

Abstract
In this tutorial review the economies of synthesis are analysed from both detailed and macroscopic perspectives, using case-studies from complex molecule synthesis. Atom, step, and redox economy are more than philosophical constructs, but rather guidelines, which enable the synthetic chemist to design and execute an efficient synthesis. Students entering the field of synthesis might find this tutorial helpful for understanding the subtle differences between these economic principles and also see real-world situations where such principles are put into practice.
Introduction
Since the dawn of organic chemistry, the synthesis of complex natural products has been the testing ground of methods and strategies in synthesis and in doing so defines the frontiers of the field.1 Endeavours in complex molecule total synthesis often have a way of yielding enormous dividends in the form of fundamental insight into selectivity principles and the outright invention of new chemistry in order to reach the target. With increasing capabilities in the power of organic synthesis the perception of what constitutes a complex target structure has changed accordingly, be it from acetic acid to adamantane to strychnine to erythronolide to maitotoxin and so on. The last century has witnessed a breathtaking development of synthetic methods and celebrated epochal syntheses of terrifyingly complex target compounds,1 and in doing so has pushed the capabilities of synthetic methodology to its limits. One culmination point was the total synthesis of palytoxin, 1,2 a compound with 64 stereogenic centres (Scheme 1).
Scheme 1.
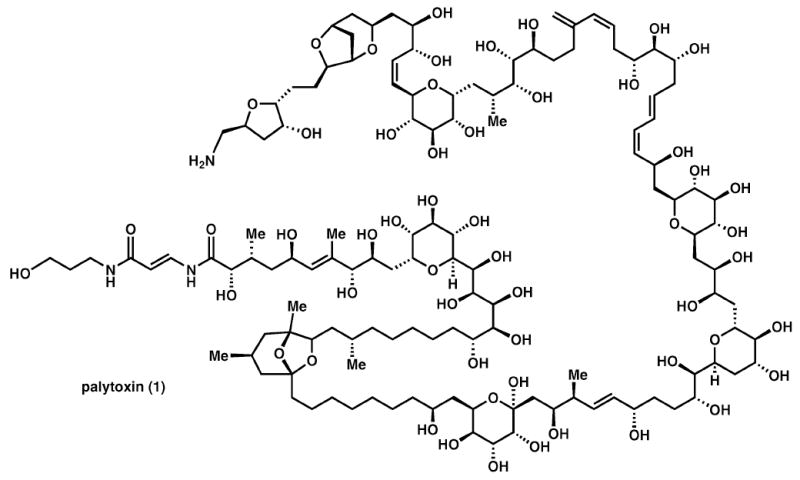
Palytoxin (1).
Palytoxin, 1, was assembled from seven building blocks in 39 steps. The synthesis of the seven building blocks has not been published yet in detail, but it can be safely guessed that this required more than 140 steps. Hence it is obvious what enormous effort, manpower, and logistics were involved.
Due to economic considerations, eventually society will not be able to support such an endeavour, unless it is of utmost importance such as the penicillin synthesis project during World War II. In that effort more than 1000 chemists in 39 laboratories joined forces.3a The synthetic strategies and methods of the 20th century reach their limits with molecules such as palytoxin, when the number of synthetic steps significantly exceeds 100. In a manufacturing setting, the production plant synthesis of Roche’s Fuzeon with 106 steps3b may well hold the record for decades to come. It must be the goal of the 21st century to accomplish more with less, and in order to do so, one has to analyse the factors that are limiting in traditional organic synthesis.
From the discussion above one realises that the number of steps in a synthesis has a paramount influence on the feasibility and practicality of a synthesis project, as it determines the manpower input, the material input, the logistics involved as well as the amount of waste and byproducts that are handled. It is for this very good reason that Wender et al. emphasise the “step economy” of synthesis projects.4 Yet this appeal has to be translated into clear guidance on how to reduce the step count, i.e. to simplify or streamline complex target syntheses.
This leads to the fundamental question: which steps in a synthesis sequence are essential and, hence, indispensable? This question has been answered by Hendrickson in 1975, when he addressed “the ideal synthesis:”5
“The ideal synthesis creates a complex molecule… in a sequence of only construction reactions involving no intermediary refunctionalisations, and leading directly to the target, not only its skeleton but also its correctly placed functionality.”
Hendrickson postulates that the skeleton forming steps are the only essential steps in synthesis. If the skeleton forming steps are executed correctly, one should be able to simultaneously set up the functionality needed for the final target or the next step, including the requisite stereocentres. All other refunctionalisation steps (e.g. protecting group management steps, non-strategic redox manipulations, etc.) should be minimised in order to simplify complex target synthesis. Indeed, the simplification of synthesis with respect to reducing the number of steps and redox manipulations, as well as molecular waste, is imperative. However, one must not forget the overall goal of increased efficiency and should keep in mind the importance of molecular yield. A balance between brevity of sequence and overall yield is critically important.
To understand how far we are away from Hendrickson’s ideal synthesis, take for example the assembly line of Kishi’s palytoxin synthesis. It involved 11 skeleton forming steps, 15 refunctionalisation steps, and 13 protecting group management steps. Kishi’s palytoxin synthesis can be used as a ruler for the state of natural product synthesis at the end of the last century. To generalise from this single example, this reveals that roughly two thirds of the steps of a synthesis are ones that reflect our inability to achieve the Hendrickson “ideal.” This is a consequence of the fact that the most sophisticated methods for many skeleton-forming reactions have not yet been developed. The current set of methods are not chemoselective enough to allow for the elimination of non-skeleton building steps.6 While new methodology is enabling in the synthesis of complex natural products, thinking about the logic and strategy required to reach the “ideal synthesis” is essential.7
The economies of synthesis are a useful conceptual framework from which to design and analyse syntheses. What follows below is a brief description of the “economic principles” underlying synthesis logic and a series of synthesis vignettes, which illustrate such economic considerations.
Step economy
The precepts of step economy teach organic chemists that minimising the number of steps leads to an efficient multistep synthesis in terms of cost and time expended to obtain the desired target. While designing and executing a target-oriented synthesis, the structure cannot be modified to accommodate the current state of the art in organic synthesis, rather “the goal structure is very clear and stubbornly inflexible.”8 Thus, the conceptual framework of step economy brings about the conclusion that achieving a total synthesis requires innovation in the form of strategy, tactics and method invention. Alternatively, in cases where there is not a goal structure, but rather a goal function, such as in some biological, medicinal or materials studies, the target can be chemically simplified to the extent that it requires a fewer number of steps to access. The judicious choice and development of reactions or sequences of reactions allows for the step-economical synthesis of a given target structure.9
Atom economy
A traditional perspective on the development of novel chemical transforms prioritises synthetic efficiency in terms of mole percent by the maximisation of theoretical product yield with respect to chemo-, regio-, diastereo-, and enantioselective control. A modification of this ideological perspective came with Trost’s concept of atom economy,10 which places an additional restriction on synthetic methodology—that is to maximise the mass efficiency of a reaction with respect to all of the reactants. When viewing organic reactions with an atom economy “filter,” the ideal reaction incorporates all of the atoms of the starting materials into the desired product by the merger of chemical building blocks, using only catalytic quantities of reagents. For example, the Diels–Alder reaction proceeds with 100% atom economy. Multiple features of a reaction come under scrutiny, including stoichiometric reagents and high molecular weight byproducts. The importance of this perspective becomes clear given the cost of pharmaceutical and material production, as well as the increasing importance of green chemistry within the context of sustainable development.11
Redox economy
Accounting for redox manipulations over the course of a synthesis can be a powerful tool to shorten the pathway to a given target molecule. The basic goal of redox economy is to minimise non-strategic redox manipulations in order to achieve an isohypsic synthesis—one which has no redox steps. When oxidation or reduction operations are used, the oxidation state of the intermediates should not fluctuate, but rather increase or decrease steadily (or exponentially) in the construction of complex molecules. The natural consequence of this is a step economic synthesis.12 Additionally, due to a lack of chemoselective methodologies, the avoidance of oxidation and reduction steps frequently results in a more atom economic synthesis, as these reactions (relative to redox neutral reactions) tend to be less atom economical. For example, in the Dess–Martin-mediated oxidation of an alcohol a reagent with a molecular weight of 424 is used to remove 2 hydrogen atoms.
Illustrative syntheses
Methyl homodaphniphyllate and proto-daphniphylline (Heathcock)
One of the most time-tested approaches to achieving an efficient synthesis is through employing a biomimetic strategy.13 These strategies frequently have the natural consequence of reducing the number of functional group interconversions necessary to achieve the desired level of complexity. An overwhelming example of how this can be a successful strategy is in Heathcock and Piettre’ synthesis of proto-daphniphylline, 10. They use a purely abiotic “network analysis” (standard retrosynthetic analysis) to methyl homodaphniphyllate, 6 (Scheme 2A), and another approach draws heavily from the presumed biosynthesis (Scheme 2B).
Scheme 2.
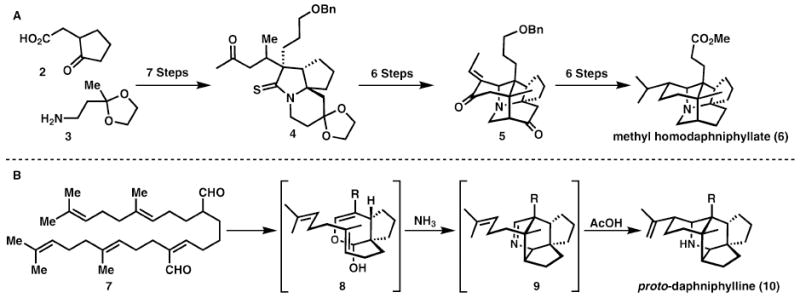
A dramatic cascade reaction en route to daphniphyllum alkaloids.
The first approach builds the skeleton layer by layer using network analysis and must address several refunctionalisation steps of the type Hendrickson teaches us to avoid. In contrast, the use of a biomimetic Michael, Diels–Alder, aza-Prins cascade leads to the most efficient synthesis of the natural product. In a single isohypsic cascade (7 → 10), two C–N bonds, four C–C bonds, and five rings are generated, underscoring the power of redox neutral (isohypsic) reactions in synthesis. This rapid generation of complexity demonstrated in Heathcock’s second generation synthesis not only allows for a general strategy for the synthesis of a diverse family of alkaloids, but also minimises the use of protecting groups. The biomimetic strategy was later applied to a number of family members, including methyl homodaphniphyllate, 6.
While protecting groups cannot always be completely eliminated in the synthesis of complex molecules, minimisation of their use is advantageous from the perspectives of both step and atom economy. The pros and cons of protecting group chemistry have recently been reviewed.6
Torreyanic acid (Porco)
Another broadly applicable biomimetic Diels–Alder strategy has been employed by Porco and co-workers in their elegant syntheses of epoxyquinoid natural products.14 One example of this Diels–Alder dimerisation event is shown in Scheme 3 with the total synthesis of torreyanic acid (17). While protecting group interchanges are employed in the synthesis of the monomer, 16, the final step proceeds with all of the functionality present in the dimeric natural product. In this convergent approach, the number of steps required is greatly diminished, relative to a hypothetical alternative strategy, as the monomeric components are identical and do not require separate syntheses.
Scheme 3.
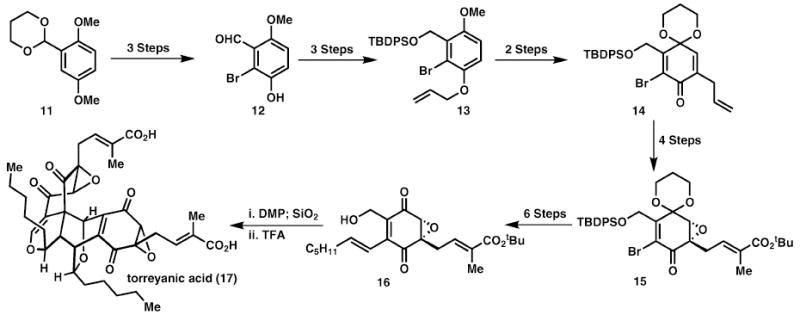
Biomimetic Diels–Alder reaction in the synthesis of epoxyquinoid natural products.
Typical of pericyclic reactions (sigmatropic rearrangements, electrocyclisations, group transfer reactions, and cycloadditions) is their atom-economical nature with all of the atoms in the starting materials typically present in the product or with loss of low molecular weight materials (CO2, N2, etc.). In a spectacular show of atom economy, no other reactants (catalyst, solvent, etc.) are necessary to effect Porco’s Diels–Alder cycloaddition. Also typical of pericyclic reactions is that the starting materials’ oxidation state usually does not change (cycloadditions and ene reactions with singlet-oxygen are obvious exceptions to this rule). Thus, striving for a redox neutral strategy can naturally guide a chemist to consider pericyclic reactions in retrosynthetic analysis.
Rugulosin (Nicolaou)
Another beautiful example of biomimetic cascade reactions is seen in Nicolaou’s total synthesis of rugulosin, 23, and related family members (Scheme 4).15 Nicolaou and co-workers were attracted to these targets by their highly congested core structures and were intrigued by the possibility of re-enacting their biosynthesis. This was ultimately achieved by first synthesising a suitably protected monomer, 22, which upon exposure to basic, oxidative conditions underwent the desired oxidative dimerisation, Michael, Michael cascade reaction. As with all biomimetic dimerisation reactions, the synthetic route is of a highly step and atom economic nature. This synthesis also provocatively shows how a daring late-stage oxidation strategy can be successful in the synthesis of complex molecules.
Scheme 4.
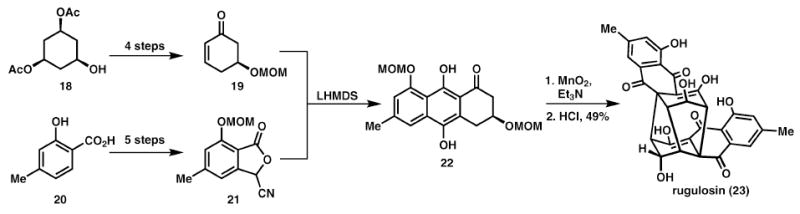
Biomimetic oxidative dimerisation in the synthesis of rugulosin.
Hirsutene (Wender and Mehta)
The arene–olefin cycloaddition reaction, popularised by Wender and coworkers16a is another example of a pericyclic reaction, which has been used to generate complexity in target-oriented synthesis (Scheme 5). In the synthesis of hirsutene, 29, a photoinduced, formal [3 + 2] cycloaddition, followed by [1,3] diradical recombination provides a useful tetracycle, 27, after deacetylation, proceeding in 22% yield along with three other diastereomers (9%). A second redox neutral transformation, a cationic dehydration event, fragments the recently formed cyclopropane to afford the hirsutene core, 28, in short order. In the final steps of the synthesis, a few refunctionalisations are necessary, deviating from the otherwise isohypsic synthesis. The combined two-step strategy of an arene–olefin cycloaddition with a strategic cyclopropane fragmentation is atom economical in that all three rings of the carbocyclic core of hirsutene are formed with only the loss of water from the starting materials. While there is one protecting group interchange the sequence is highly step economic.
Scheme 5.
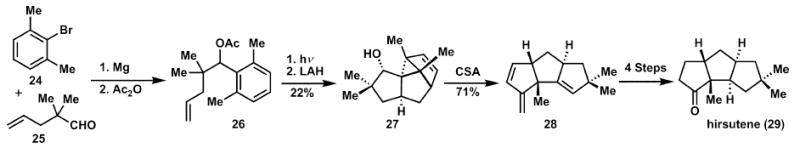
Arene–olefin cycloaddition reaction in the synthesis of hirsutene.
It is also instructive to note that compound 27 might be considered more complex than the final target. Such “overbred skeletons” are only desirable when they can be generated and converted to the target in a rapid fashion.
Mehta and co-workers16b later reported an extremely atom-economical synthesis of the tricyclic hirsutene core, 35 (Scheme 6). A remarkable sequence of events, requiring only heat, light, and solvent as reagents transpired to assemble the core structure in an isohypsic fashion. Beginning with a benzoquinone, 30, and cyclopentadiene a thermal Diels–Alder gives 32. This is followed by photochemical [2 + 2] to afford 33 and thermal cyclobutane fragmentation provides 34. Finally, a double epimerisation (via olefin isomerisation) sets the required stereochemistry to deliver the core, 35, in four steps and 28% overall yield, in the near absence of any reagents. To complete the synthesis, a mere six steps were then required to adjust the functionality present to that of hirsutene, 29.
Scheme 6.
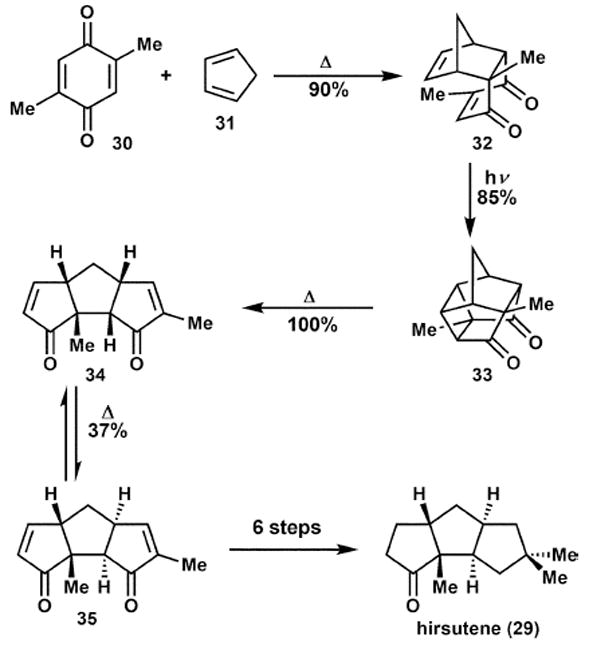
Synthesis of the hirsutene core in the near absence of reagents.
Pentalenene (Paquette)
Paquette and co-workers have developed two syntheses17 of pentalenene (37), separated by 20 years (Scheme 7). The first of these is a traditional approach proceeding in over 20 steps and involving several corrective refunctionalisations. The second-generation synthesis used only two skeletal building operations and a total of eight steps. This remarkable simplification of the synthesis was due to a cascade process in which the complex tricyclic structure, 43, was generated in one stroke from a monocyclic starting material, 38, in a sequence of electrocyclic reactions and an aldol addition. In this way a remarkably rapid increase in complexity was attained.
Scheme 7.
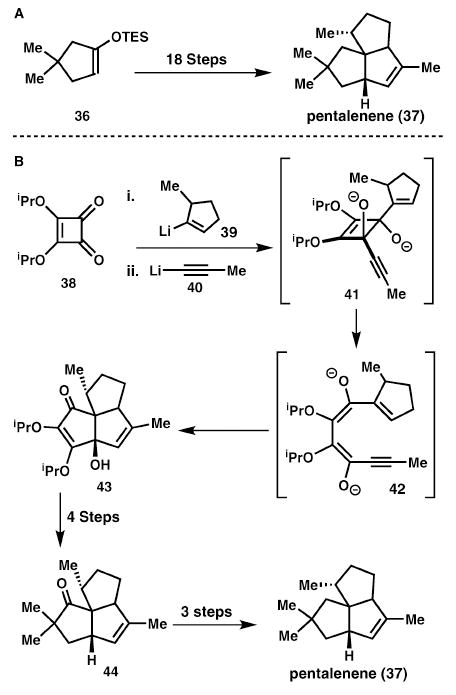
Squarate ester cascade reaction enables a rapid pentalenene synthesis.
Cascade reactions are still restricted to rather special situations, yet their application in complex target synthesis is steadily increasing,18 because they provide a truly powerful tool to rapidly generate complexity.
Methylphenidate (Matsumura and Davies)
In the total synthesis of methylphenidate (Ritalin), 48, Davies and co-workers demonstrate excellent atom, step and redox economy by the use of a C–H activation reaction (Scheme 8).19 One previous asymmetric route relied on the use of a chiral auxiliary in an aldol reaction (46 → 47), which provided material with excellent diastereoselectivity.20 However, the use of this stoichiometric reagent is costly from the perspective of atom economy and requires a number of steps to install and then later remove. An abbreviated synthesis utilised an enantioselective, rhodium(II)-catalysed intermolecular C–H insertion with phenyldiazoacetate to directly afford methylphenidate, 48, after Boc deprotection. While the diastereoselectivity is notably lower, the direct nature of this synthesis greatly enables the rapid synthesis of a diversity of analogs. When used strategically, the logic of C–H functionalisation can dramatically shorten sequences and, in some cases, even solve seemingly insurmountable challenges.
Scheme 8.
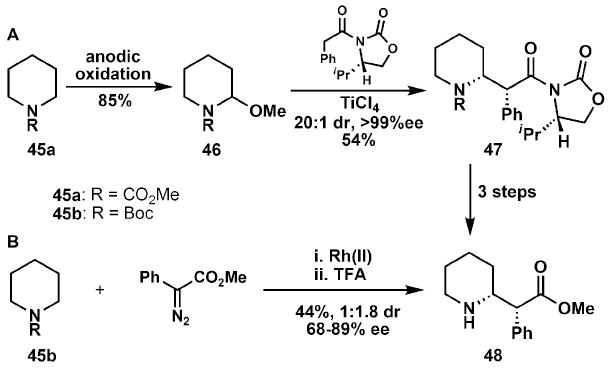
Strategic C–H oxidation in the synthesis of methylphenidate (48).
Miyakolide C6–C13 fragment (Masamune and White)
The last decade has seen major advances in the field of allylic oxidation methodology, especially palladium catalysed oxidation of alkenes to allylic alcohols and amines. The benefit of linear increase of oxidation state with simultaneous functional group installation can be illustrated by comparison of parts A and B in Scheme 9. The original synthesis21a of the intermediate 52 starts from 1,4-butanediol, 49 and subsequent differentiation of the hydroxyl groups requires a sequence involving a total of four protecting group steps.
Scheme 9.
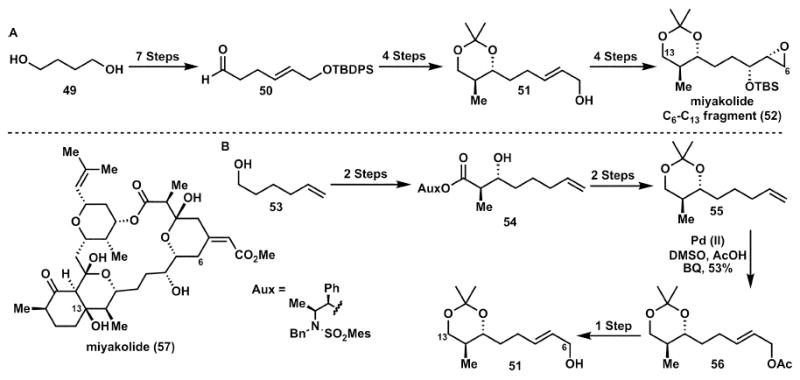
C–H oxidation used in the synthesis of the miyakolide C6–C13 fragment (51).
The alternative strategy21b in Scheme 9B starts from alcohol 53, having a terminal double bond as profunctionality for the other alcohol function. This allowed for the generation of the allylic alcohol at the end of the reaction sequence by a palladium catalysed oxidation (i.e. 55 → 56). Since this functionality was now generated after all the other steps had been completed, the differentiation problem did not exist as one alcohol was masked as an olefin. While desymmetrisation of a late stage intermediate can be a powerful tool (vide infra), in the case of Scheme 9A the use of a symmetrical starting material brought no advantage, because the desymmetrisation required a large number of steps. Hence, the alternative strategy (Scheme 9B) turned out to be more effective. Thus, as seen in the previous section C–H functionalisation can allow for significant shortening of synthetic sequences.
Davanone (Honda–Tsuchihashi and Vosburg)
A recent total synthesis of (+)-davanone, 61, from Vosburg and co-workers22 achieves step, atom and redox economy through careful synthetic design (Scheme 10). Using Bode’s thiazolium catalyst, a redox isomerisation is first used for the synthesis of a β-hydroxyester, 63. The resulting alcohol, 63, then participates in a Tsuji–Trost reaction to form the desired tetrahydrofuran, 64. After conversion of the ethyl ester to a Weinreb amide, reaction with prenyl magnesium chloride affords the enantiopure natural product, 61. The four successive isohypsic transformations provide a successful strategy for a step economic synthesis relative to the previous enantio-selective synthesis (20 steps from commercial material). Protecting groups are avoided, further simplifying the synthesis. Additionally, the reaction sequence involves two catalytic reactions, which are highly atom economical, producing only minimal byproducts.
Scheme 10.
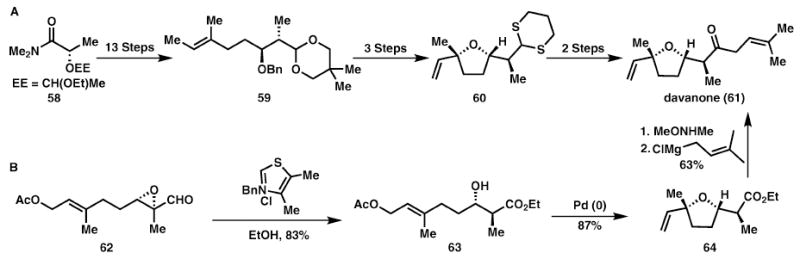
Two approaches for the total synthesis of (+)-davanone (61).
Platencin (Rutjes and Mulzer)
While transition metal processes are powerful opportunities to maximise synthesis economy, judicious reaction selection is still necessary. This can be seen by comparing the final steps of two formal syntheses of platencin, 70.23 In Rutjes’ synthesis in Scheme 11A the authors chose to form the skeletal bond by a samarium iodide reductive coupling reaction. This required protection and later deprotection of the enone, 65, as well as ozonolytic cleavage of both double bonds. Refunctionalisation of diol 66 then requires an additional three steps to reach Nicolaou’s intermediate 67 for the synthesis of platencin 70.
Scheme 11.
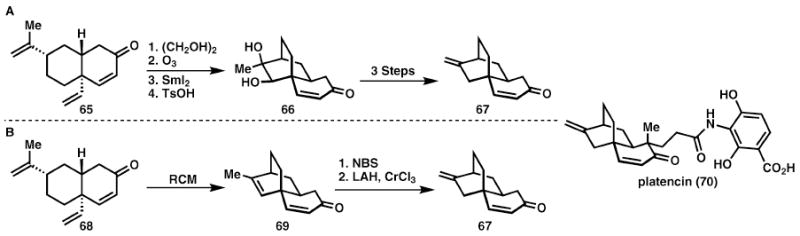
Ring-closing metathesis to synthesise platencin (70).
By opting to use ring-closing metathesis as the transition-metal catalysed bond-forming reaction (Scheme 11B), protecting groups are no longer necessary and the overall transformation to intermediate 67 can be performed in a mere three steps as opposed to seven. Overall, the Mulzer synthesis of platencin takes place in nine steps and is a profound example of step, atom, and redox economy in action.
Hemibrevetoxin B (Nakata and Nelson)
Another dramatic example of step reduction is the Nelson approach24 to hemibrevetoxin B, 79. The advanced key intermediate 72, which can be converted to 79, was synthesised by the Nakata group through an approach involving sequential ring formation requiring 29 steps (Scheme 12A). With the importance of this intermediate apparent, Nelson’s approach, which proceeds via bidirectional synthesis, followed by late-stage desymmetrisation (Scheme 12B), significantly simplified the pathway to 79. This approach relies on an acid-catalysed epoxide opening to simultaneously form both tetrahydropyran rings in 75. Net two-carbon homologation and epoxidation furnishes the key intermediate 77. Jacobsen’s epoxide opening then acts to desymmetrise 77 and after acetonide formation affords 72. This approach results in a four-fold reduction in the total number of steps required to synthesise this important intermediate, 72. The approach is guided by linearly increasing oxidation state and a general avoidance of protecting groups, leading to a highly efficient synthesis.
Scheme 12.
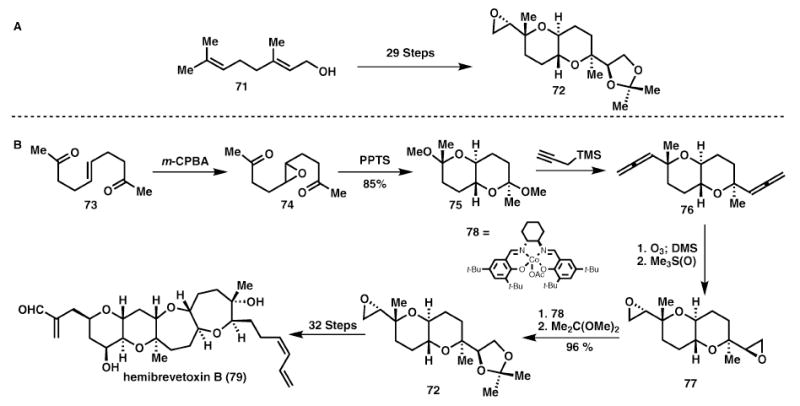
Desymmetrisation of a diepoxide en route to hemibrevetoxin B (79).
Rifamycin (Kishi and Harada–Oku)
As demonstrated in Scheme 12, a valuable strategy to reduce the number of skeleton forming steps in a synthesis is to find a hidden symmetry in the target or a precursor intermediate: such symmetry permits a bidirectional elaboration of the skeleton with an attendant reduction in the skeleton building steps.25 This can be illustrated with regard to rifamycin S, 80, in the synthesis of a portion of its ansa chain, 82. The pioneering synthesis by Nagaoka and Kishi25a synthesised the key intermediate 82 from the aldehyde 81 by chain extension (Scheme 13A). The skeleton was extended in a sequential manner, amounting to four skeleton building steps (accompanied by one strategic and eight corrective redox-operations and eight protecting group steps).
Scheme 13.
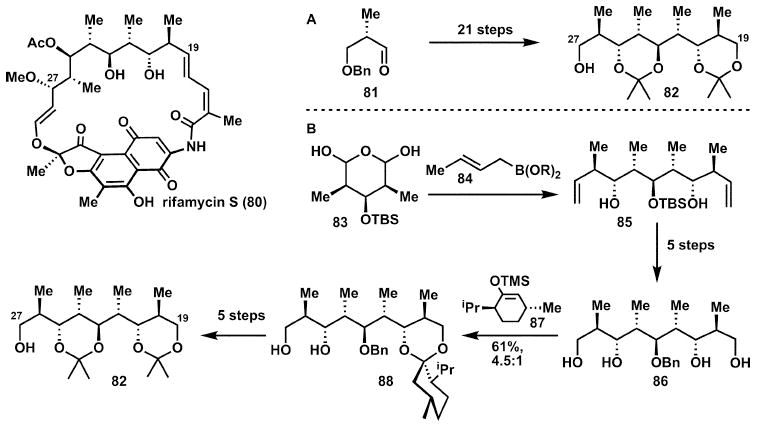
Desymmetrisation of a meso tetraol in the synthesis of rifamycin S (80).
A key to an improvement is the direct use of the latent symmetry in the intermediate 82. This provided the opportunity to elaborate the skeleton in a symmetrical manner from a central building block 83 (Scheme 13B). Two-fold simultaneous chain extension by addition of crotylboronate to 83 is the single skeleton building reaction in this novel synthesis25b of 82. Symmetry is maintained throughout a number of refunctionalisation steps until tetraol 86. At this point the symmetry of the prochiral compound 86 is broken by differentiation with the aid of a chiral auxiliary (TMS-enol ether of menthone, 87). The resulting monoacetal is then readily converted into the enantiomerically pure target 82. Note that this synthesis of 82 has only one skeleton building step, but the remaining 10 refunctionalisation steps (redox manipulations and protecting group interchanges) detract from the otherwise elegant approach.
Spirastrellolide A methyl ester (Paterson)
Paterson’s first approach to the C26–C40 bis-spiroacetal subunit of spirastrellolide A, 97, while only 11 linear steps, was plagued by a low yielding acetonide deprotection and spirocyclisation step (92 → 93) as a result of the propensity of the five-membered lactone, 92, to undergo elimination and aromatisation (Scheme 14A).26 The first generation route also suffers from the use of high molecular weight, stoichiometric reagents (DMP, [Ph3PCuH]6, etc.). An alternative strategy was employed,26 which both avoids these problems and allows for greater material throughput.
Scheme 14.
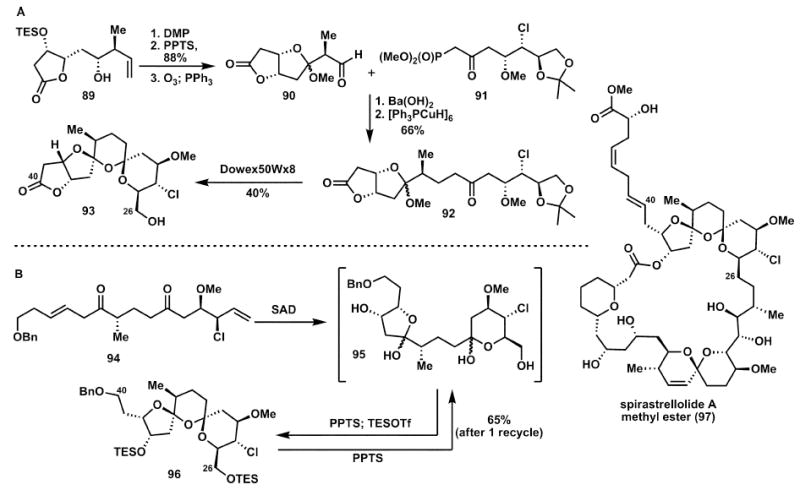
Double spirocyclisation in the total synthesis of spirastrellolide A methyl ester (97).
In Paterson’s landmark total synthesis of spirastrellolide A methyl ester, 97,26 an exponential increase in oxidation state via a double dihydroxylation event is accompanied by an exponential increase in complexity (Scheme 14B). This approach allows for the rapid formation of the tricyclic bis-spiroacetal subunit, 96, from a linear carbon chain precursor, 94, containing the requisite functionality necessary for synthesis of the natural product. While a mixture of isomers is obtained, the undesired isomers can be conveniently recycled to afford a predominance of the desired product. While the step economy of the two routes is identical, the second-generation route leads to a dramatic increase in redox and atom economy—thus a greater supply of material can be obtained.
Oseltamivir phosphate (Roche, Corey and Hayashi)
The synthetic challenge of the H5N1 influenza treatment, oseltamivir phosphate, largely relates to the development of a process-friendly route, which can directly impact the current supply problem.27 Roche has previously disclosed a highly efficient process scale route (12–13 steps, 39% overall yield) shown in Scheme 15A. However, this route has the Achilles heel of requiring a starting material, shikimic acid, 98, which is not readily available for manufacturing scale production. A number of subtle features, which are generally overlooked, become major players in the chemistry of drug production. Thus, as with most process routes to medicinal agents, the Roche process route is a masterpiece of design, engineering, and execution.
Scheme 15.
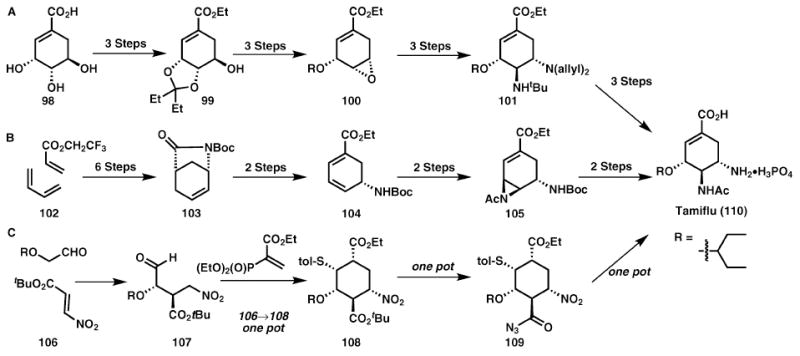
Addressing the supply problem of Tamiflu (110).
Following the potential for a life-threatening influenza epidemic in 2005, the global supply of Tamiflu came into question due to the limited available quantities of 98. While Corey’s alternative strategy27 reported in 2006, depicted in Scheme 15B is less efficient in terms of overall yield (22%), the greater availability of the starting materials makes this route more attractive. Another fantastic route reported in 2009 by Hayashi et al.27 requires only three pots from relatively cheap materials to afford the target in 57% overall yield. However within the context of process chemistry, the use of an acyl azide, 109, detracts somewhat from this otherwise brilliant approach, as these are potentially dangerous intermediates. Both of the syntheses in Schemes 15B and 15C have the clear advantage over Scheme 15A, as they are not limited by the availability of the starting materials. Several syntheses of Tamiflu have recently emerged and a very insightful review of their relative merits has been published.28
Perovskone (Majetich)
In a total synthesis of perovskone (115), Majetich demonstrates atom, step and redox economy with a spectacular biomimetic cascade (Scheme 16). After eleven steps Majetich obtains intermediate 111, which then undergoes a Diels–Alder cycloaddition with diene 112 and olefin isomerisation to arrive at 113. This tetracycle then participates in an ene reaction, followed by two acid-catalysed etherifications to afford the natural product, 115. While the details of the one-pot transformation have not yet been published, the two-pot procedure proceeds with excellent regio-, chemo-, and diastereoselectivity (74% overall). The use of an isohypsic cascade with no refunctionalisations necessary resulted in a highly efficient increase in molecular complexity.29
Scheme 16.
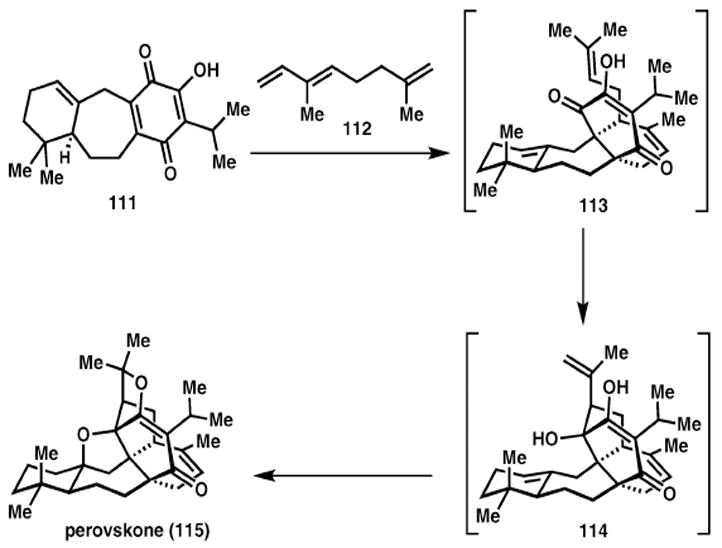
Majetich rapidly assembles the core of perovskone.
Suggested further reading
As the purpose of this review is to highlight the various aspects of the economies of synthesis in action, it cannot possibly be comprehensive. The interested reader can find several fine monographs on the topic of total synthesis for more examples that can be scrutinised through the economic “filters” delineated herein. Scheme 17 features a number of other excellent examples of syntheses that display striking levels of step, redox, or atom economy.30-37
Scheme 17.
More examples of economic total synthesis.
Conclusion
“If all the economists were laid end to end, they would not reach a conclusion”
– George Bernard Shaw
Indeed, the economies of synthesis are nice guidelines and conceptual frameworks for analysing and even designing syntheses, but that is all they are—apparent concepts to the seasoned practitioner. Thus, while all organic chemists can agree that the goal of minimising steps, waste, and redox fluctuations is an admirable one, the means to achieve this will always be the subject of debate and will always have more than one good answer. With the above quote in mind it can be said that this review does not have a truly satisfying conclusion, but instead these ideas are continuously re-evaluated during the act of organic synthesis. As the examples in this “tutorial” review demonstrate, it is the act of actual execution of such syntheses (i.e. aiming for the Hendrickson ideal) that will have the greatest impact on the practice of organic synthesis.
Biographies

Timothy Newhouse
Timothy Newhouse received his BA in Chemistry from Colby College in 2005 under the supervision of Professor Dasan M. Thamattoor. He is currently a graduate student studying the total synthesis of indole alkaloids with Professor Phil S. Baran at the Scripps Research Institute.

Phil S. Baran
Phil S. Baran was born in New Jersey in 1977 and received his undergraduate education from New York University with Professor David I. Schuster in 1997. After earning his PhD with Professor K. C. Nicolaou at the Scripps Research Institute in 2001 he pursued postdoctoral studies with Professor E. J. Corey at Harvard until 2003, at which point he began his independent career at Scripps, rising to the rank of Professor in 2008. His laboratory is dedicated to the study of fundamental organic chemistry through the auspices of natural product total synthesis.

Reinhard W. Hoffmann
Reinhard W. Hoffmann studied chemistry from 1951 to 1958 at the University of Bonn, finishing with a doctorate under the guidance of Professor B. Helferich. Two years of postdoctoral studies at the Pennsylvania State University were followed by a second postdoctorate with Professor G. Wittig at the University of Heidelberg. There he started his independent research that led to his habilitation in 1964. Three years later he was appointed as Dozent at the Technische Hochschule Darmstadt. Since 1970 he has held a position as professor of organic chemistry at the Universität Marburg (emeritus status since 2001).
Footnotes
Part of the rapid formation of molecular complexity in organic synthesis themed issue.
Contributor Information
Phil S. Baran, Email: pbaran@scripps.edu.
Reinhard W. Hoffmann, Email: rwho@chemie.uni-marburg.de.
References
- 1.(a) Nicolaou KC, Montagnon T. Molecules That Changed The World. Wiley-VCH; Weinheim: 2008. [Google Scholar]; (b) Corey EJ, Czakó B, Kürti L. Molecules and Medicine. John Wiley & Sons, Inc.; Hoboken: 2007. [Google Scholar]; (c) Nicolaou KC, Vourloumis D, Winssinger N, Baran PS. Angew Chem. 2000;112:46–126. Angew. Chem., Int. Ed., 2000, 39, 44–126. [PubMed] [Google Scholar]; (d) Nicolaou KC, Sorensen EJ. Classics in Total Synthesis. 1. Wiley-VCH; New York: 1996. [Google Scholar]; (e) Nicolaou KC, Snyder SA. Classics in Total Synthesis II. 1. Wiley-VCH; New York: 2003. [Google Scholar]
- 2.(a) Armstrong RW, Beau J-M, Cheon SH, Christ WJ, Fujioka H, Ham W-H, Hawkins LD, Jin H, Kang SH, Kishi Y, Martinelli MJ, McWhorther WW, Jr, Mizuno M, Nakata M, Stutz AE, Talamas FX, Taniguchi M, Tino JA, Ueda K, Uenishi J, White JB, Yonaga M. J Am Chem Soc. 1989;111:7530–7533. [Google Scholar]; (b) Suh EM, Kishi Y. J Am Chem Soc. 1994;116:11205–11206. [Google Scholar]
- 3.(a) Sheehan JC. The Enchanted Ring: The Untold Story of Penicillin. The MIT Press; Cambridge, MA, USA: 1982. [Google Scholar]; (b) Marx V. Chem Eng News. 2005;83(11):16–17. [Google Scholar]
- 4.(a) Wender PA, Croatt MP, Witulski B. Tetrahedron. 2006;62:7505–7511. [Google Scholar]; (b) Wender PA, Miller BL. Nature. 2009;460:197–201. doi: 10.1038/460197a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hendrickson JB. J Am Chem Soc. 1975;97:5784–5800. [Google Scholar]
- 6.(a) Hoffmann RW. Synthesis. 2006:3531–3541. [Google Scholar]; (b) Young IS, Baran PS. Nat Chem. 2009;1:193–205. doi: 10.1038/nchem.216. [DOI] [PubMed] [Google Scholar]; (c) Shenvi RA, O’Malley DP, Baran PS. Acc Chem Res. 2009;42:530–541. doi: 10.1021/ar800182r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corey EJ, Cheng X-M. The Logic of Chemical Synthesis. John Wiley & Sons, Inc.; New York: 1989. [Google Scholar]
- 8.Wilson RM, Danishefsky SJ. J Org Chem. 2007;72:4293–4305. doi: 10.1021/jo070871s. [DOI] [PubMed] [Google Scholar]
- 9.Wender PA, Verma VA, Paxton TJ, Pillow TH. Acc Chem Res. 2008;41:40–49. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- 10.Trost BM. Science. 1991;254:1471–1477. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]
- 11.Horváth IT, Anastas PT. Chem Rev. 2007;107:2167–2168. doi: 10.1021/cr078380v. [DOI] [PubMed] [Google Scholar]
- 12.Burns NZ, Baran PS, Hoffmann RW. Angew Chem. 2009;121:2896–2910. doi: 10.1002/anie.200806086. Angew. Chem., Int. Ed., 2009, 48, 2854–2867. [DOI] [PubMed] [Google Scholar]
- 13.(a) Heathcock CH. Angew Chem. 1992;104:675–691. Angew. Chem., Int. Ed. Engl., 1992, 31, 665. [Google Scholar]; (b) Piettre S, Heathcock CH. Science. 1990;248:1532–1534. doi: 10.1126/science.248.4962.1532. [DOI] [PubMed] [Google Scholar]
- 14.Li C, Lobkovsky E, Porco JA., Jr J Am Chem Soc. 2000;122:10484–10485. [Google Scholar]
- 15.Nicolaou KC, Lim YH, Papageorgiou CD, Piper JL. Angew Chem. 2005;117:8131–8135. doi: 10.1002/anie.200503678. Angew. Chem., Int. Ed., 2005, 44, 7917–7921. [DOI] [PubMed] [Google Scholar]
- 16.(a) Wender PA, Howbert JJ. Tetrahedron Lett. 1982;23:3983–3986. [Google Scholar]; (b) Mehta G, Murthy AN, Reddy DS, Reddy AV. J Am Chem Soc. 1986;108:3443–3452. [Google Scholar]
- 17.(a) Paquette LA, Annis GD. J Am Chem Soc. 1983;105:7358–7363. [Google Scholar]; (b) Paquette LA, Geng F. Org Lett. 2002;4:4547–4549. doi: 10.1021/ol020208k. [DOI] [PubMed] [Google Scholar]
- 18.Nicolaou KC, Edmonds DJ, Bulger PG. Angew Chem. 2006;118:7292–7344. doi: 10.1002/anie.200601872. Angew. Chem., Int. Ed., 2006, 45, 7134–7186. [DOI] [PubMed] [Google Scholar]
- 19.Davies HML, Hopper DW, Hansen T, Liu Q, Childers SR. Bioorg Med Chem Lett. 2004;14:1799–1802. doi: 10.1016/j.bmcl.2003.12.097. [DOI] [PubMed] [Google Scholar]
- 20.Matsumura Y, Kanda Y, Shirai K, Onomura O, Maki T. Org Lett. 1999;1:175–178. doi: 10.1021/ol9905046. [DOI] [PubMed] [Google Scholar]
- 21.(a) Yohimitsu T, Song JJ, Wang G-Q, Masamune S. J Org Chem. 1997;62:8978–8979. [Google Scholar]; (b) Fraunhoffer KJ, Bachovchin DA, White MC. Org Lett. 2005;7:223–226. doi: 10.1021/ol047800p. [DOI] [PubMed] [Google Scholar]
- 22.(a) Honda Y, Ori A, Tsuchihashi G. Chem Lett. 1987;16:1259–1262. [Google Scholar]; (b) Morrison KC, Litz JP, Scherpelz KP, Dossa PD, Vosburg DA. Org Lett. 2009;11:2217–2218. doi: 10.1021/ol900697w. [DOI] [PubMed] [Google Scholar]
- 23.(a) Nicolaou KC, Tria GS, Edmonds DJ. Angew Chem. 2008;120:1804–1807. doi: 10.1002/anie.200800066. Angew. Chem., Int. Ed., 2008, 47, 1780–1783. [DOI] [PubMed] [Google Scholar]; (b) Waalboer DCJ, Schaapman MC, van Delft FL, Rutjes FPJT. Angew Chem. 2008;120:6678–6680. Angew. Chem., Int. Ed., 2008, 47, 6576–6578. [Google Scholar]; (c) Tiefenbacher K, Mulzer J. Angew Chem. 2008;120:6294–6295. Angew. Chem., Int. Ed., 2008, 47, 6199–6200. [Google Scholar]; (d) Tiefenbacher K, Mulzer J. J Org Chem. 2009;74:2937–2941. doi: 10.1021/jo9001855. [DOI] [PubMed] [Google Scholar]
- 24.(a) Nakata T, Nomura S, Matsukura H, Morimoto M. Tetrahedron Lett. 1996;37:217–220. [Google Scholar]; (b) Holland JM, Lewis M, Nelson A. J Org Chem. 2003;68:747–753. doi: 10.1021/jo026456b. [DOI] [PubMed] [Google Scholar]
- 25.(a) Nagaoka H, Kishi Y. Tetrahedron. 1981;37:3873–3888. [Google Scholar]; (b) Harada T, Kagamihara Y, Tanaka S, Sakamoto K, Oku A. J Org Chem. 1992;57:1637–1639. [Google Scholar]; (c) Poss CS, Schreiber SL. Acc Chem Res. 1994;27:9–17. [Google Scholar]; (d) Magnuson SR. Tetrahedron. 1995;51:2167–2213. [Google Scholar]
- 26.(a) Paterson I, Anderson EA, Dalby SM, Loiseleur O. Org Lett. 2005;7:4121–4124. doi: 10.1021/ol051403c. [DOI] [PubMed] [Google Scholar]; (b) Paterson I, Anderson EA, Dalby SM, Lim JH, Maltas P, Moessner C. Chem Commun. 2006:4186–4188. doi: 10.1039/b612697a. [DOI] [PubMed] [Google Scholar]; (c) Paterson I, Anderson EA, Dalby SM, Lim JH, Genovino J, Maltas P, Moessner C. Angew Chem. 2008;120:3058–3062. doi: 10.1002/anie.200705566. Angew. Chem., Int. Ed., 2008, 47, 3016–3020. [DOI] [PubMed] [Google Scholar]; (d) Paterson I, Anderson EA, Dalby SM, Lim JH, Genovino J, Maltas Philip, Moessner C. Angew Chem. 2008;120:3063–3067. doi: 10.1002/anie.200705566. Angew. Chem., Int Ed., 2008, 47, 3021–3025. [DOI] [PubMed] [Google Scholar]
- 27.(a) Harrington PJ, Brown JD, Foderaro T, Hughes RC. Org Process Res Dev. 2004;8:86–91. [Google Scholar]; (b) Moscona A. N Engl J Med. 2005;353:1363–1373. doi: 10.1056/NEJMra050740. [DOI] [PubMed] [Google Scholar]; (c) Yeung Y-Y, Hong S, Corey EJ. J Am Chem Soc. 2006;128:6310–6311. doi: 10.1021/ja0616433. [DOI] [PubMed] [Google Scholar]; (d) Ishikawa H, Suzuki T, Hayashi Y. Angew Chem. 2009;121:1330–1333. doi: 10.1002/anie.200804883. Angew. Chem. Int. Ed., 2009, 48, 1304–1307. [DOI] [PubMed] [Google Scholar]; (e) Andraos J. Org Process Res Dev. 2009;13:161–185. [Google Scholar]
- 28.Magano J. Chem Rev. 2009;109 doi: 10.1021/cr800449m. [DOI] [PubMed] [Google Scholar]
- 29.(a) Majetich G, Zhang Y. J Am Chem Soc. 1994;116:4979–4980. [Google Scholar]; (b) Majetich G, Wang Y, Li Y, Vohs JK, Robinson GH. Org Lett. 2003;5:3847–3850. doi: 10.1021/ol035380i. [DOI] [PubMed] [Google Scholar]; (c) Hudlicky T, Reed JW. The Way of Synthesis. ch. 3. Wiley-VCH; Weinheim: 2007. pp. 485–490. [Google Scholar]
- 30.Hu Q-Y, Rege PD, Corey EJ. J Am Chem Soc. 2004;126:5984–5986. doi: 10.1021/ja048808x. [DOI] [PubMed] [Google Scholar]
- 31.Kwon S, Myers AG. J Am Chem Soc. 2005;127:16796–16797. doi: 10.1021/ja056206n. [DOI] [PubMed] [Google Scholar]
- 32.Fürstner A, Radkowski K, Peters H. Angew Chem. 2005;117:2837–2841. doi: 10.1002/anie.200462215. Angew. Chem., Int. Ed., 2005, 44, 2777–2781. [DOI] [PubMed] [Google Scholar]
- 33.(a) Nicolaou KC, Vassilikogiannakis G, Mägerlein W, Kranich R. Angew Chem. 2001;113:2543–2547. doi: 10.1002/1521-3773(20010702)40:13<2482::AID-ANIE2482>3.0.CO;2-A. Angew. Chem., Int. Ed., 2001, 40, 2482–2486. [DOI] [PubMed] [Google Scholar]; (b) Davies HML, Dai X, Long MS. J Am Chem Soc. 2006;128:2485–2490. doi: 10.1021/ja056877l. [DOI] [PubMed] [Google Scholar]
- 34.Min and S-J, Danishefsky SJ. Angew Chem. 2007;119:2249–2252. doi: 10.1002/anie.200605058. Angew. Chem., Int. Ed., 2007, 46, 2199–2202. [DOI] [PubMed] [Google Scholar]
- 35.Baxendale IR, Ley SV, Piutti C. Angew Chem. 2002;114:2298–2301. doi: 10.1002/1521-3773(20020617)41:12<2194::aid-anie2194>3.0.co;2-4. Angew. Chem., Int. Ed., 2002, 41, 2194–2197. [DOI] [PubMed] [Google Scholar]
- 36.Wasserman HH, DeSimone RW, Boger DL, Baldino CM. J Am Chem Soc. 1993;115:8457–8458. [Google Scholar]
- 37.Nicolaou KC, Sarlah D, Shaw DM. Angew Chem. 2007;119:4792–4795. doi: 10.1002/anie.200701552. Angew. Chem., Int. Ed., 2007, 46, 4708–4711. [DOI] [PubMed] [Google Scholar]