

Abstract
Although it is well established that low-voltage-activated T-type Ca2+ channels play a key role in many neurophysiological functions and pathological states, the lack of selective and potent antagonists has so far hampered a detailed analysis of the full impact these channels might have on single-cell and neuronal network excitability as well as on Ca2+ homeostasis. Recently, a novel series of piperidine-based molecules has been shown to selectively block recombinant T-type but not high-voltage-activated (HVA) Ca2+ channels and to affect a number of physiological and pathological T-type channel-dependent behaviors. Here we directly show that one of these compounds, 3,5-dichloro-N-[1-(2,2-dimethyl-tetrahydro-pyran-4-ylmethyl)-4-fluoro-piperidin-4-ylmethyl]-benzamide (TTA-P2), exerts a specific, potent (IC50 = 22 nm), and reversible inhibition of T-type Ca2+ currents of thalamocortical and reticular thalamic neurons, without any action on HVA Ca2+ currents, Na+ currents, action potentials, and glutamatergic and GABAergic synaptic currents. Thus, under current-clamp conditions, the low-threshold Ca2+ potential (LTCP)-dependent high-frequency burst firing of thalamic neurons is abolished by TTA-P2, whereas tonic firing remains unaltered. Using TTA-P2, we provide the first direct demonstration of the presence of a window component of Ca2+ channels in neurons and its contribution to the resting membrane potential of thalamic neurons and to the Up state of their intrinsically generated slow (<1 Hz) oscillation. Moreover, we demonstrate that activation of only a small fraction of the T-type channel population is required to generate robust LTCPs, suggesting that LTCP-driven bursts of action potentials can be evoked at depolarized potentials where the vast majority of T-type channels are inactivated.
Introduction
Since their discovery (Carbone and Lux, 1984; Nowycky et al., 1985), T-type Ca2+ channels have been implicated in many physiological and pathological brain states, including sleep (Contreras, 2006; Crunelli et al., 2006), nociception (Todorovic et al., 2001; Kim et al., 2003; Bourinet et al., 2005), and epilepsy (Tsakiridou et al., 1995; Kim et al., 2001; Crunelli and Leresche, 2002b; Vitko et al., 2007; Powell et al., 2009). From a mechanistic perspective, the low-threshold Ca2+ potential (LTCP), and associated high-frequency burst of action potentials, has become the hallmark for the contribution of T-type Ca2+ channels to single-cell and neuronal network excitability in different brain regions, including thalamus (Llinas and Jahnsen, 1982), neocortex (McCormick et al., 1985), and cerebellum (Llinas and Muhlethaler, 1988). However, to limit the function of T-type Ca2+ channels to LTCP generation might lead to underestimating their potential roles in (patho)physiology. Thus, the window component of the T-type Ca2+ current (IT), which relies on the few channels that are constantly opened at approximately −60 mV, could contribute to the resting membrane potential and has been suggested to be essential for the slow (<1 Hz) sleep oscillations of thalamic neurons (Hughes et al., 2002). Moreover, since T-type channels are highly expressed in dendrites (Williams and Stuart, 2000; Joksovic et al., 2005b; McKay et al., 2006; Diana et al., 2007), they could, even in the absence of an LTCP, be involved in signal amplification (Markram and Sakmann, 1994; Magee et al., 1995; Tsay et al., 2007) and synaptic plasticity (Oliet et al., 1997; Aizenman et al., 1998). In addition, the mechanism underlying LTCP-driven action potential bursts during sensory processing in the awake state (i.e., when only a small number of T-type channels is available for activation) is still unclear (Llinas and Steriade, 2006). Conclusive answers to these questions require a potent, specific, and reversible block of T-type Ca2+ channels. Whereas antisense strategies (Lambert et al., 1998; Bourinet et al., 2005) and knock-out mice (Kim et al., 2001; Anderson et al., 2005) provide such a specific block, the lack of internal controls, the variable efficiency of the antisense knockdown, and the compensatory mechanisms that may develop in knock-out mice compromise the interpretation of the resulting data. Similarly, the usefulness of the available pharmacological approaches (e.g., mibefradil, Ni2+, ethosuximide, etc.) is limited by their poor potency and selectivity (McGivern, 2006).
Here we show that 3,5-dichloro-N-[1-(2,2-dimethyl-tetrahydro-pyran-4-ylmethyl)-4-fluoro-piperidin-4-ylmethyl]-benzamide (TTA-P2), a novel piperidine derivative that inhibits recombinant T-type Ca2+ channels (Shipe et al., 2008), induces a potent, specific, and reversible block of native IT, LTCPs, and their associated high-frequency burst firing in thalamic neurons. We also provide the first direct demonstration of the contribution of the window component of IT (ITwindow) to the resting membrane potential and the slow (<1 Hz) oscillations of thalamic neurons. Moreover, we demonstrate that activation of only a small fraction of the T-type channel population is required to generate robust LTCPs, suggesting that LTCP-driven bursts of action potentials can be evoked at depolarized potentials where the vast majority of T-type channels are inactivated.
Materials and Methods
All procedures involving experimental animals were performed in accordance with the European Union Council Directive 86-609, the United Kingdom Animals (Scientific Procedures) Act 1986, and local ethics committee guidelines. All efforts were made to minimize animal suffering and the number of animals used.
Rat and mouse slice preparation and recordings.
Brains were excised from 5- to 20-d-old Wistar rats or 15- to 30-d-old C57Blc6 CaV3.1−/− and wild-type mice (Kim et al., 2001). A block of tissue containing the thalamus was removed and placed in a cold (<4°C) oxygenated (95% O2/5% CO2) solution of artificial CSF (aCSF; in mm): 125 NaCl, 2.5 KCl, 0.4 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, 20 glucose, 5 Na-pyruvate, and 1 kynurenic acid (pH 7.3; osmolarity, 310 mOsm). The block of tissue was glued, ventral surface uppermost, to the stage of a vibroslice (HM650V; Microm), and 200- to 280-μm-thick horizontal slices containing the ventrobasal nucleus (VB) and the nucleus reticularis thalami (NRT) were prepared using the internal capsule and the medial lemniscus as landmarks. Slices (three to four per hemisphere) were stored in an oxygenated incubation chamber containing aCSF of the above composition, but without kynurenic acid and with 2 mm CaCl2, for at least 1 h before being transferred to the recording chamber, where they were perfused (1.5 ml/min) continuously with an oxygenated recording solution of the same composition. Experiments were conducted either at room temperature for voltage-clamp recordings or at 32°C for current-clamp recordings.
Using the patch-clamp technique (Optopatch amplifier, Cairn; Clampex 10, Molecular Devices), whole-cell recordings were performed in neurons of the VB or NRT visualized under Nomarski optics (60× lens; BX51WI; Olympus). Current and membrane potential recordings were filtered by a four-pole Bessel filter set at a corner frequency of 2 kHz, digitalized at 10 kHz, and later analyzed using Igor 6 (Wavemetrics).
Except when specifically mentioned, Ca2+ currents were recorded in 7- to 10-d-old rats. For voltage-clamp experiments, electrodes were filled with the following solution: 110 mm CsCl, 1 mm CaCl2, 5 mm MgCl2, 10 mm EGTA, 10 mm HEPES, 4 mm Na-ATP, 15 mm phosphocreatine, and 50 U/ml creatine phosphokinase (pH 7.3; osmolarity, 305 mOsm). To isolate Ca2+ currents, the following were added to the extracellular solution: 10 mm tetraethylamoniumchloride, 1 mm 4-aminopyridine, 0.5 μm tetrodotoxin, and 2 mm CsCl. ITwindow recording was obtained with a similar extracellular solution, except for the addition of 1 μm nifedipine to suppress the small high-voltage-activated (HVA) currents evoked at the end of the slow depolarizing ramp. Leak currents were subtracted off-line using templates constructed from the current induced by a 10 mV hyperpolarizing step applied from −100 mV at the end of each recording. In every experiment, values of access resistance ranged from 4 to 6 MΩ. At least 70% of the cell capacitance and series resistance were compensated.
Current-clamp recordings were performed in 8- to 20-d-old rats and 15- to 30-d-old mice. To record membrane potential, electrodes were filled with the following solution: 140 mm potassium-methanesulfonate, 0.1 mm CaCl2, 5 mm MgCl2, 1 mm EGTA, 10 mm HEPES, 4 mm Na-ATP, 15 mm phosphocreatine, and 50 U/ml creatine phosphokinase (pH 7.3; osmolarity, 290 mOsm). Access resistance was monitored by measuring responses to small hyperpolarizing current pulses from resting membrane potential, and only data obtained from pipettes with access resistances of 9–13 MΩ with <20% change during the experiment were included in this study. When required to analyze the ITwindow contribution to the resting membrane potential, a depolarizing DC of 75 ± 29 pA (n = 11) and 85 ± 17 pA (n = 6) was used to set the membrane potential of thalamocortical (TC) and nucleus reticularis neurons to −60 mV from their original resting membrane potential of −74 ± 4 mV and −85 ± 17 mV, respectively.
Synaptic responses were induced using a glass pipette (6–8 μm tip diameter) filled with the extracellular medium. This stimulating electrode was positioned 25–60 μm from the recorded neuron to evoke IPSPs/IPSCs or in the medial lemniscus to evoke sensory EPSPs/EPSCs. The stimulus consisted of a 20–200 μs pulse of 10–40 V amplitude. Isolation of GABAA IPSPs/IPSCs and glutamatergic EPSPs/EPSCs was achieved by the addition of 50 μm dl-APV plus 10 μm CNQX and 1 μm SR95531, respectively, to the perfusion medium. Synaptic currents were recorded with electrodes filled with solutions of the following compositions (in mm): for IPSC, 128 potassium-methanesulfonate, 12 CsCl, 0.5 EGTA, 10 HEPES, 3 MgCl2, 0.1 CaCl2, 4 Na-ATP, 0.4 Na-GTP, and 15 phosphocreatine; for EPSC, 150 KCl, 1 EGTA, 10 HEPES, 4.6 MgCl2, 0.1 CaCl2, 4 Na-ATP, and 0.4 Na-GTP.
Cat slice preparation and recordings.
Slices of cat thalamus were prepared as described previously (Hughes et al., 2002). Briefly, 6- to 8-week-old cats were deeply anesthetized with a mixture of O2 and NO2 (2:1) and 5% isoflurane, a wide craniotomy was performed, and the brain was removed. Sagittal slices of the thalamus (300–400 μm), including the medial geniculate body (MGB), were prepared and incubated at 35°C for 1 h before being maintained at room temperature. For recording, slices were transferred to a submersion chamber on the stage of an Olympus BX61WI microscope and perfused continuously with gassed (95% O2/5% CO2) aCSF containing (in mm) 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 25 NaHCO3, and 25 d-glucose (pH 7.3; 305 mOsm) at 34 ± 0.2°C. Somatic whole-cell patch-clamp recordings (3–5 MΩ) were performed (Multiclamp 700B; Molecular Devices) on visually identified MGB neurons (IR-Dodt contrast videomicroscopy; Luigs & Neumann) with an internal pipette solution containing (in mm) 115 potassium-gluconate, 20 KCl, 2 Mg-ATP, 2 Na-ATP, 0.3 Na-GTP, 10 Na-phosphocreatine, and 10 HEPES (pH 7.3; 290 mOsm), supplemented with Alexa Fluor 594 (50 μm). Slow (<1 Hz) sleep oscillations were induced by bath application of 50 μm (±)-1-aminocyclopentane-trans-1,3-dicarboxylic acid (trans-ACPD) (Hughes et al., 2002), followed by small steady-state current injection (80–150 pA) to bring the neuron into the voltage region of expression of these oscillations. Data were acquired using pClamp 10 software (Molecular Devices), digitized at 20 kHz, and filtered at 5 kHz. At the end of each experiment, a z-series (1 μm) of two-photon excitation fluorescence images (512 × 512 pixels, 400× magnification) was captured using a Prairie Ultima II two-photon microscope (Prairie Technologies) fitted with a Chameleon Ultra II laser (Coherent) tuned to 810 nm (<4 mW power at sample). A maximum-intensity z-series projection was then constructed off-line using MetaMorph 7.1 (Molecular Devices) to confirm the TC neuron morphology of the recorded cell.
Drugs and data analysis.
TTA-P2 [compound (S)-5 in Fig. 3 of Shipe et al. (2008); Merck] was made up as a 10 mm stock solution in dimethylsulfoxide (DMSO), kept at −20°C until use, and diluted for experiments so that the final concentration of DMSO was ≤0.03%.
Figure 3.
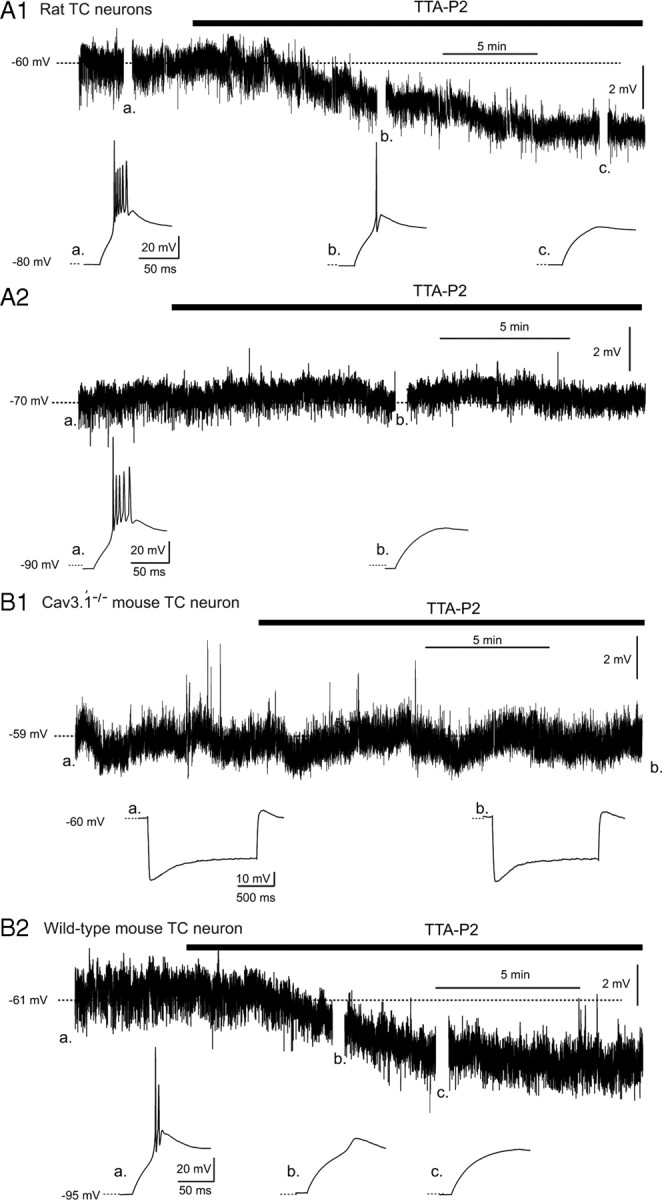
Effect of TTA-P2 on the resting membrane potential of TC neurons. A1, TTA-P2 (1 μm) induces a small hyperpolarization in a Wistar rat TC neuron held at −60 mV (top trace), concomitantly with the block of the LTCP evoked at the end of a hyperpolarizing step (bottom traces, with a–c marking the corresponding breaks in the top trace). A2, TTA-P2 (1 μm) does not hyperpolarize a TC neuron held at −70 mV (top trace) but still blocks the rebound LTCP (bottom traces, with a and b marking the corresponding breaks in the top trace). B1, TTA-P2 (1 μm) has no effect on the membrane potential of a TC neuron from a Cav3.1−/− mouse recorded at −59 mV. Note the absence of a rebound LTCP after the hyperpolarizing steps and the lack of effect of TTA-P2 on the depolarizing sag of the hyperpolarizing response. B2, TTA-P2 (1 μm) induces a small hyperpolarization in a TC neuron from a wild-type mouse held at −61 mV (top trace), concomitantly with the block of the rebound LTCP (bottom traces, with a–c marking the corresponding breaks in the top trace).
TTX was obtained from Latoxan, dl-APV was from Fluka, and CNQX was from Tocris. All other chemicals were purchased from Sigma.
Concentration–response curves were generated from the percentage reduction in the peak T-type Ca2+ current amplitude evoked at −50 mV. Quantitative comparison of action potential characteristics (threshold, amplitude, half-width) was performed on the first action potential of the spike trains evoked by a 200 pA depolarizing step from a holding potential of −60 mV. Quantitative data in the text and figures are given as mean ± SD.
Results
Effect of TTA-P2 on TC neurons
T-type Ca2+ currents (IT) and HVA Ca2+ currents, similar to those described previously (Guyon and Leresche, 1995), were evoked in TC neurons of the VB before, during, and after bath application of TTA-P2. As illustrated in Figure 1, A and B, application of 1 and 3 μm TTA-P2 resulted in a full block (96 ± 1%) of IT (control, 1135 ± 306 pA; TTA-P2, 45 ± 21 pA; p < 0.001, paired Student's t test; n = 7), whereas the compound had no effect on the amplitude of the HVA Ca2+ currents (control, 1182 ± 211 pA; TTA-P2, 1072 ± 177 pA; p > 0.05, paired Student's t test; n = 7) recorded in the same TC neurons. The maximal effect was achieved within 3–4 min from the beginning of TTA-P2 application (Fig. 1A2), and a full recovery of IT could be observed after ∼50 min of wash-out (Fig. 1B). The block of IT by TTA-P2 was dose dependent with an IC50 of 22 nm (Fig. 1C). A maximal reduction of IT by TTA-P2 could be induced even in the absence of channel activation (Fig. 1D), indicating the absence of a use-dependent block. I–V relationships were constructed from IT evoked in control conditions and in the presence of 25 nm TTA-P2 using slices obtained from very young animals [postnatal day 5 (P5) to P6] to optimize the voltage control. TTA-P2 induced a clear decrease in current amplitude with no apparent shift in the voltage dependence of channel activation and steady-state inactivation (Fig. 1E) (n = 5). Therefore, we conclude that the TTA-P2-induced decrease of IT is not attributable to a significant shift in the activation/inactivation properties of the current.
Figure 1.

TTA-P2 fully and reversibly blocks T-type Ca2+ current without affecting HVA Ca2+ currents in VB TC neurons. A, IT were evoked every 20 s by step depolarization (200 ms long) to −50 mV after a 1 s hyperpolarizing prepulse to −100 mV. HVA Ca2+ currents were similarly evoked by step depolarization to 10 mV from the −60 mV holding potential. A1, Traces illustrate IT (●, top traces) and HVA Ca2+ currents (○, bottom traces) recorded in the same VB neuron under control condition (a) and in the presence of 3 μm TTA-P2 (b, 2 min application; c, 5 min application). A2, The peak amplitude of IT (●) and HVA Ca2+ currents (○) are plotted against time. TTA-P2 produced a 95% block of IT without any effect on the HVA Ca2+ currents. B1, B2, Same protocols as in A1 and A2 in a different VB TC neuron. A full recovery of the amplitude of IT was obtained after 55 min of wash-out of 1 μm TTA-P2. C, Dose–response curve of the effect of TTA-P2 on the amplitude of IT. Data were fitted with the following equation: y = ymax/(1 + IC50/x)n, where IC50 = 22 nm, ymax = 94.5%, and n = 1.2. D, Same protocols as in A1 and A2. Interruption of the stimulating protocols during the first 8 min of 1 μm TTA-P2 application did not preclude the block of IT. E, Activation and inactivation properties of IT were estimated in the same neuron under control condition and after 20 min of 25 nm TTA-P2 application. The neuron was maintained at −60 mV between protocols. E1, I–V curves were constructed by successive step depolarizations from −80 to −45 mV (2.5 mV increments) preceded by a 1 s hyperpolarizing prepulse to −100 mV. Left traces illustrate currents evoked at the various potentials under control conditions (○) and in the presence of TTA-P2 (●). Despite the strong decrease in current amplitude induced by TTA-P2 application, the apparent voltage dependence of channel activation appears similar in control condition (○) and in the presence of TTA-P2 (●). E2, Normalized steady-state inactivation curves. Inactivation of T-type Ca2+ channels was induced using a 1 s prepulse of increasing potential (from −100 to −60 mV with 2.5 mV increments), and the resulting channel availability was estimated from the normalized current amplitude measured at −50 mV. Note the lack of any significant change in the presence of TTA-P2.
Precise measurement of the voltage dependence of Cav3. channel activation and inactivation in recombinant systems demonstrated that in a narrow range of voltages where T-type channels can open, a fraction of the channel population is not inactivated, therefore generating a small tonic current called the window current (ITwindow) (Perez-Reyes, 2003). To test the ability of TTA-P2 to suppress ITwindow, experiments were performed on older rats (P19–P20) that express a higher density of T-type channels and therefore display an ITwindow of measurable amplitude. ITwindow was evoked using voltage ramps slow enough to achieve steady-state equilibrium between activation and inactivation of the channels during depolarization (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). ITwindow evoked with 10-s-long ramps from −100 to −40 mV had a mean amplitude of 26 ± 7 pA (n = 5) and was fully blocked by 1 μm TTA-P2 (Fig. 2), similarly to the results obtained for the transient component of IT.
Figure 2.

Effect of TTA-P2 on ITwindow of VB TC neurons. A1, The window T-type Ca2+ current evoked by a 10-s-long depolarizing voltage ramp from −100 to −40 mV preceded by a 1 s hyperpolarizing prepulse to −100 mV is fully blocked by 1 μm TTA-P2. A2, The voltage dependence of ITwindow is presented. Note that the maximum amplitude of the current occurs around −63 mV.
Having established that TTA-P2 potently blocks both the transient and tonic components of IT in TC neurons without affecting the HVA Ca2+ currents, we then investigated its action on neuronal excitability. Under current-clamp conditions, we observed that 1–3 μm TTA-P2 produced a 3.1 ± 0.5 mV (n = 11) hyperpolarization of TC neurons held at −60 mV (Fig. 3A1) that could be compensated by injecting a depolarizing DC of 23 ± 5 pA (n = 11), a value very similar to the amplitude of ITwindow (26 ± 7 pA) directly measured under voltage-clamp conditions (Fig. 2). Such hyperpolarization was not observed when the neurons were held at −70 mV (Fig. 3A2) (i.e., outside the voltage range of ITwindow) (Perez-Reyes, 2003). To confirm that the TTA-P2-induced hyperpolarization resulted from the T-type channel block, and not from unspecific actions on other membrane channels, similar experiments were performed in Cav3.1−/− mice TC neurons that lack IT (Kim et al., 2001). Indeed, no hyperpolarization was observed in Cav3.1−/− TC neurons during wash-in of TTA-P2 (n = 6) (Fig. 3B1), whereas in TC neurons from wild-type mice, TTA-P2 induced a membrane hyperpolarization of 3.4 ± 0.4 mV (n = 5) (Fig. 3B2). As an additional control, we tested the effect of TTA-P2 on Cav3.1−/− NRT neurons, which, because of their selective expression of Cav3.2 and Cav3.3 channels, show a robust LTCP (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). As expected, TTA-P2 did hyperpolarize NRT neurons of Cav3.1−/− mice (3.2 ± 0.6 mV; n = 4) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Together, these data demonstrate that the TTA-P2-induced hyperpolarization is caused by the block of ITwindow and indicate a contribution of this current to the resting membrane potential of TC neurons, as suggested previously on the basis of indirect evidence (Crunelli et al., 2005). Therefore, in all following experiments under the current-clamp condition, the TTA-P2-induced hyperpolarization was compensated by steady-current injection when required.
TTA-P2 at concentrations that abolished IT (1 or 3 μm) fully blocked the LTCP evoked after a hyperpolarizing step (1.5 s long at −100 mV; holding potential, −60 mV), that deinactivated the entire T-type channel population (n = 11) (Fig. 4A; see also Fig. 3). As shown in Figure 4Ba, TTA-P2 did not affect the depolarizing sag associated with these transient hyperpolarizations, suggesting a lack of effect on the h current.
Figure 4.
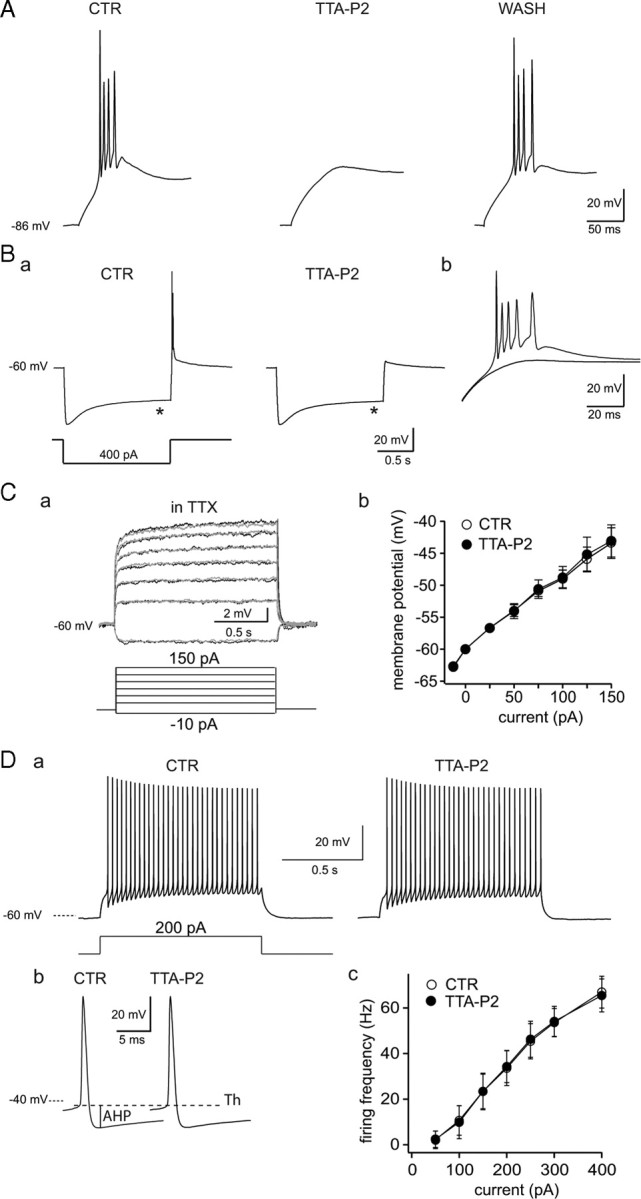
Effect of TTA-P2 on rat TC neuron excitability. A, Typical example of the reversible block by TTA-P2 (3-min-long application, 1 μm) of the rebound LTCP evoked at the end of a 1.5-s-long hyperpolarizing step in a rat TC neuron. Ba, The block of the rebound LTCP by 1 μm TTA-P2 was not associated with a change in the depolarizing sag (*) of the hyperpolarizing response to an intracellular current step. Bb, The membrane potential responses observed after repolarization in both conditions are superimposed at a larger time scale. Ca, Superimposed voltage traces illustrate the lack of effect of 1 μm TTA-P2 on membrane potential steps induced by injection of depolarizing currents (control, gray traces; TTA-P2, black traces). Cb, An I–V plot for similar experiments as in a from four TC neurons. Da, Typical example of the tonic firing observed in control condition and after 1 μm TTA-P2 application (same cell as in B). A similar number of action potentials (n = 32) is evoked during a 200 pA step depolarization in both control and TTA-P2 conditions. The first action potential recorded in control and in TTA-P2 conditions are enlarged in Db. Note the lack of effect of TTA-P2 on the threshold (Th), amplitude, half-width, and afterhyperpolarization (AHP) of the action potential. The graph in Dc presents the frequency–current plot from six neurons. CTR, Control.
Except for the small fraction that generates the window current, T-type Ca2+ channels are inactivated at −60 mV (Perez-Reyes, 2003). Thus, they should not contribute to the neuronal responses evoked by step depolarization from membrane potentials > −60 mV, and hence selective block of T-type channels should have no effect under these experimental conditions. Having established that 1 μm TTA-P2 in the presence of TTX had no effect on the input resistance of TC neurons when assessed with depolarizing current pulses of increasing intensities (Fig. 4C), the properties of single action potentials and tonic firing were compared in the control condition and in the presence of the antagonist. Application of 1 μm TTA-P2 had no effect on the threshold (control, −45.9 ± 2.6 mV; TTA-P2, −45.8 ± 2.6 mV; p > 0.05, paired Student's t test; n = 6), amplitude (control, 74 ± 6.6 mV; TTA-P2, 73.7 ± 6.6 mV; p > 0.05, paired Student's t test; n = 6), half-width (control, 0.6 ± 0.1 ms; TTA-P2, 0.6 ± 0.1 ms; p > 0.05, paired Student's t test; n = 6), and afterhyperpolarization (control, 13.1 ± 2.2 mV; TTA-P2, 13.3 ± 1.8 mV; p > 0.05, paired Student's t test; n = 6) of the action potential (Fig. 4Db), or on the frequency of tonic firing (Fig. 4Da,Dc). These results are strengthened by the lack of effect of 1 μm TTA-P2 on both the fast and persistent Na+ currents underlying tonic action potential firing (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Together, therefore, these data confirm the selectivity of TTA-P2, indicating a lack of action on tonic firing and apparent input resistance but a potent effect on T-type Ca2+ channel-dependent LTCP-mediated burst firing of TC neurons.
Effect of TTA-P2 on NRT neurons
Since TC neurons have been shown to specifically express the Cav3.1 T-type channel isoform, whereas the GABAergic neurons of NRT express both Cav3.2 and Cav3.3 isoforms (Talley et al., 1999), the effect of TTA-P2 was tested on IT of NRT neurons. Confirming that TTA-P2 potently interacts with all three T-type Ca2+channel isoforms (Shipe et al., 2008), a 97 ± 1% (n = 6) decrease in the amplitude of IT recorded in rat NRT neurons was observed after application of 1 μm TTA-P2, whereas the HVA Ca2+ current in the same neurons was unaffected (Fig. 5A).
Figure 5.
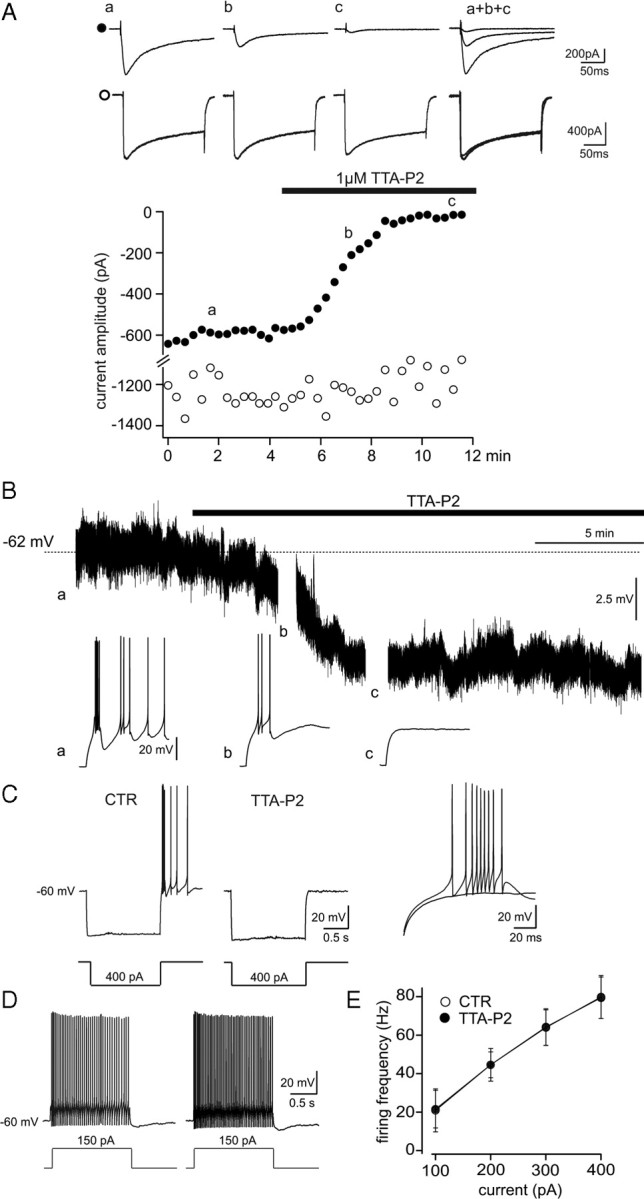
Effect of TTA-P2 on NRT neurons. A, Same protocol as in Figure 1A was used to record the IT (●, top traces) and HVA Ca2+ currents (○, bottom traces) in an NRT neuron. TTA-P2 (1 μm) produced a 97% block of IT without a concomitant decrease in the amplitude of the HVA Ca2+ current. B, Continuous voltage record from an NRT neuron showing that 1 μm TTA-P2 induces a small hyperpolarization concomitantly with the block of the rebound LTCP evoked by an hyperpolarizing step (breaks in the trace) before (a) and after a 4 and 8 min wash-in (b, c) of the antagonist. C, D, Same protocols as in Figure 4, B and D, were used to study the effect of 1 μm TTA-P2 on NRT neuron excitability. C, The block of the rebound LTCP by TTA-P2. E, The graph presents the frequency–current plot from nine neurons. CTR, Control.
Current-clamp experiments showed that application of TTA-P2 hyperpolarized NRT neurons held at −60 mV by 5 ± 2.2 mV (n = 6) (Fig. 5B). Such hyperpolarization was compensated by injection of a 30 ± 1 pA depolarizing DC (n = 6). Similarly to what had been observed in TC neurons, TTA-P2 suppressed the somatically evoked LTCPs (Fig. 5C) but did not affect the tonic firing frequency (Fig. 5D,E) or action potential properties: threshold (control, −47.7 ± 3.6 mV; TTA-P2, −48.1 ± 2.5 mV; p > 0.05, paired Student's t test; n = 5), amplitude (control, 79.3 ± 5.6 mV; TTA-P2, 83.1 ± 4.7 mV; p > 0.05, paired Student's t test; n = 5), half-width (control, 0.3 ± 0.1 ms; TTA-P2, 0.3 ± 0.1 ms; p > 0.05, paired Student's t test; n = 5), and afterhyperpolarization (control, 19.6 ± 3.2 mV; TTA-P2, 19.6 ± 2.3 mV; p > 0.05, paired Student's t test; n = 5).
Effect of TTA-P2 on synaptic responses and intrinsic oscillations
Having established the selectivity and potency of TTA-P2 and its ability to block LTCPs without any effect on single or tonic action potential firing evoked by somatic current injections, we then started to investigate its potential use in network excitability by testing its action on excitatory and inhibitory synaptic currents. As shown in Figure 6Aa, perfusion of the slice with TTA-P2 (1 μm) did not change the amplitude (control, 193 ± 14 pA; TTA-P2, 192 ± 6 pA; p > 0.05, paired Student's t test; n = 5), rise time (10–90% rise time; control, 1.8 ± 0.2 ms; TTA-P2, 1.6 ± 0.1 ms; p > 0.05, paired Student's t test; n = 5), and decay (control, τ1 = 8.3 ± 0.8 ms, τ2 = 43.5 ± 12.4 ms; TTA-P2, τ1 = 7.9 ± 0.7 ms, τ2 = 44.6 ± 14.3 ms; p > 0.05, paired Student's t test; n = 5) of the GABAA receptor-mediated IPSCs evoked in TC neurons by electrical stimulation of the NRT. Similarly, 1 μm TTA-P2 had no effect on the amplitude (control, 415 ± 17 pA; TTA-P2, 413 ± 16 pA; p > 0.05, paired Student's t test; n = 5), rise time (10–90% rise time; control, 0.6 ± 0.05 ms; TTA-P2, 0.5 ± 0.05 ms; p > 0.05, paired Student's t test; n = 5), and decay (control, τ1 = 1.7 ± 0.5 ms; τ2 = 9.3 ± 2.5 ms; TTA-P2, τ1 = 1.9 ± 0.4 ms, τ2 = 8.9 ± 1.8 ms; p > 0.05, paired Student's t test; n = 5) of the glutamatergic EPSCs evoked by stimulation of the lemniscal afferents to TC neurons (Fig. 6Ab). Since this novel piperidine derivative had no effect on GABAergic and glutamatergic synaptic currents, we then investigated its action on the characteristic burst firing evoked by these two types of synaptic potentials. As shown in Figure 6B, application of TTA-P2 at 20 nm (IC50) delayed the occurrence of the rebound LTCP evoked by the NRT-elicited IPSP without affecting its associated burst firing (n = 5). This result provides the first demonstration that IT participates in the early phase of membrane repolarization after an IPSP and, as a consequence, that the kinetics of this repolarization is highly sensitive to the number of T-type Ca2+ channels available for activation. A full suppression of the rebound LTCP could only be achieved using 50 nm TTA-P2 (n = 5), a concentration that blocks 70% of IT (see dose–response curve in Fig. 1C). The need to block a large proportion of IT to substantially affect synaptically evoked LTCPs was further emphasized by similar experiments on the EPSPs. Indeed, since LTCPs triggered by glutamatergic EPSPs have been shown to occur during tonic or transient hyperpolarization in TC neurons (Steriade et al., 1997), we tested the effect of TTA-P2 on the sensory EPSP evoked by stimulation of the lemniscal fibers in TC neurons maintained at slightly hyperpolarized potentials (−70 mV) (Fig. 6C). In this condition, a concentration of TTA-P2 (50 nm) that blocks 70% of IT was necessary to decrease the number of action potentials in the LTCP burst, but robust burst firing was still present when only 20% of the T channels were available (i.e., in the presence of 100 nm TTA-P2) (Fig. 6C). The full block of the LTCP could only be achieved using 1 μm (n = 5) (Fig. 6C). Together, these results indicate that the number of T-type Ca2+ channels expressed in TC neurons widely exceeds the number of channels required to elicit an LTCP by synaptic potentials.
Figure 6.

Effect of TTA-P2 on synaptically evoked LTCPs. A, Lack of effect of 1 μm TTA-P2 on evoked IPCSs (a; averages of 8 IPSCs) and EPSCs (b; averages of 7 EPSCs) recorded from two TC neurons. B, In a TC neuron maintained at −60 mV, rebound LTCPs were evoked by stimulations of the NRT at 20 s intervals in control condition and during successive applications of 20 and 50 nm TTA-P2. Ba, A concentration of 20 nm TTA-P2 had no effect on the high-frequency firing, whereas 50 nm TTA-P2 fully blocked the LTCP. However, as clearly seen on the superimposed traces in Bb, 20 nm TTA-P2 slowed the membrane repolarization and delayed the LTCP occurrence by 110 ms. C, In a TC neuron maintained at −70 mV, LTCPs were evoked by repetitive stimulations of the lemniscal pathway every 20 s under control conditions and while successively applying increasing concentrations of TTA-P2 (20, 50, and 100 nm and 1 μm). Note that a reduction in the LTCP-associated firing is observed with TTA-P2 concentrations ≥50 nm, which block >70% of the IT (see Fig. 1C). The full block of the LTCP evoked by the sensory EPSP required a TTA-P2 concentration that abolishes IT (i.e., 1 μm).
TC neurons are capable of generating intrinsic membrane potential oscillations that are key to a number of sleep rhythms (Crunelli et al., 2005, 2006), including the delta and slow (<1 Hz) sleep oscillations. Whereas the former activity mainly requires the interaction of T-type Ca2+ and h currents for its generation, the Up and Down states of the latter oscillation have been suggested to result from the switching on and off of ITwindow on the basis of dynamic-clamp experiments (Hughes et al., 2002, 2008) and the relatively higher potency of Ni2+ compared with Cd2+ (Williams et al., 1997). The slow oscillation was induced in TC neurons of the MGB in cat thalamic slices by bath application of the metabotropic glutamate receptor agonist trans-ACPD, as described previously (Hughes et al., 2002). Application of TTA-P2 (3 μm) abolished the slow (<1 Hz) oscillation by progressively shortening the duration, and decreasing the amplitude, of the Up state (Fig. 7A,B), such that after a few minutes in the presence of this antagonist the activity expressed by the recorded TC neurons resembled a delta oscillation. This activity too was eventually abolished after an additional few minutes of perfusion with TTA-P2. This pattern of events was observed in all TC neurons tested (n = 5), and recovery of the slow and delta oscillations occurred within 25–45 min after the beginning of the antagonist wash-out (Fig. 7C).
Figure 7.

Block of intrinsic thalamic oscillations by TTA-P2. A, Bath application of trans-ACPD (50 μm) produces a rapid depolarization of a cat MGB TC neuron resulting in continuous tonic firing of action potentials. Hyperpolarization of the cell by steady-state somatic current injection brings about the slow (<1 Hz) sleep oscillation (II). Application of TTA-P2 (3 μm) progressively decreases the duration of the slow oscillation Up state (III and IV) until it eventually resembles the delta oscillation (V), which also is abolished after a few additional minutes of drug exposure (VI). B, Superimposition of the Up states marked by the asterisks (*) in A shows the progressive reduction of their amplitude and duration during the continuous presence of TTA-P2. The inset shows a maximum-intensity z-series reconstruction of the Alexa Fluor 594-filled MGB neuron from which the data illustrated in A and B were collected. C, Reversible block by TTA-P2 of the slow and delta oscillations in a different cat MGB neuron. The break in the trace is 20 min.
Discussion
The use of a potent, selective T-type Ca2+ channel antagonist has enabled the discovery of novel roles for these channels in the modulation of single-neuron excitability. In particular, the present results provide the first direct demonstration of the contribution of ITwindow to the resting membrane potential of thalamic neurons and to the Up state of their intrinsic slow (<1 Hz) sleep oscillation and show that only a small fraction of the T-type Ca2+ channel population is sufficient to generate robust LTCPs in these neurons.
Pharmacological profile of TTA-P2
Our experiments show that TTA-P2 fully blocks IT in TC and NRT neurons. Since TC neurons specifically express the Cav3.1 isoform whereas NRT neurons express Cav3.3 and Cav3.2 (Talley et al., 1999), these results are in agreement with previous data on recombinant Cav3.1, Cav3.2, and Cav3.3 channels that showed an identical potency of TTA-P2 on all three isoforms (Shipe et al., 2008). The similarity of the TTA-P2 IC50 value for native T-type channels in TC neurons (22 nm) and recombinant channels expressed in HEK cells (93 nm for Cav3.1) (Shipe et al., 2008) demonstrates that the potency of TTA-P2 does not depend on potential splice variant difference between native and heterologous systems and/or the presence of still unknown auxiliary subunits in TC neurons (Perez-Reyes, 2003). Our results also demonstrate that the effects of TTA-P2 on the intrinsic and synaptic excitability of TC and NRT neurons specifically involve T-type Ca2+ channel activation, since this antagonist does not modify the input resistance, the depolarizing sag present in the voltage response to negative current pulses, the transient and persistent Na+ currents, the action potential properties, and tonic firing.
The pharmacological profile of TTA-P2 therefore is superior to existing putative T-type Ca2+ channel blockers. One of the oldest and possibly best established blockers of T-type Ca2+ channels is the divalent cation Ni2+ (Fox et al., 1987) that was shown to be selective for low voltage over HVA Ca2+ channels in cells expressing the Cav3.2 isoform (Lee et al., 1999). However, for a substantial block of the other two Cav3 isoforms, a concentration of Ni2+ that already affects HVA currents is required (Zamponi et al., 1996). The antihypertensive mibefradil and the antiepileptics based on succinimides have also been used extensively in recombinant and native preparations, but the potency and selectivity of these drugs are not high and vary according to cell type and recording conditions (McGivern, 2006). With an IC50 value in the micromolar range, mibefradil is an effective T-type Ca2+ channel blocker but also inhibits HVA Ca2+ and Na+ channels, as well as voltage-gated and ATP-sensitive K+ channels (Mishra and Hermsmeyer, 1994; Bezprozvanny and Tsien, 1995; Nilius et al., 1997; Viana et al., 1997; Gomora et al., 1999; Liu et al., 1999). The use of ethosuximide and α-methyl-α-phenylsuccinimide have provided contrasting results, from a maximum 50% block of IT when used in millimolar concentrations (Coulter et al., 1989; Gomora et al., 2001) to no effect but with inhibition of the persistent Na+ current and Ca2+-activated K+ currents (Leresche et al., 1998; Crunelli and Leresche, 2002a). Finally, U-92032 (Porcello et al., 2003) and some anesthetic and antipsychotic drugs (Herrington et al., 1991; Takenoshita and Steinbach, 1991; Todorovic et al., 2000; Joksovic et al., 2005a; Orestes et al., 2008) also block IT but concurrently affect many other targets. Thus, the poor specificity and/or potency of these antagonists have so far hampered the interpretation of studies on neuronal excitability that have used these compounds. Instead, the use of the selective and potent TTA-P2 opens a novel avenue to investigate the role of T-type Ca2+ currents in single-cell excitability, synaptic physiology, and neuronal network activities. In particular, it should allow precise dissection of physiological mechanisms such as the amplification of synaptic inputs by T-type Ca2+ channels (Markram and Sakmann, 1994; Magee et al., 1995; Tsay et al., 2007), the IT-dependent component of synaptic plasticity (Oliet et al., 1997; Aizenman et al., 1998; Uebele et al., 2009), and the relative contribution of cortical and thalamic T-type Ca2+ channels to the pathophysiology of absence seizures (Vitko et al., 2007; Ernst et al., 2009; Powell et al., 2009).
Novel contributions of T-type Ca2+ channels to single-neuron excitability
By using the novel compound TTA-P2, the present results have already allowed us to consolidate and enlarge our current understanding of the contribution of T-type Ca2+ channels to single-neuron excitability.
First, we have directly demonstrated that there are sufficient numbers of T-type Ca2+ channels expressed in thalamic neurons to provide a robust redundancy mechanism for LTCPs. Previous studies in thalamic slices had suggested that a full block of LTCPs requires a marked reduction in the number of available T channels. However, difficulties with the potency and specificity of the various antagonists used in these studies (i.e., U-92032, ethosuximide, methylphenylsuccinimide, mibefradil, and valproate) did preclude a direct measurement of the minimal number of T channels required to evoke an LTCP in various physiological situations (Porcello et al., 2003; Broicher et al., 2007), whereas a computational model indicated that LTCPs could still be evoked when the T conductance was decreased by 50% (Broicher et al., 2007). Our data directly show that LTCPs and their associated burst firing could still be readily evoked when up to 70% of the channels are blocked. Indeed, application of increasing concentrations of TTA-P2 demonstrates that only when <30% of the T-type channel population is available for activation, the GABAergic inputs occurring at depolarized potentials are unable to generate rebound LTCPs. Similarly, in physiological conditions more favorable to T-type channel deinactivation (i.e., slightly hyperpolarized potentials), lemniscal EPSP triggers a strong LTCP and associated high-frequency firing when only 20% of the channels are available. These data, together with the demonstration of the physiological impact of the potentiation of IT that occurs at depolarized potentials in TC neurons (Bessaih et al., 2008), explain why high-frequency bursts of action potentials can be recorded in awake animals when TC neurons are depolarized and most of the T-type Ca2+ channels are inactivated (Llinas and Steriade, 2006).
Second, we have directly demonstrated the presence of a window component of a Ca2+ current in neurons and its contribution to their resting membrane potential, a role that was proposed more than a decade ago on the basis of indirect in vitro experiments (Williams et al., 1997) using the dynamic-clamp technique (Hughes et al., 1999) in thalamic neurons. As the window component is an inherent biophysical property of all three Cav3 isoforms, this result suggests a similar control by T-type Ca2+ channels of the resting membrane potential of other neuronal types that possess this class of membrane channels (Perez-Reyes, 2003). Although it is, at present, difficult to precisely ascertain the extent of this contribution to the “resting” membrane potential in vivo, and in particular in nonanesthetized freely moving animals, its role under conditions of progressively increasing membrane hyperpolarization, such as it occurs in thalamic and cortical neurons during the behavioral shift from an attentive waking state to relaxed wakefulness and then to early sleep stages (Crunelli et al., 2006), should not be underestimated. A similar contribution of ITwindow to the membrane potential may also be of significance in pathological states, such as absence seizures, in which neuronal hyperpolarization has been highlighted as one of the important underlying cellular abnormalities (Polack et al., 2007) together with an increase in IT (Tsakiridou et al., 1995; Ernst et al., 2009).
Third, we have shown that the Up state of the slow (<1 Hz) oscillation of thalamic neurons depends on IT. The biophysical mechanism of this slow oscillation in TC (and NRT) neurons requires the membrane potential bistability that is brought about by the interaction of ITwindow and the leak current (Hughes et al., 2002; Crunelli et al., 2005; Zhu et al., 2006), such that the Up and Down states are defined by ITwindow being switched on and off, respectively. The Ca2+ entry via the T-type channels activates ICAN, which in turn determines the duration of the Up state. As predicted by previous dynamic-clamp experiments (Hughes et al., 1999; Crunelli et al., 2005), the selective block of IT by TTA-P2 shortens the duration and decreases the amplitude of the Up state, eventually leading to a delta oscillation, which is still evoked by the interaction between the remaining vast majority of T-type Ca2+ and h channels. Thus, the present experiments provide the first direct demonstration of a role for the window component of a Ca2+ current in a physiologically relevant brain activity, the slow (<1 Hz) oscillation.
In conclusion, our results clearly demonstrate that TTA-P2 is a potent and selective T-type Ca2+ channel blocker that should prove a valuable tool for identifying the precise contribution of these channels to the excitability of single cells, as we have shown here for thalamic neurons, and to the dynamical output of neuronal networks in physiological and pathological conditions. Since TTA-P2 easily crosses the blood–brain barrier after oral dosing (Shipe et al., 2008), its use would not be restricted to in vitro analysis but could be easily extended to behavioral studies in freely moving animals.
Footnotes
This work was supported by the ANR-06-Neuro, Centre National de la Recherche Scientifique LEA 528 (Thalamic function in health and disease states), National Honor Scientist Support Program from the Korean Government, and the Wellcome Trust (Grant 71436).
References
- Aizenman CD, Manis PB, Linden DJ. Polarity of long-term synaptic gain change is related to postsynaptic spike firing at a cerebellar inhibitory synapse. Neuron. 1998;21:827–835. doi: 10.1016/s0896-6273(00)80598-x. [DOI] [PubMed] [Google Scholar]
- Anderson MP, Mochizuki T, Xie J, Fischler W, Manger JP, Talley EM, Scammell TE, Tonegawa S. Thalamic Cav3.1 T-type Ca2+ channel plays a crucial role in stabilizing sleep. Proc Natl Acad Sci U S A. 2005;102:1743–1748. doi: 10.1073/pnas.0409644102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessaih T, Leresche N, Lambert RC. T current potentiation increases the occurrence and temporal fidelity of synaptically evoked burst firing in sensory thalamic neurons. Proc Natl Acad Sci U S A. 2008;105:11376–11381. doi: 10.1073/pnas.0801484105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Tsien RW. Voltage-dependent blockade of diverse types of voltage-gated Ca2+ channels expressed in Xenopus oocytes by the Ca2+ channel antagonist mibefradil (Ro 40-5967) Mol Pharmacol. 1995;48:540–549. [PubMed] [Google Scholar]
- Bourinet E, Alloui A, Monteil A, Barrere C, Couette B, Poirot O, Pages A, McRory J, Snutch TP, Eschalier A, Nargeot J. Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J. 2005;24:315–324. doi: 10.1038/sj.emboj.7600515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broicher T, Seidenbecher T, Meuth P, Munsch T, Meuth SG, Kanyshkova T, Pape HC, Budde T. T-current related effects of antiepileptic drugs and a Ca2+ channel antagonist on thalamic relay and local circuit interneurons in a rat model of absence epilepsy. Neuropharmacology. 2007;53:431–446. doi: 10.1016/j.neuropharm.2007.05.030. [DOI] [PubMed] [Google Scholar]
- Carbone E, Lux HD. A low voltage-activated, fully inactivating Ca channel in vertebrate sensory neurones. Nature. 1984;310:501–502. doi: 10.1038/310501a0. [DOI] [PubMed] [Google Scholar]
- Contreras D. The role of T-channels in the generation of thalamocortical rhythms. CNS Neurol Disord Drug Targets. 2006;5:571–585. doi: 10.2174/187152706779025526. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Huguenard JR, Prince DA. Characterization of ethosuximide reduction of low-threshold calcium current in thalamic neurons. Ann Neurol. 1989;25:582–593. doi: 10.1002/ana.410250610. [DOI] [PubMed] [Google Scholar]
- Crunelli V, Leresche N. Block of thalamic T-type Ca(2+) channels by ethosuximide is not the whole story. Epilepsy Curr. 2002a;2:53–56. doi: 10.1046/j.1535-7597.2002.00024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci. 2002b;3:371–382. doi: 10.1038/nrn811. [DOI] [PubMed] [Google Scholar]
- Crunelli V, Toth TI, Cope DW, Blethyn K, Hughes SW. The “window” T-type calcium current in brain dynamics of different behavioural states. J Physiol. 2005;562:121–129. doi: 10.1113/jphysiol.2004.076273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Cope DW, Hughes SW. Thalamic T-type Ca2+ channels and NREM sleep. Cell Calcium. 2006;40:175–190. doi: 10.1016/j.ceca.2006.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana MA, Otsu Y, Maton G, Collin T, Chat M, Dieudonne S. T-type and L-type Ca2+ conductances define and encode the bimodal firing pattern of vestibulocerebellar unipolar brush cells. J Neurosci. 2007;27:3823–3838. doi: 10.1523/JNEUROSCI.4719-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst WL, Zhang Y, Yoo JW, Ernst SJ, Noebels JL. Genetic enhancement of thalamocortical network activity by elevating α1g-mediated low-voltage-activated calcium current induces pure absence epilepsy. J Neurosci. 2009;29:1615–1625. doi: 10.1523/JNEUROSCI.2081-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox AP, Nowycky MC, Tsien RW. Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurones. J Physiol. 1987;394:149–172. doi: 10.1113/jphysiol.1987.sp016864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomora JC, Enyeart JA, Enyeart JJ. Mibefradil potently blocks ATP-activated K(+) channels in adrenal cells. Mol Pharmacol. 1999;56:1192–1197. doi: 10.1124/mol.56.6.1192. [DOI] [PubMed] [Google Scholar]
- Gomora JC, Daud AN, Weiergraber M, Perez-Reyes E. Block of cloned human T-type calcium channels by succinimide antiepileptic drugs. Mol Pharmacol. 2001;60:1121–1132. [PubMed] [Google Scholar]
- Guyon A, Leresche N. Modulation by different GABAB receptor types of voltage-activated calcium currents in rat thalamocortical neurones. J Physiol. 1995;485:29–42. doi: 10.1113/jphysiol.1995.sp020710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington J, Stern RC, Evers AS, Lingle CJ. Halothane inhibits two components of calcium current in clonal (GH3) pituitary cells. J Neurosci. 1991;11:2226–2240. doi: 10.1523/JNEUROSCI.11-07-02226.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes SW, Cope DW, Toth TI, Williams SR, Crunelli V. All thalamocortical neurones possess a T-type Ca2+ “window” current that enables the expression of bistability-mediated activities. J Physiol. 1999;517:805–815. doi: 10.1111/j.1469-7793.1999.0805s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes SW, Cope DW, Blethyn KL, Crunelli V. Cellular mechanisms of the slow (1 Hz) oscillation in thalamocortical neurons in vitro. Neuron. 2002;33:947–958. doi: 10.1016/s0896-6273(02)00623-2. [DOI] [PubMed] [Google Scholar]
- Hughes SW, Lorincz M, Cope DW, Crunelli V. NeuReal: an interactive simulation system for implementing artificial dendrites and large hybrid networks. J Neurosci Methods. 2008;169:290–301. doi: 10.1016/j.jneumeth.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joksovic PM, Brimelow BC, Murbartian J, Perez-Reyes E, Todorovic SM. Contrasting anesthetic sensitivities of T-type Ca2+ channels of reticular thalamic neurons and recombinant Ca(v)3.3 channels. Br J Pharmacol. 2005a;144:59–70. doi: 10.1038/sj.bjp.0706020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joksovic PM, Bayliss DA, Todorovic SM. Different kinetic properties of two T-type Ca2+ currents of rat reticular thalamic neurones and their modulation by enflurane. J Physiol. 2005b;566:125–142. doi: 10.1113/jphysiol.2005.086579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Song I, Keum S, Lee T, Jeong MJ, Kim SS, McEnery MW, Shin HS. Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking alpha(1G) T-type Ca(2+) channels. Neuron. 2001;31:35–45. doi: 10.1016/s0896-6273(01)00343-9. [DOI] [PubMed] [Google Scholar]
- Kim D, Park D, Choi S, Lee S, Sun M, Kim C, Shin HS. Thalamic control of visceral nociception mediated by T-type Ca2+ channels. Science. 2003;302:117–119. doi: 10.1126/science.1088886. [DOI] [PubMed] [Google Scholar]
- Lambert RC, McKenna F, Maulet Y, Talley EM, Bayliss DA, Cribbs LL, Lee JH, Perez-Reyes E, Feltz A. Low-voltage-activated Ca2+ currents are generated by members of the CavT subunit family (α1G/H) in rat primary sensory neurons. J Neurosci. 1998;18:8605–8613. doi: 10.1523/JNEUROSCI.18-21-08605.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Gomora JC, Cribbs LL, Perez-Reyes E. Nickel block of three cloned T-type calcium channels: low concentrations selectively block alpha1H. Biophys J. 1999;77:3034–3042. doi: 10.1016/S0006-3495(99)77134-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leresche N, Parri HR, Erdemli G, Guyon A, Turner JP, Williams SR, Asprodini E, Crunelli V. On the action of the anti-absence drug ethosuximide in the rat and cat thalamus. J Neurosci. 1998;18:4842–4853. doi: 10.1523/JNEUROSCI.18-13-04842.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JH, Bijlenga P, Occhiodoro T, Fischer-Lougheed J, Bader CR, Bernheim L. Mibefradil (Ro 40-5967) inhibits several Ca2+ and K+ currents in human fusion-competent myoblasts. Br J Pharmacol. 1999;126:245–250. doi: 10.1038/sj.bjp.0702321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Jahnsen H. Electrophysiology of mammalian thalamic neurones in vitro. Nature. 1982;297:406–408. doi: 10.1038/297406a0. [DOI] [PubMed] [Google Scholar]
- Llinas R, Muhlethaler M. Electrophysiology of guinea-pig cerebellar nuclear cells in the in vitro brain stem-cerebellar preparation. J Physiol. 1988;404:241–258. doi: 10.1113/jphysiol.1988.sp017288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas RR, Steriade M. Bursting of thalamic neurons and states of vigilance. J Neurophysiol. 2006;95:3297–3308. doi: 10.1152/jn.00166.2006. [DOI] [PubMed] [Google Scholar]
- Magee JC, Christofi G, Miyakawa H, Christie B, Lasser-Ross N, Johnston D. Subthreshold synaptic activation of voltage-gated Ca2+ channels mediates a localized Ca2+ influx into the dendrites of hippocampal pyramidal neurons. J Neurophysiol. 1995;74:1335–1342. doi: 10.1152/jn.1995.74.3.1335. [DOI] [PubMed] [Google Scholar]
- Markram H, Sakmann B. Calcium transients in dendrites of neocortical neurons evoked by single subthreshold excitatory postsynaptic potentials via low-voltage-activated calcium channels. Proc Natl Acad Sci U S A. 1994;91:5207–5211. doi: 10.1073/pnas.91.11.5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Connors BW, Lighthall JW, Prince DA. Comparative electrophysiology of pyramidal and sparsely spiny stellate neurons of the neocortex. J Neurophysiol. 1985;54:782–806. doi: 10.1152/jn.1985.54.4.782. [DOI] [PubMed] [Google Scholar]
- McGivern JG. Pharmacology and drug discovery for T-type calcium channels. CNS Neurol Disord Drug Targets. 2006;5:587–603. doi: 10.2174/187152706779025535. [DOI] [PubMed] [Google Scholar]
- McKay BE, McRory JE, Molineux ML, Hamid J, Snutch TP, Zamponi GW, Turner RW. Ca(V)3 T-type calcium channel isoforms differentially distribute to somatic and dendritic compartments in rat central neurons. Eur J Neurosci. 2006;24:2581–2594. doi: 10.1111/j.1460-9568.2006.05136.x. [DOI] [PubMed] [Google Scholar]
- Mishra SK, Hermsmeyer K. Selective inhibition of T-type Ca2+ channels by Ro 40-5967. Circ Res. 1994;75:144–148. doi: 10.1161/01.res.75.1.144. [DOI] [PubMed] [Google Scholar]
- Nilius B, Prenen J, Kamouchi M, Viana F, Voets T, Droogmans G. Inhibition by mibefradil, a novel calcium channel antagonist, of Ca(2+)- and volume-activated Cl- channels in macrovascular endothelial cells. Br J Pharmacol. 1997;121:547–555. doi: 10.1038/sj.bjp.0701140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowycky MC, Fox AP, Tsien RW. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature. 1985;316:440–443. doi: 10.1038/316440a0. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Orestes P, Bojadzic D, Chow R, Todorovic SM. Mechanisms and functional significance of inhibition of neuronal T-type calcium channels by isoflurane. Mol Pharmacol. 2008;75:542–554. doi: 10.1124/mol.108.051664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003;83:117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A, Charpier S. Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. J Neurosci. 2007;27:6590–6599. doi: 10.1523/JNEUROSCI.0753-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcello DM, Smith SD, Huguenard JR. Actions of U-92032, a T-type Ca2+ channel antagonist, support a functional linkage between I(T) and slow intrathalamic rhythms. J Neurophysiol. 2003;89:177–185. doi: 10.1152/jn.00667.2002. [DOI] [PubMed] [Google Scholar]
- Powell KL, Cain SM, Ng C, Sirdesai S, David LS, Kyi M, Garcia E, Tyson JR, Reid CA, Bahlo M, Foote SJ, Snutch TP, O'Brien TJ. A Cav3.2 T-type calcium channel point mutation has splice-variant-specific effects on function and segregates with seizure expression in a polygenic rat model of absence epilepsy. J Neurosci. 2009;29:371–380. doi: 10.1523/JNEUROSCI.5295-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipe WD, Barrow JC, Yang ZQ, Lindsley CW, Yang FV, Schlegel KA, Shu Y, Rittle KE, Bock MG, Hartman GD, Tang C, Ballard JE, Kuo Y, Adarayan ED, Prueksaritanont T, Zrada MM, Uebele VN, Nuss CE, Connolly TM, Doran SM, et al. Design, synthesis, and evaluation of a novel 4-aminomethyl-4-fluoropiperidine as a T-type Ca2+ channel antagonist. J Med Chem. 2008;51:3692–3695. doi: 10.1021/jm800419w. [DOI] [PubMed] [Google Scholar]
- Steriade M, Jones EG, McCormick DA. Thalamus. Oxford: Elsevier Science; 1997. [Google Scholar]
- Takenoshita M, Steinbach JH. Halothane blocks low-voltage-activated calcium current in rat sensory neurons. J Neurosci. 1991;11:1404–1412. doi: 10.1523/JNEUROSCI.11-05-01404.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. 1999;19:1895–1911. doi: 10.1523/JNEUROSCI.19-06-01895.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic SM, Perez-Reyes E, Lingle CJ. Anticonvulsants but not general anesthetics have differential blocking effects on different T-type current variants. Mol Pharmacol. 2000;58:98–108. doi: 10.1124/mol.58.1.98. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Jevtovic-Todorovic V, Meyenburg A, Mennerick S, Perez-Reyes E, Romano C, Olney JW, Zorumski CF. Redox modulation of T-type calcium channels in rat peripheral nociceptors. Neuron. 2001;31:75–85. doi: 10.1016/s0896-6273(01)00338-5. [DOI] [PubMed] [Google Scholar]
- Tsakiridou E, Bertollini L, de Curtis M, Avanzini G, Pape HC. Selective increase in T-type calcium conductance of reticular thalamic neurons in a rat model of absence epilepsy. J Neurosci. 1995;15:3110–3117. doi: 10.1523/JNEUROSCI.15-04-03110.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsay D, Dudman JT, Siegelbaum SA. HCN1 channels constrain synaptically evoked Ca2+ spikes in distal dendrites of CA1 pyramidal neurons. Neuron. 2007;56:1076–1089. doi: 10.1016/j.neuron.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uebele VN, Nuss CE, Santarelli VP, Garson SL, Barrow JC, Stauffer SR, Koblan KS, Renger JJ, Aton S, Seibt J, Dumoulin M, Jha SK, Coleman T, Frank MG. T-type calcium channels regulate cortical plasticity in-vivo. Neuroreport. 2009;20:257–262. doi: 10.1097/WNR.0b013e3283200111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viana F, Van den Bosch L, Missiaen L, Vandenberghe W, Droogmans G, Nilius B, Robberecht W. Mibefradil (Ro 40-5967) blocks multiple types of voltage-gated calcium channels in cultured rat spinal motoneurones. Cell Calcium. 1997;22:299–311. doi: 10.1016/s0143-4160(97)90068-3. [DOI] [PubMed] [Google Scholar]
- Vitko I, Bidaud I, Arias JM, Mezghrani A, Lory P, Perez-Reyes E. The I–II loop controls plasma membrane expression and gating of Ca(v)3.2 T-type Ca2+ channels: a paradigm for childhood absence epilepsy mutations. J Neurosci. 2007;27:322–330. doi: 10.1523/JNEUROSCI.1817-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR, Stuart GJ. Action potential backpropagation and somato-dendritic distribution of ion channels in thalamocortical neurons. J Neurosci. 2000;20:1307–1317. doi: 10.1523/JNEUROSCI.20-04-01307.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR, Toth TI, Turner JP, Hughes SW, Crunelli V. The “window” component of the low threshold Ca2+ current produces input signal amplification and bistability in cat and rat thalamocortical neurones. J Physiol. 1997;505:689–705. doi: 10.1111/j.1469-7793.1997.689ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E, Snutch TP. Nickel block of a family of neuronal calcium channels: subtype- and subunit-dependent action at multiple sites. J Membr Biol. 1996;151:77–90. doi: 10.1007/s002329900059. [DOI] [PubMed] [Google Scholar]
- Zhu L, Blethyn KL, Cope DW, Tsomaia V, Crunelli V, Hughes SW. Nucleus- and species-specific properties of the slow (<1 Hz) sleep oscillation in thalamocortical neurons. Neuroscience. 2006;141:621–636. doi: 10.1016/j.neuroscience.2006.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]