

Abstract
Previous study has shown that a peroxidase is present in the mature eggs of Aedes aegypti mosquitoes, and the enzyme is involved in the formation of a rigid and insoluble chorion by catalyzing chorion protein crosslinking through dityrosine formation. In this study, chorion peroxidase was solubilized from egg chorion by 1% SDS and 2 M urea and purified by various chromatographic techniques. The enzyme has a relative molecular mass of 63,000 as estimated by SDS–PAGE. Spectral analysis of the enzyme revealed the presence of the Soret band with a λmax at 415 nm, indicating that chorion peroxidase is a hemoprotein. Treatment of the native enzyme with H2O2 in excess in the absence of reducing agents shifted the Soret band from 415 to 422 nm, and reduction of the native enzyme with sodium hydrosulfite under anaerobic conditions changed the Soret band from 415 to 446 nm. These results show that the chorion peroxidase behaves similarly to other peroxidases under oxidative and reductive conditions, respectively. Compared to other peroxidases, the chorion peroxidase, however, is extremely resistant to denaturing agents, such as SDS and organic solvents. For example, chorion peroxidase remained active for several weeks in 1% SDS, while horseradish peroxidase irreversibly lost all its activity in 2 h under the same conditions. Comparative analysis between mosquito chorion peroxidase and horseradish peroxidase showed that the specific activity of chorion peroxidase to tyrosine was at least 100 times greater than that of horseradish peroxidase to tyrosine. Chorion peroxidase is also capable of catalyzing polypeptide and chorion protein crosslinking through dityrosine formation during in vitro assays. Our data suggest that the characteristics of the chorion peroxidase in mosquitoes closely reflect its functions in chorion formation and hardening.
Keywords: Aedes aegypti, chorion peroxidase, dityrosine, protein crosslinking
The chorion of newly oviposited mosquito eggs is white and soft, but turns black and becomes rigid and insoluble 2–3 h after oviposition in a moist environment. These changes, termed chorion hardening in our previous reports, increase tremendously the resistance of the eggs to desiccation and other environmental adversities. Floodwater mosquitoes, such as Aedes aegypti, lay their eggs on substrates at the edge of water, and the eggs hatch only after being flooded into water following adequate rainfall. Therefore, the time for these eggs to survive in the environment before hatching varies enormously depending on weather conditions. Consequently, the ability to resist desiccation is critical for the survival of these eggs before being flooded for hatching.
Two interrelated biochemical events, phenol oxidase/ dopa decarboxylase catalyzed chorion melanization and peroxidase-mediated chorion protein crosslinking reaction, contribute to the formation of a rigid and protective chorion in A. aegypti (1, 2). In peroxidase catalyzed protein crosslinking, the enzyme catalyzes oxidation of tyrosine residues on proteins to tyrosine radicals which interact to form dityrosine, thereby leading to chorion protein crosslinking (3). The involvement of peroxidase in chorion protein crosslinking in A. aegypti is based on the progressive increase of peroxidase activity in the developing eggs, the localization of the enzyme in the chorion layer of the mature eggs, and the detection of dityrosine in the hydrolysate of the hardened egg chorion (2).
Although the presence of a specific chorion peroxidase and its involvement in chorion protein crosslinking have been established, the biochemical characteristics of the enzyme remain poorly understood. Peroxidase activity has been detected in Drosophila eggs (4–7). However, no other information about the Drosophila egg peroxidase is available. To understand the chorion peroxidase, we initiated the purification and characterization of the enzyme from A. aegypti eggs. In this report, we describe the techniques used for the isolation of chorion peroxidase and characteristics of the enzyme in relation to its biological functions in chorion formation and hardening in A. aegypti mosquitoes.
MATERIALS AND METHODS
Chemicals
Diaminobenzidine, EDTA, guaiacol, H2O2, horseradish peroxidase (HRP,2 EC 1.11.1.7), β-ME, PMSF, sodium hydrosulfite, tropolone, L-tyrosine, angiotensin II (Asp-Arg-Val-Tyr-Ile-His- Pro-Phe), BSA, and quaternary ammonium cellulose (Q-cellulose) were from Sigma (St. Louis, MO). Hydroxyapatite, polyvinylidene difluoride (PVDF sheet, 0.2 mm), 10% acrylamide ready gel, and Coomassie blue R-250 were purchased from Bio-Rad (Hercules, CA). Dityrosine was synthesized by a described method (3).
Mosquito rearing, blood feeding, and ovary collection
Aedes aegypti black-eyed Liverpool strain mosquitoes were reared as described previously (1). Blood feeding of female mosquitoes and ovary dissection were the same as described in our previous report (2).
Purification of Chorion Peroxidase
Chorion isolation and solubilization
About 25,000 ovary pairs (dissected from female mosquitoes 3 days after a blood meal) were homogenized using a Polytron homogenizer (England) in Buffer A (10 mM phosphate, 1 mM PMSF, and 5 mM EDTA, pH 6.5). EDTA at the applied concentration in Buffer A had no negative effect on chorion peroxidase activity. The homogenate was centrifuged at 1500g for 10 min and supernatant, containing released proteins, lipids, and other components from oocytic materials, was separated. The chorion sediments were washed three times with Buffer A and treated with 1% SDS and 2 M urea prepared in Buffer A. The mixture was sonicated for 5 min and centrifuged at 120,000g for 20 min at 2°C to obtain supernatant. The chorion sediments were treated another three times by the same procedure, and supernatants, containing released chorion peroxidase and other chorion proteins, were pooled, dialyzed against Buffer A, and then used as the initial material for chorion peroxidase purification.
Peroxidase activity assay
Chorion peroxidase activity was assayed primarily with guaiacol and H2O2 as substrates (8). Typically, 1 ml of a reaction mixture containing 6 mM guaiacol, 0.8 mM H2O2, and varying amounts of solubilized chorion proteins or pooled active peroxidase fractions was incubated at 30°C. Absorbance increase at 435 nm due to guaiacol oxidation was continuously monitored for 3 min using a Hitachi Model U-2001 double-beam spectrophotometer equipped with a water-jacketed six-cell positioned (San Jose, CA). Peroxidase activity was defined arbitrarily as units of absorbance increase at 435 nm in 1 ml of reaction mixture per minute measured in a cuvette with d 5 1 cm. Protein content was determined using the colorimetric method of Lowry (9).
Q-cellulose column chromatography
A Q-cellulose column (2.5 × 50 cm) was equilibrated with 10 mM phosphate buffer (pH 6.5) at 4°C, and the dialyzed chorion protein sample (approximately 150 ml) was applied to the column. Proteins were eluted with 600 ml of a linear NaCl gradient (0–500 mM) prepared in Buffer A. Active fractions were pooled and concentrated using a stirred cell and a YM 30 membrane with molecular weight cutoff at 30,000 (Millipore).
Hydroxyapatite chromatography
The concentrated peroxidase fractions were chromatographed on a hydroxyapatite column (1.5 × 10 cm) that was equilibrated with Buffer A, and proteins were eluted with 300 ml of a linear NaCl gradient (0–500 mM). The peroxidase fractions were pooled and concentrated using a 15-ml concentrator with a 30,000 molecular weight cutoff membrane (Millipore).
Mono-Q chromatography
A Mono-Q anion exchange column (7 × 65 mm, Pharmacia) was equilibrated with 10 mM phosphate buffer (pH 6.5) using a Hitachi gradient HPLC system. Concentrated peroxidase fractions (0.4 ml) were injected into the column. Proteins were eluted with a linear NaCl gradient (0–500 mM) during a 60-min period at a flow rate of 1 ml min−1. The fractions corresponding to individual protein peaks (monitored at 280 nm) were collected manually, and attention was paid to avoid overlap among the different protein peaks. The fraction with peroxidase activity was concentrated and rechromatographed using the same Mono-Q column.
Electrophoresis
Purified chorion peroxidase was mixed with treatment buffer (1:1) with or without 2% β-ME for 15 min and then analyzed using SDS–PAGE with 10% acrylamide gel. Some enzyme was also heated in boiling water for 5 min in the presence or absence of β-ME prior to electrophoresis to determine the effect of heat treatment on its migration in polyacrylamide gel. After electrophoresis, the gel was cut vertically into sections that were stained either with Coomassie blue or with 5 mM tropolone or 5 mM diaminobenzidine in the presence of 1 mM H2O2. The effects of heat and β-ME treatment on the mobility of the chorion peroxidase in the gels were evaluated by the migration distances of the treated enzyme in comparison with those of untreated enzymes after electrophoresis.
Characterization of Chorion Peroxidase
N-Terminal AA sequence and AA composition
The partial Nterminal AA sequence of chorion peroxidase was determined using an Applied Biosystems Procise (Department of Biochemistry, Michigan State University). The AA composition was determined at the Biotechnology Center of the University of Illinois (Champaign, IL).
Spectral analysis
The spectrum of the native enzyme in both the UV and visible regions was measured using a Hitachi Model U-2001 double-beam spectrophotometer. Changes of redox states in chorion peroxidase during its oxidation and reduction by H2O2 and sodium hydrosulfite, respectively, were based on spectral shifts of the Soret band and other minor peaks in the visible region as described for HRP (10, 11).
Optimum reaction conditions
The optimum pH and H2O2 concentration for chorion peroxidase were determined using guaiacol as a substrate. Guaiacol, H2O2, and chorion peroxidase were prepared in different buffers, including 100 mM phosphate buffer (pH 6–7.5), Tris buffer (pH 8–8.5), and 2-amino-2-methanol/1-propanol buffer (pH 9–10). Guaiacol and peroxidase were mixed prior to the addition of H2O2, and the reaction was initiated by adding 0.1 ml of H2O2 (8 mM) into 0.9 ml of guaiacol/peroxidase mixture. The optimum concentration for H2O2 was assayed by mixing 0.1 ml of H2O2 solution (1–40 mM) into 0.9 ml of guaiacol/peroxidase mixture prepared in Tris buffer (pH 8). For both assays, peroxidase activity was based on absorbance increase at 435 nm during 3-min incubation at 30°C. The amount of chorion peroxidase used in the reaction mixture was 0.2 µg, and the final concentration of guaiacol in the reaction mixture was 8 mM.
Resistance to SDS
Purified chorion peroxidase was treated with 1.5% SDS in 100 mM phosphate buffer (pH 8). At 1, 5, 10, 20, 30, and 120 min after SDS treatment, 0.2 ml of the treated enzyme wasmixed with 0.8 ml of substrate preparation containing 9.25 mM guaiacol and 1 mM H2O2. The enzyme activity was monitored spectrophotometrically at 435 nm. The effect of SDS on chorion peroxidase activity was based on absorbance increase at 435 nm during 3-min incubation in the reaction mixtures in comparison with that recorded for the untreated enzyme. SDS was not removed from the treated enzyme. Therefore, the final reaction mixture contained 0.3% of SDS. The effect of SDS on HRP was determined in the same manner, and the difference in resistance to SDS between the chorion peroxidase and HRP was compared.
Substrate specificities
The commonly used peroxidase substrates, including guaiacol, diaminobenzidine, and tyrosine, also were the substrates of chorion peroxidase. Among these substrates, guaiacol and tyrosine were used as representatives, and the specific activities of the chorion peroxidase to these two compounds were determined and compared to those of HRP to guaiacol and tyrosine under identical assay conditions. Production of dityrosine during chorion peroxidase and HRP catalyzed tyrosine oxidation was based on the increase in absorbance at 315 nm, where the dityrosine product had an absorption coefficient of 6.3 × 10−3 M−1 cm−1 (12).
Peroxidase-mediated protein crosslinking through dityrosine formation
The ability of chorion peroxidase to catalyze protein crosslinking through dityrosine formation was determined using angiotensin II (a tyrosine-containing polypeptide), BSA, and some isolated chorion protein that was devoid of peroxidase activity. A 1-ml reaction mixture consisting of 1 or 2 µg of chorion peroxidase, 0.8 mM H2O2, and 1.0 mg of angiotensin II or 5 mg of chorion protein or BSA was incubated at 30°C. Peptide or protein crosslinking through dityrosine formation was based on the detection of dityrosine in the hydrolysates using HPLC-ED after the above reaction mixtures were hydrolyzed in 6 M HCl at 110°C for 20 h. Identification of dityrosine in the hydrolysate was based on both its retention time and oxidation potentials during HPLC-ED analysis in comparison with those generated using dityrosine standard under identical conditions (2).
RESULTS
Purification of chorion peroxidase
Chorion isolation and chorion peroxidase solubilization
When dissected mature eggs were homogenized briefly in Buffer A containing 5 mM EDTA, the oocytic materials within the chorion, including proteins, lipids, and other components for embryo development, were released into the supernatant. Homogenization broke the intact chorion into pieces; however, this process did not result in a substantial solubilization of chorion peroxidase, because the majority of the enzyme activity (90%) remained associated with the chorion pieces.
Treatment of chorion sediments with 1% SDS and 2 M urea in Buffer A plus sonication was effective to solubilize the enzyme. The majority of the enzyme activity (>90%) was released after the chorion sediments were treated for four times using 1% SDS and 2Murea with sonication.
Purification of chorion peroxidase
Purification of chorion peroxidase from the solubilized chorion proteins was achieved by Q-cellulose, hydroxyapatite, and Mono-Q column chromatographies. Chorion peroxidase activity was eluted within broad gradient ranges from 165 to 430 mM and from 100 to 185 mM NaCl during conventional Q-cellulose and hydroxyapatite chromatographies, respectively. After hydroxyapatite chromatography, the concentrated peroxidase fractions were separated by Mono-Q anion exchange HPLC. Two peaks, one major with a retention time of 14.8 min and one minor with a retention time of 10.3 min, showed peroxidase activity (Fig. 1A). Due to the limited amount of sample available, no further effort was made to purify the minor peroxidase peak. After the second step of Mono-Q column chromatography, peroxidase became the only detected protein (Fig. 1B). The chorion peroxidase was purified 128-fold, with a yield of 35.4% (Table I).
FIG. 1.
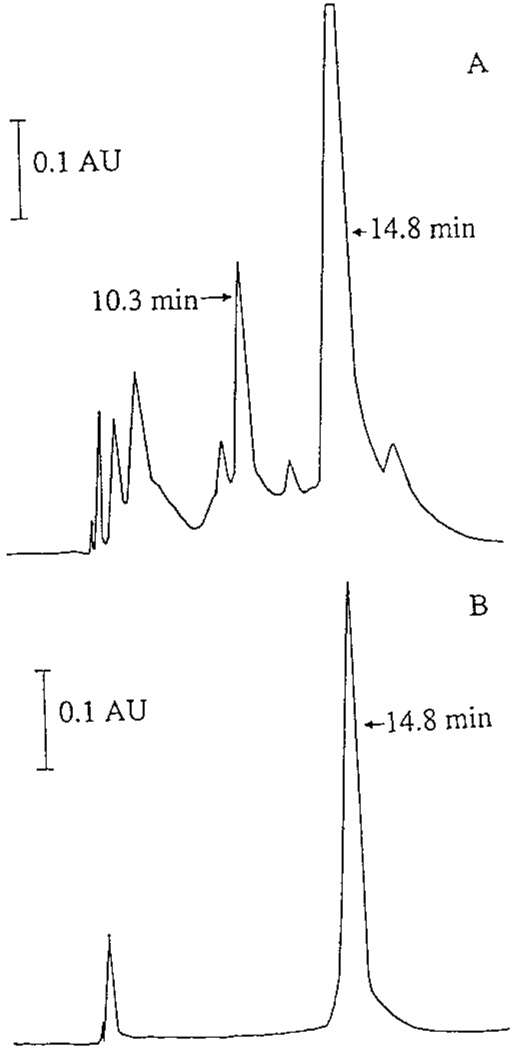
Mono-Q anion exchange chromatography. Concentrated peroxidase fractions, obtained after conventional Q-cellulose and hydroxyapatite chromatographies, were separated by anion exchange HPLC with a Mono-Q column (7 × 65 mm) using a linear NaCl gradient. The peroxidase peak was collected, concentrated, and rechromatographed on the same Mono-Q column under identical conditions. Chromatogram A shows the protein elution profile of the sample during the first Mono-Q column chromatography. Chromatogram B illustrates the protein profile of the concentrated peroxidase during the second Mono-Q column chromatography. Both the 14.8- min major peak and the 10.3-min minor peak in A show peroxidase activity (see Results for details), but only the major peroxidase peak (14.8 min) was isolated and used for subsequent enzyme characterization.
ABLE I.
Purification of Chorion Peroxidase from Mosquito Eggs
| Fraction | Volume (ml) | Protein(s) a mg/mg) |
Specific activity b |
Fold | Recovery (%) |
|---|---|---|---|---|---|
| Solubilized chorion proteins |
150.0 | 2.70 | 5 | 1 | 100.0 |
| QA-cellulose fraction |
30.0 | 0.46 | 115 | 23 | 78.4 |
| Hydroxyapatite fraction |
20.0 | 0.28 | 245 | 49 | 67.8 |
| First Mono-Q raction |
2.8 | 1.00 | 340 | 68 | 47.1 |
| Second Mono- Q fraction |
1.4 | 0.80 | 640 | 128 | 35.4 |
Measurement of protein(s) in each fraction was performed by the method of Lowry (9), using bovine serum albumin as a standard.
The specific activity of the chorion peroxidase is expressed as units of absorbance of tetraguaiacol min−1 mg−1 of protein.
SDS–PAGE analysis of the purified chorion peroxidase, treated in boiling water in the presence of β-ME prior to electrophoresis, showed the enzyme with an Mr of 63,000 (Fig. 2, lane A2). Heat treatment of the enzyme under reducing conditions also completely abolished its activity because the peroxidase band was not tained positive for peroxidase activity (not shown). In contrast, the enzyme was stained easily with tropolone, diaminobenzidine, guaiacol, and dopa in the presence of H2O2 (Fig. 2, lane B1) when it was not reduced and heated prior to electrophoresis. In addition, the untreated enzyme migrated faster on the gel than the heated and reduced enzyme (see Fig. 2, lanes A2 and B1). Treatment of β-ME alone (i.e., without heating) did not completely abolish its activity because the peroxidase band on the polyacrylamide gel remained detectable by substrate staining, but it did not reach the same staining intensity (Fig. 2, lane B2) as that of unreduced enzyme (Fig. 2, lane B1). β-ME treatment also reduced the mobility of the enzyme in the gel during electrophoresis.
FIG. 2.
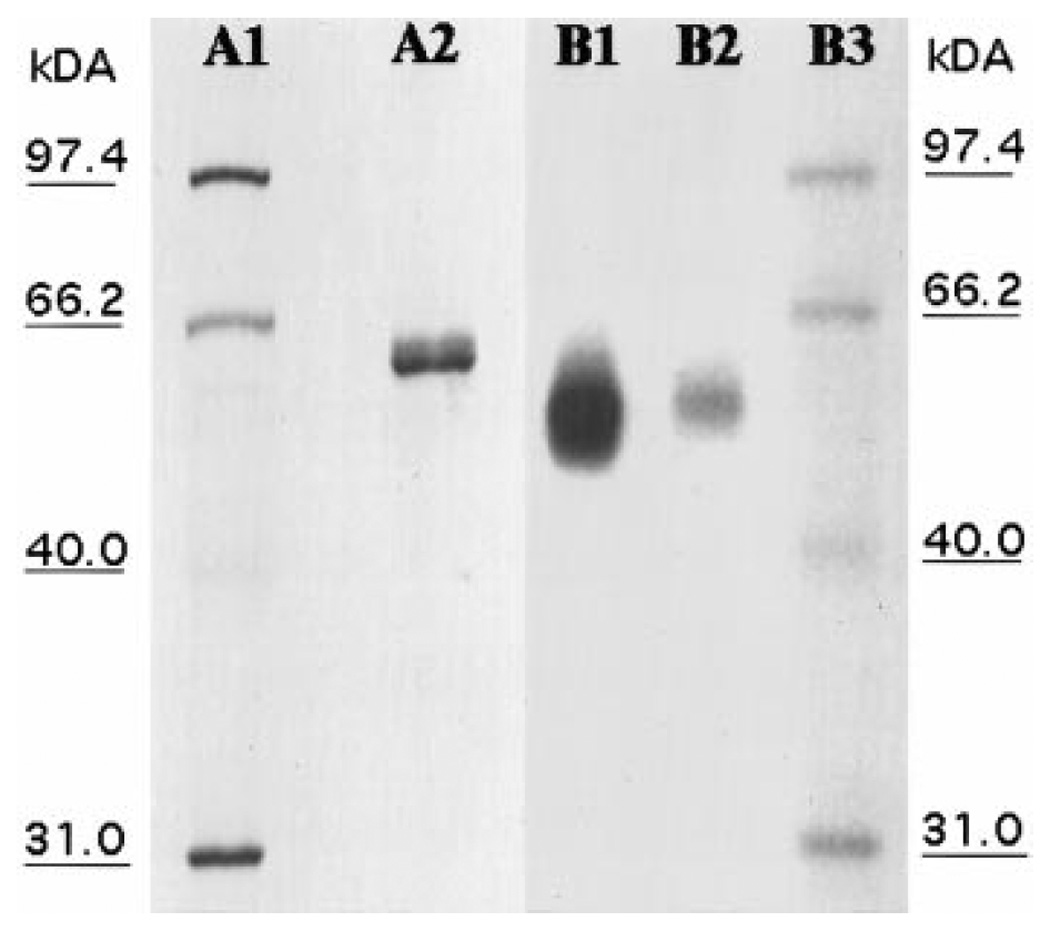
SDS–PAGE analysis. Purified chorion peroxidase was analyzed using SDS–PAGE with either Coomassie blue or substrate staining as described under Materials and Methods. Lanes A1 and A2 are protein molecular mass standards and purified chorion peroxidase, respectively, stained with Coomassie blue. Lanes B1 and B2 are purified chorion peroxidase stained with dopa in the presence of H2O2. Lane A2, with heated and reduced enzyme; lane B1, with untreated enzyme; lane B2, with b-ME reduced enzyme.
Characterization of Chorion Peroxidase
Spectral characteristics
Spectral analysis of chorion peroxidase showed two major absorption peaks with λmax at 280 and 415 nm (Fig. 3A), respectively, and four minor peaks at 495, 545, 582, and 648 nm (Fig. 3B), respectively. The detection of the 415-nm peak, commonly termed the Soret band, suggests that chorion peroxidase is a heme-containing protein. Addition of H2O2 caused a shift of the Soret band from 415 to 422 nm (Fig. 4A) and the disappearance of the 495- and 648-nm peaks with concomitant formation of two new minor peaks at 546 and 586 nm. The spectral shift of the Soret band in chorion peroxidase was similar to that observed for HRP under identical assay conditions, in which the Soret band of HRP was shifted from 403 to 417 nm (not shown). Reduction of the native chorion peroxidase by sodium hydrosulfite shifted the Soret band from 415 to 446 nm under anaerobic conditions (Fig. 4B), which was also similar to that observed for HRP under the same conditions (not shown).
FIG. 3.
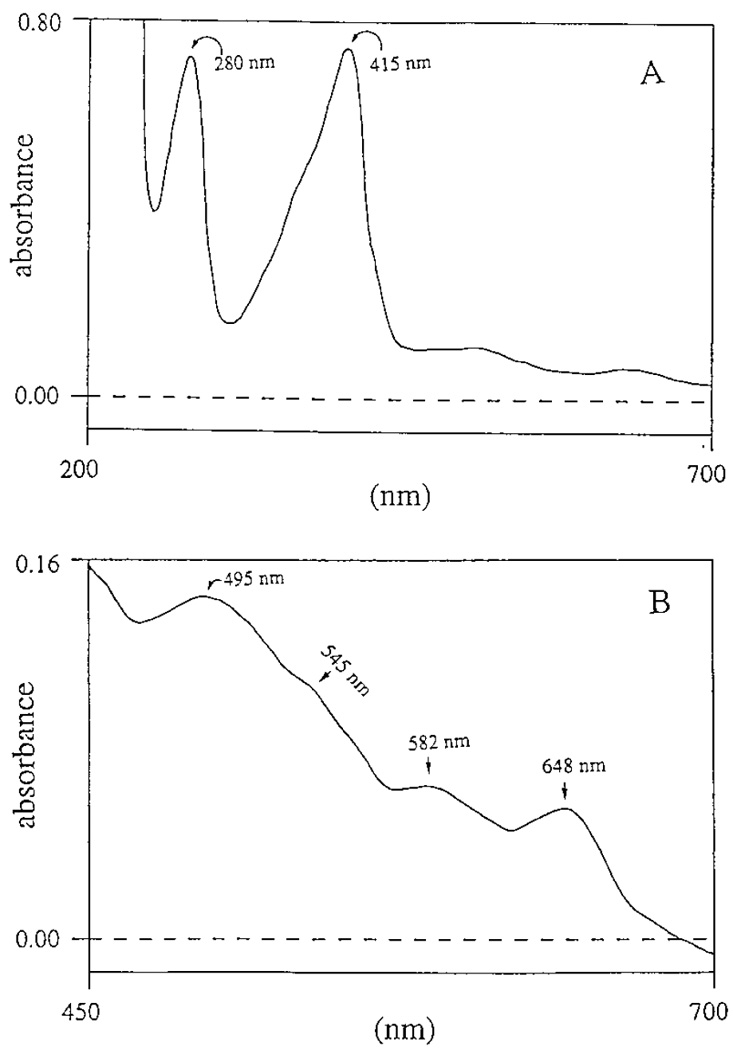
Spectrum of the native chorion peroxidase. (A) Spectrum of the native chorion peroxidase in both the UV and visible regions; (B) minor peaks at 495, 545, 582, and 648 nm in the visible region at high sensitivity.
FIG. 4.
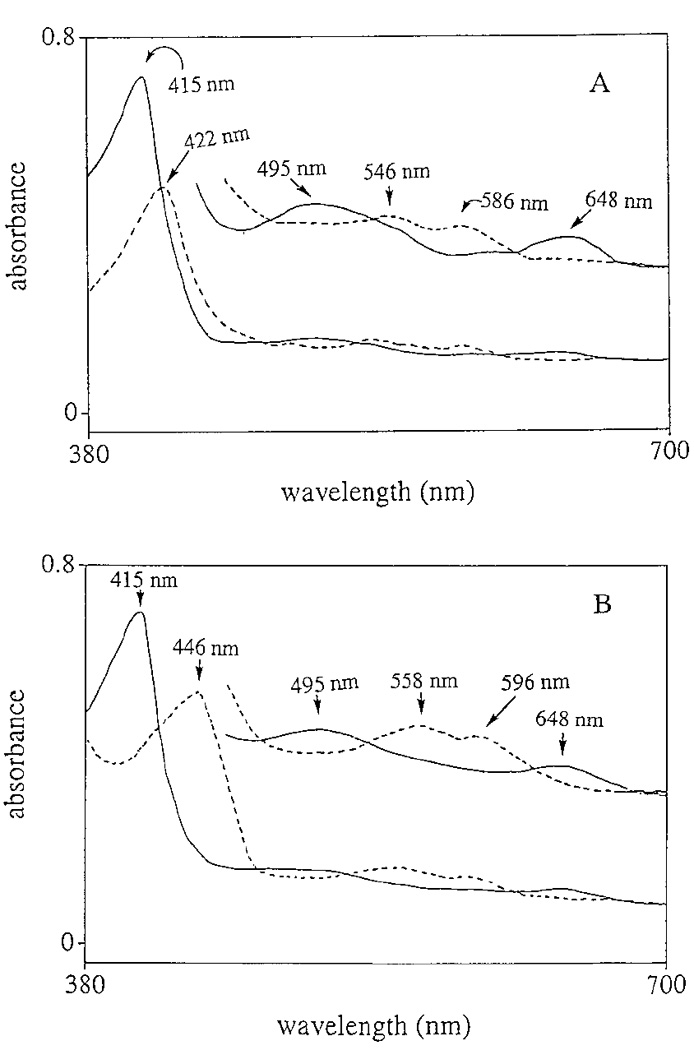
Spectral changes of chorion peroxidase in relation to its redox states. (A) Spectral shift of the Soret band from 415 to 422 nm, with the concomitant disappearance of the 495- and 648-nm minor peaks and formation of two new minor peaks at 546 and 586 nm for chorion peroxidase, after mixing 5.4 µM chorion peroxidase with 6.2 mM H2O2 in 0.1 M acetate buffer (pH 5.4). (B) Spectral shift of the Soret band from 415 to 446 nm and disappearance of the minor 495- and 648-nm peaks and concomitant formation of two new minor peaks at 558 and 596 nm for chorion peroxidase after the enzyme (5.4 mM) was treated with 2 mM sodium hydrosulfite in 0.1 M acetate (pH, 4.0) at anaerobic conditions. The solid line illustrates the spectrum of the native enzyme and the dashed line shows the spectral change of the enzyme in the presence of H2O2 or sodium hydrosulfite, respectively. Inserts in A and B show the spectral changes of the minor peaks at higher sensitivity.
Partial N-terminal AA sequence and AA composition
The partial N-terminal AA sequence of the chorion peroxidase, obtained by Edman digestion, was -LPNVPPNNLS- YRTI-DE. This partial sequence showed essentially no similarity to N-terminal sequences of HRP (13), sea urchin ovoperoxidase (14), and the derived AA sequence of a putative peroxidase from Drosophila (15). Residues 1, 12, and 17 of the N-terminal sequence of the chorion peroxidase were not identified. There were fewer Ser, Ala, Arg, Val, and Phe residues and more Lys, Tyr, and Gly residues in chorion peroxidase than in HRP and sea urchin ovoperoxidase. Table II shows the amino acid composition of chorion peroxidase in comparison with those reported for HRP (13) and sea urchin ovoperoxidase (14), respectively.
TABLE II.
Amino Acid Composition of Chorion Peroxidase from A. aegypti, Sea Urchin Ovoperoxidase, and Horseradish Peroxidase.
Optimal H2O2 concentration and pH for chorion peroxidase activity
Chorion peroxidase displayed its highest activity at pH 8 with guaiacol as a reducing agent (Fig. 5A). Therefore, the subsequent enzyme assays were conducted in Tris buffer at pH 8. Oxidation of guaiacol by chorion peroxidase was not observed in the absence of H2O2, but a high concentration of H2O2 greatly decreased its specific activity (Fig. 5B). The optimal concentration of H2O2 was around 0.8 mM for chorion peroxidase (see Fig. 5B).
FIG. 5.
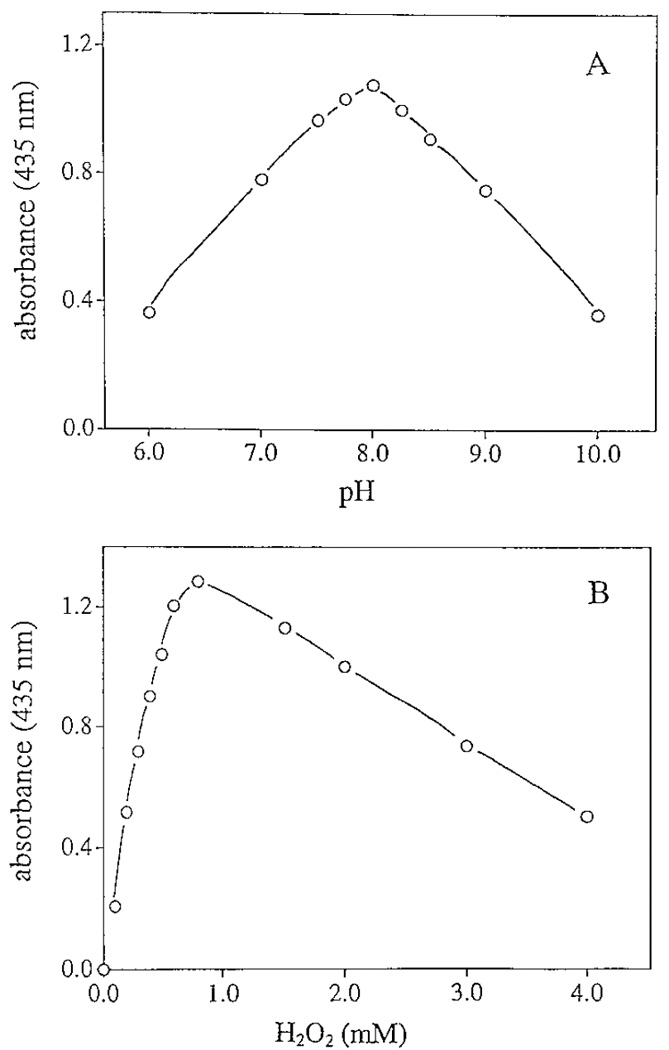
Effect of pH and H2O2 concentration on chorion peroxidase activity. The reaction mixtures containing guaiacol, H2O2, and chorion peroxidase were prepared as described under Materials and Methods. (A) Peroxidase activity under different pH conditions; (B) peroxidase activity in the presence of different concentrations of H2O2.
Resistance to SDS and other denaturing agents
Chorion peroxidase was resistant to SDS. The released peroxidase remained active for a long time in 1% SDS solution. For example, no noticeable decrease in peroxidase activity was observed when solubilized chorion protein remained in 1% SDS and 2 M urea for months at 4°C (not shown). However, when the purified chorion peroxidase was treated with 1.5% SDS for 1–120 min prior to mixing with guaiacol and H2O2, some decrease in enzyme activity was observed in the SDS treated enzyme as compared to the activity level of the untreated enzyme (Fig. 6A). Interestingly, when treated briefly (≤5 min) with SDS prior to mixing with substrate preparation, the enzyme displayed a relatively large decrease (about 15%) in activity, while prolonged incubation of the enzyme in SDS prior to activity assay did not result in further decreases in activity (see Fig. 6A). The enzyme remained active after having been in 1.5% SDS solution for days at room temperature. In contrast, when HRP was treated with 1.5% SDS, rapid inactivation of HRP became apparent within minutes and the decrease in activity correlated with the time of SDS exposure (Fig. 6B). Dialysis of the SDS-treated HRP did not lead to any recovery of its activity (not shown). Chorion peroxidase was also resistant to methanol and acetic acid. After SDS–PAGE (without boiling and β-ME treatment of the enzyme) and a staining (0.1% Coomassie blue, 40% methanol, and 5% acetic acid in water) and destaining process (50% methanol and 5% acetic acid in water), the Coomassie blue stained peroxidase band on the gel was overtaken rapidly by the brown-colored oxidation products upon mixing with tropolone and H2O2 (not shown).
FIG. 6.
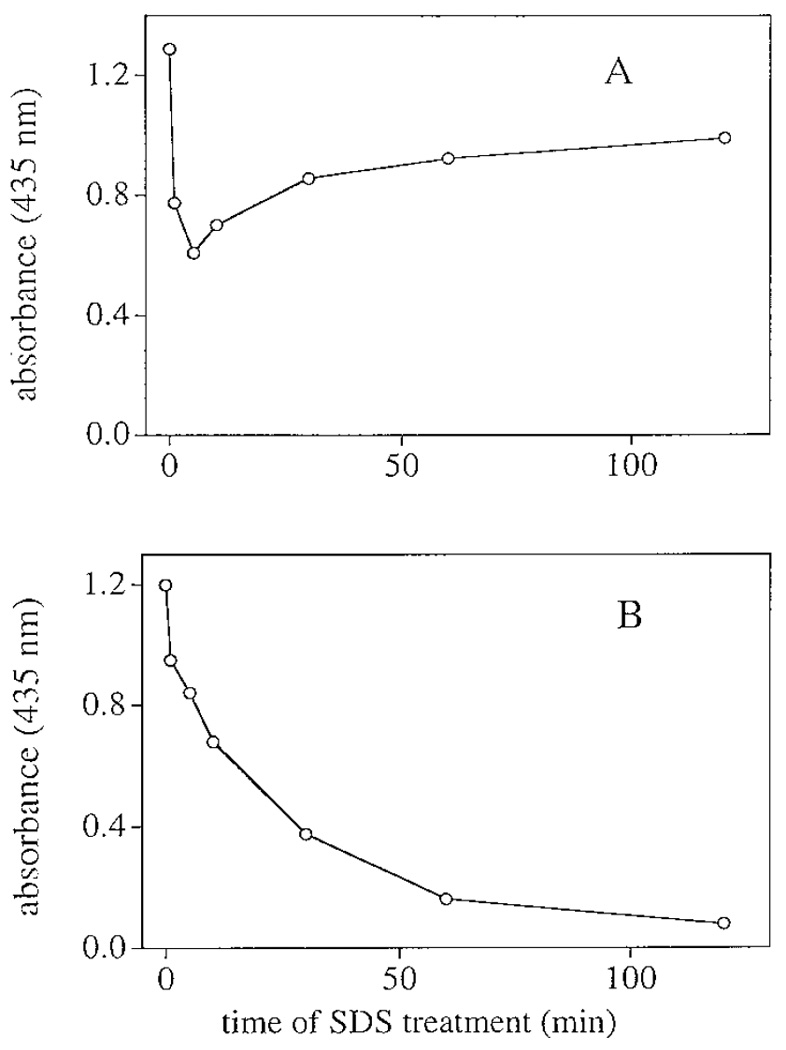
Effect of SDS on chorion peroxidase and HRP activities. Details for the treatment of the enzymes with SDS and the subsequent enzyme activity assays are described under Materials and Methods. A and B illustrate the change of enzyme activity in relation to the time of SDS exposure for chorion peroxidase and HRP, respectively. The x-axis indicates time period of SDS treatment prior to enzyme activity assay.
Substrate specificity
Chorion peroxidase responded actively to commonly used peroxidase substrates, such as guaiacol, diaminobenzidine, and tyrosine. The specific activities of chorion peroxidase were determined with guaiacol and tyrosine as reducing agents and compared to those of HRP to these two compounds under identical assay conditions (Figs. 7A and 7B). The calculated specific activity of chorion peroxidase to guaiacol (128 U min−1 mg−1) was in the same order of magnitude as that of HRP (66 U min−1 mg−1). In contrast, the specific activity of chorion peroxidase to tyrosine (µmol min−1 mg−1) was two orders of magnitude greater than that of HRP to tyrosine (0.05 µmol min−1 mg−1).
FIG. 7.
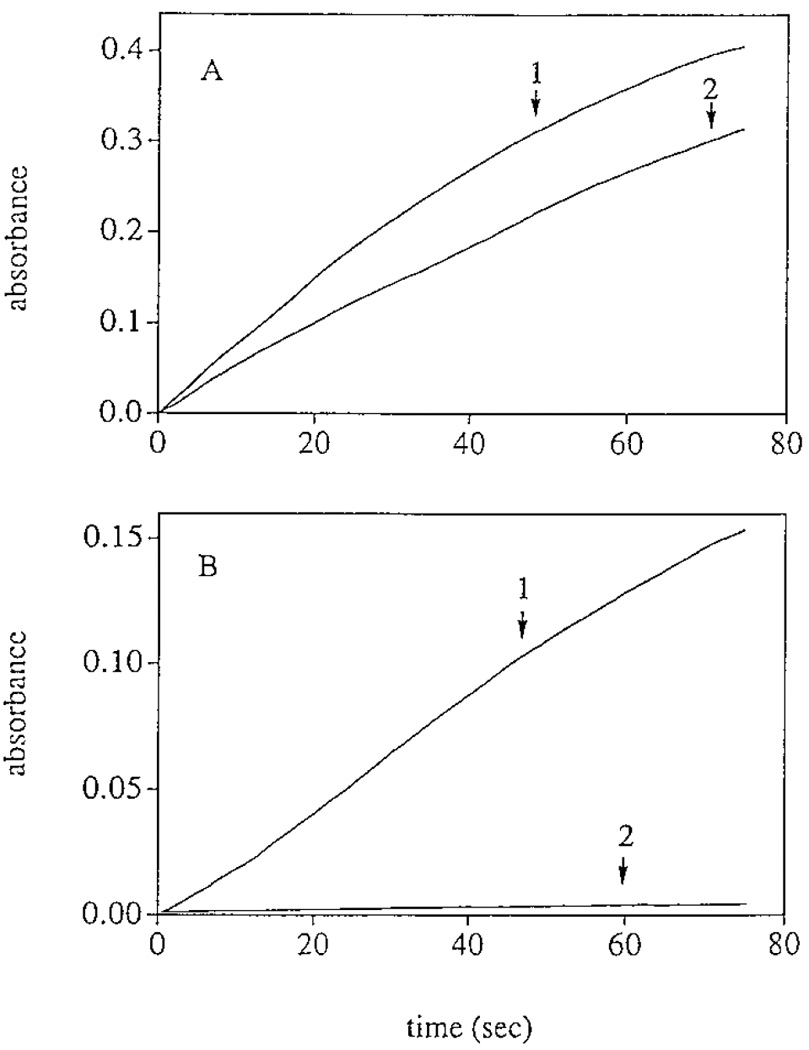
Specific activities of chorion peroxidase and HRP to guaiacol and tyrosine. (A) Absorbance increase at 435 nm in 1 ml of reaction mixture containing 6 mM guaiacol, 0.8mM H2O2, and 0.2 µg of chorion peroxidase (line 1) or 0.4 µg of HRP (line 2). (B) Absorbance increase in 1 ml of reaction mixture containing 1.4 mM tyrosine, 0.8 mM H2O2, and 1 µg of chorion peroxidase (line 1) or 2 µg of HRP (line 2). The amounts of chorion peroxidase and HRP used in A and B, respectively, were different, but the ratio of chorion peroxidase to HRP (1:2) in A was the same as in B.
Peroxidase-mediated protein crosslinking through dityrosine formation
Both tyrosine and dityrosine were electrochemically active, but they were resolved completely under the applied HPLC-ED conditions (Fig. 8A). When angiotensin II was incubated at 30°C in the presence of chorion peroxidase and H2O2, treated with 0.4 M trifluoroacetic acid for 15 min after incubation to stop the reaction, lyophilized under vacuum, hydrolyzed in 6 M HCl at 110°C, and analyzed using HPLC-ED, dityrosine was detected in the hydrolysate and the amount of dityrosine was approximately proportional to the incubation times (Figs. 8B–8D). When BSA or isolated chorion protein, after being incubated at 30°C for 1 h in the presence of chorion peroxidase and H2O2, was analyzed using procedures identical to those for angiotensin II reaction mixtures, dityrosine was also detected in the hydrolysates (Figs. 8E and 8F). In contrast, essentially no dityrosine was detected in a chorion protein reaction mixture in the absence of H2O2 (not shown).
FIG. 8.
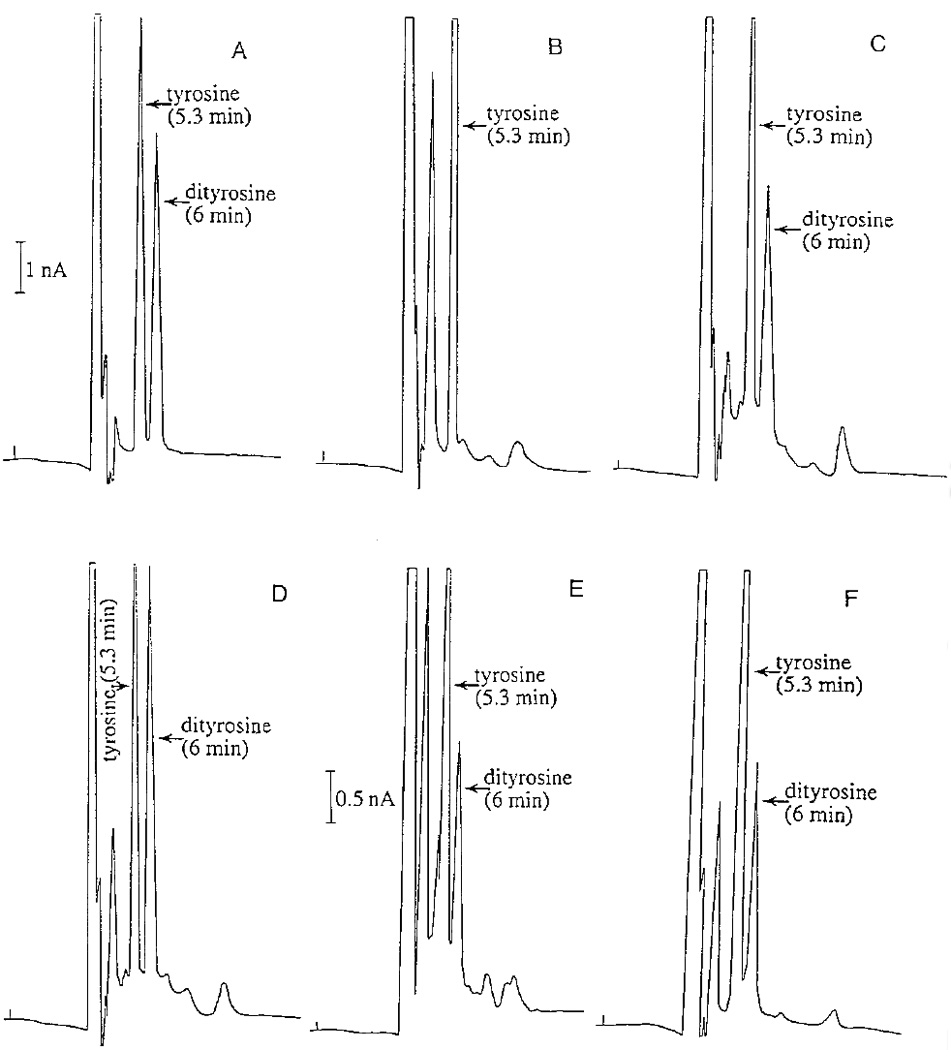
Protein crosslinking through dityrosine formation. Angiotensin II, BSA, or isolated chorion protein was mixed with purified chorion peroxidase and H2O2 and incubated at 30°C. Protein crosslinking through dityrosine formation was based on detection of dityrosine in the hydrolysate of the reaction mixtures. Chromatograms illustrate the detection of tyrosine and dityrosine standards (A), the relative amounts of dityrosine in the hydrolysates of angiotensin II reaction mixtures at 0 (B), 5 (C), and 15 min (D) after incubation, and the amounts of dityrosine in the hydrolysate of BSA reaction mixture (E) and chorion protein reaction mixture during 60 min incubation (F), respectively.
DISCUSSION
Data from our previous study demonstrated the presence of a peroxidase within the chorion of A. aegypti eggs and the involvement of the enzyme in chorion protein crosslinking during chorion hardening (2). However, little is known about the biochemical characteristics of this enzyme. In this study, mosquito chorion peroxidase was successfully purified, and some basic biochemical and biophysical characteristics of the enzyme, such as its hemoprotein nature, relative molecular mass, optimal reaction conditions, partial N-terminal AA sequence and AA composition, formation of intermediates in the presence of H2O2 and hydrosulfite, substrate specificity, and resistance to denaturing agents, were established. Peroxidase-catalyzed eggshell or chorion hardening is likely to be a critical event in all oviparous insects, yet no peroxidase from the eggs of any insects has been purified and characterized. Therefore, data generated in this study provide the basis for future research related to mosquito chorion peroxidase or egg peroxidase from other insects.
The detection of the 415-nm Soret band indicated the enzyme was a hemoprotein, and the requirement of H2O2 as the oxidizing agent for the enzyme established the peroxidatic nature of the reactions it catalyzes. HRP is the most extensively studied enzyme in biochemistry. In the catalytic cycle, the resting ferric HRP is oxidized by H2O2 to an active form, designated compound I, that is two oxidizing equivalents above its resting state (16). Two sequential one-electron oxidations of reducing agents by the enzyme, involving an intermediate species termed compound II, return the enzyme to its resting form (16–18). Formation of compound I and compound II for HRP was mainly based on spectral shift of the Soret band in the presence of H2O2 (16). It was difficult to detect compound I because it was unstable and decomposed rapidly to compound II (17–18). With H2O2 in excess, compound I and/or II was/were further oxidized to compound III (17). The lmax of the Soret band was similar between compound II and compound III in HRP except for the difference of their extinction coefficients (16). Therefore, the spectral shift from 415 to 422 nm for chorion peroxidase during H2O2 treatment was likely due to accumulation of both compound II and compound III in the reaction mixture. The similarity in spectral shift between chorion peroxidase and HRP during H2O2 oxidation suggests that the same catalytic cycle for HRP applies to chorion peroxidase mediated reactions.
An exceptional physical characteristic of the mosquito chorion peroxidase is its resistance toward denaturing agents such as SDS and organic solvents. Internal hydrophobic interaction of proteins is one of the key attractive forces maintaining their three-dimensional structure. Detergents and organic solvents interact with hydrophobic residues, which disrupt the tertiary structure of the protein molecules, leading to their denaturation. Compared to HRP, chorion peroxidase is extremely resistant to detergents and organic solvents. Although some decrease in activity was observed in SDS-treated peroxidase, it seemed apparent that the decrease in activity was not due to peroxidase inactivation because prolonged incubation of the enzyme in SDS did not lead to further decrease in its activity. If the activity decrease was due to inactivation of the chorion peroxidase, a steady decline in activity would have been observed as seen for HRP (Fig. 5B). SDS was not removed from the treated enzyme prior to mixing with substrate preparation during the assays. The presence of SDS (0.3% final concentration) likely increased greatly the viscosity of the reaction mixture, which might affect the turnover of the enzyme, thereby decreasing its specific activity.
Resistance of chorion peroxidase to denaturing conditions was also supported by results from SDS–PAGE of the enzyme. SDS denatures proteins and also binds strongly to protein, with approximately one SDS molecule per two amino acid residues (or 1.4 g of SDS/g of protein). Because the charge density and conformation are essentially constant for all proteins, their separation in SDS–PAGE is due basically to the sieving effect of the gel. Undoubtedly, SDS binds to chorion peroxidase, but the enzyme is far from being completely denatured or linearized by SDS because of its easy detection by substrate staining after SDS–PAGE. However, it seems that boiling of the enzyme in the presence of both β-ME and SDS leads to a complete denaturation of the chorion peroxidase, which is supported by the disappearance of the enzyme activity and the detection of a much sharper band of the treated enzyme. Disulfide bonds are important for maintaining the three-dimensional structures of peroxidases (19), which may also be true for chorion peroxidase. This might explain the effects of β-ME on the partial denaturation and inactivation of chorion peroxidase.
Another distinctive characteristic of the chorion peroxidase as compared to HRP is its high specific activity toward tyrosine. For example, there is about a twofold difference between chorion peroxidase and HRP to guaiacol (a commonly used substrate for peroxidase assay), but the specific activity of the chorion peroxidase to tyrosine is more than 100-fold greater than that of the same HRP to tyrosine (see Fig. 6). The high activity of chorion peroxidase to tyrosine, the ability of this enzyme to catalyze polypeptide and chorion protein crosslinking through dityrosine formation, and the detection of dityrosine in the hardened egg chorion (2) provide strong evidence for the active role of the chorion peroxidase in chorion hardening through dityrosine formation in A. aegypti eggs. In mosquitoes, peroxidase-catalyzed chorion protein crosslinking occurs in the environment because dityrosine is not detected from the dissected or newly oviposited eggs. In contrast, protein crosslinking in Drosophila eggs occurs prior to oviposition (3–6). Therefore, the environment for mosquito chorion peroxidase to display its function is different from that of other peroxidases, including Drosophila egg peroxidase. Consequently, environmental pressures or selection might explain why chorion peroxidase is extremely resistant to harsh conditions, such as its strong resistance to denaturation by SDS and organic solvents. However, we are unable to provide a clear explanation about the mechanism for the strong resistance of mosquito chorion peroxidase to denaturing agents at this time due to difficulties in obtaining an adequate amount of enzyme for further structural analysis. We also have no precise answer for the extremely high activity of the enzyme to tyrosine as compared to other peroxidases. The unambiguous answers for these unique characters of the chorion peroxidase rely ultimately on a thorough understanding of the detailed structure of the enzyme, including its primary sequence, its posttranslation modification, and its three-dimensional structure.
ACKNOWLEDGMENT
This work is supported by NIH Grant AI 37789.
Footnotes
Abbreviations used: AA, amino acid; β-ME, mercaptoethanol; BSA, bovine serum albumin; EDTA, ethylenediaminetetraacetic acid; HRP, horseradish peroxidase; PMSF, phenylmethylsulfonyl fluoride.
REFERENCES
- 1.Li J, Christensen BM. Insect Biochem. Mol. Biol. 1993;23:739–748. [Google Scholar]
- 2.Li J, Hodgeman BA, Christensen BM. Insect Biochem. Mol. Biol. 1996;26:309–317. doi: 10.1016/0965-1748(95)00099-2. [DOI] [PubMed] [Google Scholar]
- 3.Heinecke JW, Li W, Daehnke HL, III, Goldstein JA. J. Biol. Chem. 1993;268:4069–4077. [PubMed] [Google Scholar]
- 4.Giorgi F, Deri P. Histochemistry. 1976;48:325–334. doi: 10.1007/BF00499249. [DOI] [PubMed] [Google Scholar]
- 5.Margaritis LH. In: Comprehensive Insect Physiology, Biochemistry, and Pharmacology. Kerkut GA, Gilbert LI, editors. Vol. 1. Oxford: Pergamon; 1985. pp. 202–205. [Google Scholar]
- 6.Margaritis LH. Tissue Cell. 1985;17:553–560. doi: 10.1016/0040-8166(85)90031-x. [DOI] [PubMed] [Google Scholar]
- 7.Mindrinos MN, Petri WH, Galanopoulos VK, Lombard MF, Margaritis LH. Wilhelm Roux’s Arch. 1980;189:187–196. doi: 10.1007/BF00868677. [DOI] [PubMed] [Google Scholar]
- 8.Bergmeryer HU, Gawehn K, Grossl M. In: Methods of Biochemical Analysis. Bergmeyer HU, editor. New York: Academic Press; 1974. pp. 494–495. [Google Scholar]
- 9.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 10.Yamazaki I, Piette LH. Biochim. Biophys. Acta. 1963;77:47–64. doi: 10.1016/0006-3002(63)90468-2. [DOI] [PubMed] [Google Scholar]
- 11.George P. J. Biol. Chem. 1953;201:427–434. [PubMed] [Google Scholar]
- 12.Bayse GS, Michaels AW, Warrison M. Biochim. Biophys. Acta. 1972;284:34–42. doi: 10.1016/0005-2744(72)90043-5. [DOI] [PubMed] [Google Scholar]
- 13.Welinder KG. Can. J. Biochem. 1972;50:63–90. doi: 10.1139/o72-009. [DOI] [PubMed] [Google Scholar]
- 14.Nomura K, Suzuki N. Arch. Biochem. Biophys. 1995;319:525–534. doi: 10.1006/abbi.1995.1327. [DOI] [PubMed] [Google Scholar]
- 15.Ng SW, Wiedemann M, Komteramm R, Petersen G. Biochim. Biophys. Acta. 1992;1171:224–228. doi: 10.1016/0167-4781(92)90127-l. [DOI] [PubMed] [Google Scholar]
- 16.Paul KG. In: The Enzymes. Boyer PD, Lardy H, Myrback K, editors. Vol. 8. New York: Academic Press; 1963. pp. 227–275. [Google Scholar]
- 17.Dunford HB. In: Peroxidases in Chemistry and Biology. Everse J, Everse KE, Grisham MB, editors. Vol. 2. Boca Raton, FL: CRC Press; 1991. pp. 1–24. [Google Scholar]
- 18.Chance B. Arch. Biochem. 1949;22:224–252. [PubMed] [Google Scholar]
- 19.Zeng J, Fenna RE. J. Mol. Biol. 1992;226:185–207. doi: 10.1016/0022-2836(92)90133-5. [DOI] [PubMed] [Google Scholar]