in failing hearts of animal models.
Mitochondrial DNA (mtDNA) mutations lead to cardiomyopathy in humans and restoration of mtDNA content prevents the development of heart failure in mouse models.
What New Information Does This Article Contribute?
Impaired mitochondrial biogenesis in human heart failure, evidenced by depletion of mtDNA and mtDNA-encoded proteins, is attributable to reduced mtDNA replication.
Impaired mitochondrial biogenesis in human heart failure occurs in the absence of PGC-1 down-regulation but is associated with decreased expression of ERRα and its down-stream targets.
Mitochondrial dysfunction has been observed in a variety of cardiac diseases, including heart failure. Previous studies in mice suggested that down regulation of the transcriptional co-activator PGC-1α contributes to the impaired mitochondrial biogenesis and dysfunction in the failing heart. In this study, we find that mitochondrial biogenesis in human failing hearts is impaired. However, such impairment is not due to decreased expression of the PGC-1 family of proteins or reduced transcription of mtDNA. Rather, defects in the replication mechanism of the mtDNA contribute to the depletion of mtDNA and decreased level of mtDNA encoded proteins in the failing heart. Furthermore, we find down regulation of the ERRα gene in human failing hearts as well as reduced expression of several mitochondrial genes that are under the transcriptional control of ERRα. Together, these observations suggest novel mechanisms for impaired mitochondrial biogenesis in human failure and provide an important basis for future development of mitochondria-targeted therapies.
Supplementary Material
Acknowledgments
We thank Dr. Paul D. Allen and Dr. Judyth K. Gwathmey for providing non-failing heart samples, and Dr. Zoltan Arany for providing the PGC-1α knock-out and PGC-1α over-expressing mouse tissues and helpful discussions.
Sources of Funding
This study is supported by grants from the National Heart Lung and Blood Institute to Dr. Tian.
Non-standard Abbreviations and Acronyms
- ACEI
Angiotensin Converting Enzyme Inhibition
- ANT1
Adenine nucleotide translocator 1
- ATP6
ATP Synthase 6
- BNP
Brain Natriuretic Peptide
- CKMT2
Creatine Kinase, Mitochondrial 2
- COI
Cytochrome C Oxidase I
- COX5B
Cytochrome C Oxidase, subunit Vb
- CS
Citrate Synthase
- CYCS
Cytochrome c Somatic
- CYPD
Cyclophilin D
- CYTB
Cytochrome B
- DDR
Discoidin domain receptor
- ERR
Estrogen related receptor
- ETC
Electron Transport Chain
- GAPDH
Glyceraldehyde-3-Phosphate Dehydrogenase
- HF
Heart Failure
- LV
Left Ventricle
- MCAD
Medium-Chain Acyl-CoA Dehydrogenase
- NDUFS4
NADH dehydrogenase (ubiquinone) Fe-S protein 4
- mtDNA
mitochondrial DNA
- nDNA
nuclear DNA
- ND1
NADH Dehydrogenase 1
- NF
Non-Failing
- NRF
Nuclear Respiratory Factor
- NRTIs
Nucleoside Reverse Transcriptase Inhibitors
- PCR
Polymerase Chain Reaction
- PEO1
Progressive External Ophthalmoplegia 1
- PGC-1
Peroxisome Proliferator Activated Receptor Gamma Coactivator 1
- POLG
Polymerase gamma, mitochondrial
- POLRMT
Polymerase RNA, mitochondrial
- PRC
PGC-1 Related Coactivator
- SDHA
Succinate Dehydrogenase complex, subunit A, Flavoprotein
- SERCA2
Sarco-Endoplasmic Reticulum Calcium ATPase 2
- TFAM
Transcription Factor A, Mitochondrial
- TFB1/2M
Transcription Factor B1/2, Mitochondrial
- 8-OH-dG
8-hydroxydeoxyguanosin
Footnotes
Disclosures
None.
References
- 1.Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BH, Hewett TE, Robbins J, Houser SR, Molkentin JD. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–2444. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marin-Garcia J, Goldenthal MJ. Mitochondrial centrality in heart failure. Heart Fail Rev. 2008;13:137–150. doi: 10.1007/s10741-007-9079-1. [DOI] [PubMed] [Google Scholar]
- 3.Uldry M, Yang W, St-Pierre J, Lin J, Seale P, Spiegelman BM. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 2006;3:333–341. doi: 10.1016/j.cmet.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 4.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 5.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 6.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc Natl Acad Sci U S A. 2006;103:10086–10091. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garnier A, Fortin D, Delomenie C, Momken I, Veksler V, Ventura-Clapier R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol. 2003;551:491–501. doi: 10.1113/jphysiol.2003.045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, Rybkin, Shelton JM, Manieri M, Cinti S, Schoen FJ, Bassel-Duby R, Rosenzweig A, Ingwall JS, Spiegelman BM. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 9.Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, Medeiros DM, Kovacs A, Kelly DP. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008;22:1948–1961. doi: 10.1101/gad.1661708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morgunov I, Srere PA. Interaction between citrate synthase and malate dehydrogenase. Substrate channeling of oxaloacetate. J Biol Chem. 1998;273:29540–29544. doi: 10.1074/jbc.273.45.29540. [DOI] [PubMed] [Google Scholar]
- 11.Brown TA, Clayton DA. Release of replication termination controls mitochondrial DNA copy number after depletion with 2′,3′-dideoxycytidine. Nucleic Acids Res. 2002;30:2004–2010. doi: 10.1093/nar/30.9.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gensler S, Weber K, Schmitt WE, Perez-Martos A, Enriquez JA, Montoya J, Wiesner RJ. Mechanism of mammalian mitochondrial DNA replication: import of mitochondrial transcription factor A into isolated mitochondria stimulates 7S DNA synthesis. Nucleic Acids Res. 2001;29:3657–3663. doi: 10.1093/nar/29.17.3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kriukiene E. Domain organization and metal ion requirement of the Type IIS restriction endonuclease MnlI. FEBS Lett. 2006;580:6115–6122. doi: 10.1016/j.febslet.2006.09.075. [DOI] [PubMed] [Google Scholar]
- 14.Lee DY, Clayton DA. Properties of a primer RNA-DNA hybrid at the mouse mitochondrial DNA leading-strand origin of replication. J Biol Chem. 1996;271:24262–24269. doi: 10.1074/jbc.271.39.24262. [DOI] [PubMed] [Google Scholar]
- 15.Tsutsui H, Ide T, Kinugawa S. Mitochondrial oxidative stress, DNA damage, and heart failure. Antioxid Redox Signal. 2006;8:1737–1744. doi: 10.1089/ars.2006.8.1737. [DOI] [PubMed] [Google Scholar]
- 16.Tsutsui H, Ide T, Shiomi T, Kang D, Hayashidani S, Suematsu N, Wen J, Utsumi H, Hamasaki N, Takeshita A. 8-oxo-dGTPase, which prevents oxidative stress-induced DNA damage, increases in the mitochondria from failing hearts. Circulation. 2001;104:2883–2885. doi: 10.1161/hc4901.101347. [DOI] [PubMed] [Google Scholar]
- 17.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 18.Finck BN, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation. 2007;115:2540–2548. doi: 10.1161/CIRCULATIONAHA.107.670588. [DOI] [PubMed] [Google Scholar]
- 19.Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–535. doi: 10.1161/01.res.88.5.529. [DOI] [PubMed] [Google Scholar]
- 20.L’Ecuyer T, Sanjeev S, Thomas R, Novak R, Das L, Campbell W, Heide RV. DNA damage is an early event in doxorubicin-induced cardiac myocyte death. Am J Physiol Heart Circ Physiol. 2006;291:H1273–1280. doi: 10.1152/ajpheart.00738.2005. [DOI] [PubMed] [Google Scholar]
- 21.Lewis W. Defective mitochondrial DNA replication and NRTIs: pathophysiological implications in AIDS cardiomyopathy. Am J Physiol Heart Circ Physiol. 2003;284:H1–9. doi: 10.1152/ajpheart.00814.2002. [DOI] [PubMed] [Google Scholar]
- 22.Wang J, Wilhelmsson H, Graff C, Li H, Oldfors A, Rustin P, Bruning JC, Kahn CR, Clayton DA, Barsh GS, Thoren P, Larsson NG. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat Genet. 1999;21:133–137. doi: 10.1038/5089. [DOI] [PubMed] [Google Scholar]
- 23.Li H, Wang J, Wilhelmsson H, Hansson A, Thoren P, Duffy J, Rustin P, Larsson NG. Genetic modification of survival in tissue-specific knockout mice with mitochondrial cardiomyopathy. Proc Natl Acad Sci U S A. 2000;97:3467–3472. doi: 10.1073/pnas.97.7.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 25.Lebrecht D, Walker UA. Role of mtDNA lesions in anthracycline cardiotoxicity. Cardiovasc Toxicol. 2007;7:108–113. doi: 10.1007/s12012-007-0009-1. [DOI] [PubMed] [Google Scholar]
- 26.Ikeuchi M, Matsusaka H, Kang D, Matsushima S, Ide T, Kubota T, Fujiwara T, Hamasaki N, Takeshita A, Sunagawa K, Tsutsui H. Overexpression of mitochondrial transcription factor a ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation. 2005;112:683–690. doi: 10.1161/CIRCULATIONAHA.104.524835. [DOI] [PubMed] [Google Scholar]
- 27.Williams MD, Van Remmen H, Conrad CC, Huang TT, Epstein CJ, Richardson A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J Biol Chem. 1998;273:28510–28515. doi: 10.1074/jbc.273.43.28510. [DOI] [PubMed] [Google Scholar]
- 28.Matsushima S, Ide T, Yamato M, Matsusaka H, Hattori F, Ikeuchi M, Kubota T, Sunagawa K, Hasegawa Y, Kurihara T, Oikawa S, Kinugawa S, Tsutsui H. Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. 2006;113:1779–1786. doi: 10.1161/CIRCULATIONAHA.105.582239. [DOI] [PubMed] [Google Scholar]
- 29.Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res. 2008;79:208–217. doi: 10.1093/cvr/cvn098. [DOI] [PubMed] [Google Scholar]
- 30.Sebastiani M, Giordano C, Nediani C, Travaglini C, Borchi E, Zani M, Feccia M, Mancini M, Petrozza V, Cossarizza A, Gallo P, Taylor RW, d’Amati G. Induction of mitochondrial biogenesis is a maladaptive mechanism in mitochondrial cardiomyopathies. J Am Coll Cardiol. 2007;50:1362–1369. doi: 10.1016/j.jacc.2007.06.035. [DOI] [PubMed] [Google Scholar]
- 31.Garnier A, Zoll J, Fortin D, N’Guessan B, Lefebvre F, Geny B, Mettauer B, Veksler V, Ventura-Clapier R. Control by circulating factors of mitochondrial function and transcription cascade in heart failure: a role for endothelin-1 and angiotensin II. Circ Heart Fail. 2009;2:342–350. doi: 10.1161/CIRCHEARTFAILURE.108.812099. [DOI] [PubMed] [Google Scholar]
- 32.Anderson R, Prolla T. PGC-1alpha in aging and anti-aging interventions. Biochim Biophys Acta. 2009;1790:1059–1066. doi: 10.1016/j.bbagen.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ling C, Poulsen P, Carlsson E, Ridderstrale M, Almgren P, Wojtaszewski J, Beck-Nielsen H, Groop L, Vaag A. Multiple environmental and genetic factors influence skeletal muscle PGC-1alpha and PGC-1beta gene expression in twins. J Clin Invest. 2004;114:1518–1526. doi: 10.1172/JCI21889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sihag S, Cresci S, Li AY, Sucharov CC, Lehman JJ. PGC-1alpha and ERRalpha target gene downregulation is a signature of the failing human heart. J Mol Cell Cardiol. 2009;46:201–212. doi: 10.1016/j.yjmcc.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, Medeiros DM, Valencik ML, McDonald JA, Kelly DP. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res. 2004;94:525–533. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.