

Abstract
Understanding the evolution of drug resistance in malaria is a central area of study at the intersection of evolution and medicine. Antimalarial drug resistance is a major threat to malaria control and directly related to trends in malaria attributable mortality. Artemisinin combination therapies (ACT) are now recommended worldwide as first line treatment for uncomplicated malaria, and losing them to resistance would be a disaster for malaria control. Understanding the emergence and spread of antimalarial drug resistance in the context of different scenarios of antimalarial drug use is essential for the development of strategies protecting ACTs. In this study, we review the basic mechanisms of resistance emergence and describe several simple equations that can be used to estimate the probabilities of de novo resistance mutations at three stages of the parasite life cycle: sporozoite, hepatic merozoite and asexual blood stages; we discuss the factors that affect parasite survival in a single host in the context of different levels of antimalarial drug use, immunity and parasitaemia. We show that in the absence of drug effects, and despite very different parasite numbers, the probability of resistance emerging at each stage is very low and similar in all stages (for example per-infection probability of 10−10–10−9 if the per-parasite chance of mutation is 10−10 per asexual division). However, under the selective pressure provided by antimalarial treatment and particularly in the presence of hyperparasitaemia, the probability of resistance emerging in the blood stage of the parasite can be approximately five orders of magnitude higher than in the absence of drugs. Detailed models built upon these basic methods should allow us to assess the relative probabilities of resistance emergence in the different phases of the parasite life cycle.
Keywords: artemisinin, bottleneck, de novo resistance, drug resistance, life cycle, malaria, population genetics, resistance emergence
Introduction
Mathematical modelling serves as a valuable tool for understanding the growth and evolution of populations. When populations of parasites are considered, such models can yield insight into how diseases spread, how pathogens evolve, and what control strategies and medical interventions would be most effective at reducing parasite prevalence and minimizing the chance of undesirable evolutionary outcomes. An important example of such an undesirable outcome is drug treatment driving the evolution of drug-resistant parasites, a process that in the long-term can render entire classes of drugs ineffective against a particular disease. Malaria parasites have evolved resistance to all known antimalarial drugs except the artemisinsins. Recently, however, even the artemisinin-class drugs have encountered ‘tolerant’ malaria parasites (White 2008) showing prolonged parasite clearance times in patients. This does not yet amount to full-blown resistance and fortunately appears to be confined to Western Cambodia. However, the concern is that further evolution of this resistant phenotype or the emergence of a novel fully resistant phenotype and its subsequent spread may render useless this last bastion of antimalarial treatment. Sadly, there are currently no suitable alternative drugs nearing the final stages of development by pharmaceutical companies.
Most mathematical models of drug resistance in malaria have focused on the spread of resistance and assumed that multiple copies of a resistance mutation were already present in the parasite population (Cross and Singer 1991; Levin 2002; Koella and Antia 2003; Yeung et al. 2004). Examining spread is useful when resistance has already arisen in an area, however, it is also very important to look at the emergence of resistance to a drug to which resistance has not yet appeared. This is particularly pertinent for artemisinin-class drugs as these are the only drugs that show no evidence of overt resistance in vivo (Barnes and White 2005). Artemisinin-based combination therapies (ACTs) are recommended worldwide as first-line treatment for Plasmodium falciparum malaria because they are highly effective with almost no side effects (Newton et al. 2003; Adjuik et al. 2004; Ashley and White 2005; Dondorp et al. 2005; Hutagalung et al. 2006; Smithuis et al. 2006; World Health Organisation 2006). Combination therapies in general are used specifically to prevent the emergence of resistance. If ACTs were lost to resistance, global malaria control efforts would be seriously harmed and significant excess malaria morbidity and mortality would occur.
The benefit of combination therapy is twofold. First, if two drugs with different modes of action are co-formulated in a combination therapy, the parasite's probability of inheriting resistance mutations to both drugs is the product of the probabilities of inheriting resistance mutations to each drug (White and Pongtavornpinyo 2003); this is usually a very small number. An exception to this rule occurs when one resistance mechanism, such as an efflux pump, confers resistance to many types of drugs simultaneously. Second, if resistance requires two or more genetic changes, the obligate sexual recombination that occurs in the Plasmodium life cycle can break apart the gene combination(s) required to encode resistance to both components of the combination therapy, thus slowing, or even preventing, the establishment and spread of resistance (Curtis and Otoo 1986; Dye and Williams 1997; Hastings 1997).
If ACTs are to be deployed in an area where there is no resistance to either combination of drugs or perhaps some resistance to the partner drug, it would be helpful to be able to predict the interval of time before artemisinin resistance is likely to emerge given information on entomological, epidemiological and pharmacological factors. Cost-effectiveness analysis based on these models can be a powerful tool to inform national drug policy decisions, in particular in helping to persuade people that the higher initial cost of ACTs will eventually be offset by its longer useful therapeutic life (Institute of Medicine, 2004; Yeung et al. 2004).
In this study, we extend previous methods (Hastings 2004; Pongtavornpinyo 2006) and look at this early stage of resistance evolution, often referred to as resistance emergence, and consider at which stage of the parasite life cycle – mosquito, liver, or blood – this emergence is most likely to occur. We present the basic evolutionary equations that can be used to assess the relative probabilities of resistance emergence in these three stages. For very accurate estimates, we would rely on dynamical models detailing host immunity, pharmacokinetics, fitness variation for different levels of resistance, and details on gametocyte switching rates.
Methods
We consider resistance emergence independently at each stage of the parasitic life cycle. The probability of emergence is the combined probabilities of two events: the probability of any mutant occurring, and the probability of that mutant surviving in the population of parasites in a single host. Fixation of resistant phenotypes can occur at the bottlenecks separating the stages of the parasites’ life cycle. The genetic bottleneck for sporozoites entering a human host from a mosquito and for gametocytes in the blood stage being sampled by a mosquito is very small (the order of one to ten parasites), whereas the bottleneck experienced by hepatic merozoites emerging from the liver is less severe and highly dependent on the amount of residual drug present in the bloodstream. Note that drugs are only present in the bloodstream and thus do not affect resistance emergence and evolution during the mosquito stage or liver stage.
Sporozoites
It is estimated that approximately 6–10 sporozoites (and on rare occasions, >100) are injected into a human host during one mosquito bite. In this study, it is assumed that an infection derives from a single sporozoite, the other sporozoites being lost due to stochastic effects or the actions of host immunity. Resistance can emerge when a sporozoite spontaneously mutates inside a mosquito, resulting in a resistant phenotype, and is later the lone survivor of the mosquito-liver bottleneck and the sole founder of the population of parasites in the liver.
Let μ represent the per-parasite probability that a mutation will occur during parasite replication. The same hypothetical resistance mutation is assumed in each parasite stage (i.e. the value of μ is constant across different stages) and is proportional to the DNA polymerase error rate during mitotic division. Let s be the number of generations from the gametocyte stage taken during blood meal to the sporozoites number in the mosquito's salivary gland (s∼ 10).
Because we assume that a single sporozoite founds the population of parasites in the liver, the probability that this parasite mutated during s generations of replication in the mosquito is:
| (1) |
This is also the probability that such a mutation occurs and goes to fixation, because a single parasite founds the infection. Therefore,
| (2) |
which is s × μ when μ is small; if we assume that μ = 10−10 then, μs = 10−9 (Gatton et al. 2001; Paget-McNicol and Saul 2001; White and Pongtavornpinyo 2003).
Hepatic merozoites
When hepatic (liver-stage) merozoites leave the liver the newly invaded blood cells may be exposed to residual drug levels from a patient's previous treatment, and thus might be sufficient to kill either the entire parasite population (including emerging resistant mutants) or just the sensitive parasites (a patient would not be given drugs at this stage of the infection as he or she would not yet be ill). If a newly acquired infection emerges from the liver during a particular drug's selective window (Watkins and Mosobo 1993; Stepniewska and White 2008), resistant parasites will survive while sensitive parasites will continue to be eliminated.
Let h be the number of mitotic divisions required by a single hepatic schizont (initial stage of liver infection) to produce 105 parasites in the liver and to be released into the blood. Note that h in this section is not the number of generations of parasite replication (as was the parameter s in the previous section). The parameter h is on the order of 105–106, and
| (3) |
where μ is again 10−10; μh is between 10−5 and 10−4 and represents the probability that a resistance mutation occurs at some point during parasite replication in the liver, in the absence of drugs.
The survival of the emerging resistant merozoites is affected by any residual slowly eliminated drugs that are present in the blood as the parasites leave the liver for the bloodstream. Focusing on ACT treatment, we discuss the emergence of artemisinin resistance in the presence of residual artemisinin. In general, the residual drug factor can be incorporated into population-genetic equations by assuming two periods with different concentrations or levels of drug. Initially, there is a high level of drug, a concentration high enough to kill both resistant and sensitive parasites. Subsequently, there is a low level, i.e. a level that falls into the selective window killing sensitive parasites but allowing the survival of resistant parasites. When there is a low level of residual drugs, selection takes place.
We consider the situation where there exists some residual partner drug. Let γ be the probability that the emerged merozoites already have partner drug resistance. Let α be the probability that the hepatic merozoites encounter artemisinin drug levels in the artemisinin selective window (a low level of artemisinin). From a human population perspective, α is equivalent to the proportion of people with residual artemisinin within the selective window. For artemisinins, α is usually zero or if it exists is extremely small because the elimination half-life of the artemisinin is only 1 h in comparison with the ≥48 h asexual cycle of the malaria parasites (Stepniewska and White 2008).
Let FAB be the probability of fixation of the double-resistant in the presence of both artemisinin and the partner drug. In our scenario of complete resistance, FAB is probably close to 1. The probability of artemisinin-resistance emerging and fixing from the hepatic merozoite stage is
| (4) |
where the 10−5 term in the right-hand side represents the probability of random fixation in a population of size 105. Note that (1−α) represents the probability that there is abundant artemisinin or no artemisinin; in the first scenario, artemisinin kills all parasites so fixation probability is zero while in the second scenario artesunate has equal killing rates for partner drug resistant and sensitive parasites so fixation probability is 10−5. In equation (4), we therefore assume that the extremely short half-life of artemisinin ensures that the frequency of scenario 1 is negligible compared to scenario 2. We assume that if there is no resistance to the partner drug, an artemisinin-resistant parasite would have a negligible chance of surviving; this assumption can of course be relaxed. The probability in equation (4), assuming a 1/100 chance of being in the artemisinin selective window and a high chance of pre-existing partner-drug resistance, is on the order of 10−7. If α < 10−6, then the probability in equation (4) is dominated by random fixation and is of the order 10−10.
In the absence of any residual drugs, there is no bottleneck and therefore the probability of resistance emerging is just the probability that a resistance mutation occurs at some point and then goes to fixation (for this example, the probability of this event would be 10−10).
Blood-stage parasites
Here, we consider emergence of resistant parasites during blood-stage infection, assuming that these parasites did not encounter residual drug when entering the bloodstream from the liver. The parasites may encounter drugs at this stage if their population size crosses the pyrogenic threshold (108) causing the patient to be ill and to be treated with an ACT. We do not consider the case of presumptive treatment for a nonmalarial fever or intermittent presumptive treatment given to infants or pregnant women. In order to be transmitted to a mosquito feeding on a host, malaria parasites need to develop into gametocytes; gamotocytaemia in P. falciparum infections is delayed with respect to blood-stage parasitaemia. In this stage of the infection, we calculate the probability (Pb) that resistance emerges and the probability that resistant gametocytes are produced and sampled by a mosquito during a blood meal. Mosquito sampling can be viewed as the fixation event in the blood stage of the infection, and the bottleneck here is again quite small. Pb is calculated after 10 cycles of blood-stage replication (∼20 days)
The emergence of resistance in blood-stage parasites occurs when the genetic changes (mutation or gene duplication) which confer antimalarial drug resistance occur spontaneously and independently of antimalarial drugs. Hereafter, we use the term mutation to include any genetic change conferring resistance. For resistance to spread the resistant, mutant parasites must survive both antimalarial drug effects and host-defence mechanisms. A simple model of de novo resistance in the blood-stage infection was developed (see Supporting Information) which describes the stages of reproduction in the blood after hepatic merozoites are released from the liver and multiply to a density which can start gametocyte production (see Fig. 1). For the first cycle, the probability of mutation occurring among the hepatic merozoites invading the red blood cell (∼105) is calculated. For all other cycles, the probability of mutation occurring among the blood-stage parasites in each cycle is calculated. Once mutation occurs, both the sensitive and resistant parasites would multiply over the succeeding cycles, depending on the multiplication factor (m). The parasite multiplication rate every 48 h in the asexual life cycle is determined mainly by the efficiency of merogony or merozoite invasion (White et al. 1992); in our model, it is also affected by antimalarial drugs and host immunity. We assume that the effect of treatment on killing parasites, the effect of host immunity on suppressing parasites and the multiplication of the parasites are acting simultaneously in a single cycle. The bottleneck at the blood stage is identified as the chance of transmitting resistance through to the sexual stages.
Figure 1.

The various stages at which resistance can emerge during the blood stage. The model is implemented in Microsoft Excel.
We define μt as the probability that a mutant emerges from blood stage at cycle t when the population size is pt. In each cycle, the probability of exactly one mutant occurring in cycle t is
| (5) |
which is about pt × μ if we assume that μ2 is small. For the blood stage, we again assume that at most one resistant mutant can emerge.
The probability of resistance first emerging at cycle t is
| (6) |
We consider the possibility of switching to gametocytes for cycles t = 1 to t = 10. It is assumed that the resistant and sensitive parasites have equal chances of switching to gametocytes, independently of antimalarial treatment but dependent upon the parasite density. Although previous studies have shown that parasites can facultatively alter investment in gametocyte production in response to drugs (Buckling et al. 1997) and many other risk factors for gametocyte carriage (Price et al. 1999), we do not include such complications and only model a binary switching rate.
Letting r10,k and p10,k be the numbers of resistant and total parasites, respectively, at cycle 10 given that a mutant parasite emerged first at cycle k, we can calculate these numbers for any cycle k and condition on the cycle that resistance first appears. For example, if an artemisinin-resistant mutant parasite emerges first at cycle 10, the probability that one of these mutants develops into a gametocyte and is transmitted is
| (7) |
The numbers of resistant (rt) and sensitive (st) parasites are calculated in a standard discrete-generation population-genetic framework where rt depends on rt−1, st depends on st−1, and pt = rt + st, as described in the Supporting Information. The probability of any mutation occurring during the blood stage and surviving to transmit is just the sum of the probabilities of resistance emerging in any cycle, multiplied by the chance of those resistant progeny switching to gametocytes and infecting a mosquito; we write this as
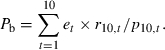 |
(8) |
The details of the dynamic model are given in Appendix B. As an example, when μ = 10−10 and the parasite multiplication rate m = 10, the chance of resistance emerging in a nonimmune person from a low-transmission setting without treatment effect (Pb) is 5.39 × 10−10.
Alternatively, we derive a simple calculation of the probability of resistance emerging at the blood stage by looking at the time of treatment. Let b be the number of cycles required for the parasites to multiply in number from the first cycle to the parasite level (v) where treatment is given (b∼ 6). The probability that any single parasite (after b cycles of reproduction) contains a resistance mutation is
| (9) |
so that an infection with v parasites has a probability v × μb (providing v × μb << 1) of containing a spontaneous resistance mutation. Let τ be the chance of an infection being treated and ρR be the chance of a resistant parasite surviving treatment, the host-immune response and reaching the density that gametocytes are sufficiently produced for transmission. Then, the probability that a given infection of biomass v contains a resistant mutation that survives in the blood stage is
| (10) |
Further details are given in Appendix C. In the Results section, we focus on blood-stage emergence using the dynamic model only.
Results
To calculate the probability of resistance emerging in the sporozoite and hepatic merozoite stages, we used equations (2) and (4) while a dynamic model (available in the on-line Supporting Information; see Fig. 1) was used to calculate the resistance emergence probability at the asexual blood stage (equation 8). Several particular situations of resistance emergence, in addition to the basic scenarios outlined in the methods, merit closer attention.
Consider the example of two extreme transmission intensity settings. In a low-transmission setting, treatment becomes important as infected hosts have little immunity and are therefore more likely to experience malaria symptoms and seek treatment. The effects of antimalarials and host immunity on the emergence of resistance occur mainly in the blood stage. In a high-transmission setting, immunity reduces the likelihood of symptoms but many people will receive presumptive antimalarials whenever they experience febrile symptoms due to other causes. This occurs because fevers in such areas are assumed to be due to malaria and most people in high-transmission settings carry malaria parasites. In this case, the chance of resistance emerging from the mutant merozoite seeing residual drug level when released from the liver is much greater, although the numbers of parasites exposed to residual levels and their individual survival probabilities are much lower.
To explore resistance emergence in the blood stage, the baseline scenario is defined as the case where treatment is absent. Figure 2 shows the parasite biomass in the absence of treatment for the low- and high-transmission setting. The parameter estimates for these scenarios, given in Appendix D, are set such that the parasite-time curves are similar to what could be observed in the field. In the baseline scenarios, the main difference between the high-transmission and low-transmission scenarios is the presence of host immunity. The effect of immunity incorporated into this model acts mainly to reduce the parasite multiplication rate. The reduction in multiplication rate leads to a slow increase or even a decrease in the parasite population, equally for sensitive and resistant parasites. This results in a smaller chance of mutant parasites developing into gametocytes and passing through the transmission bottleneck. In the low-transmission setting, the chance of resistance emerging in the blood stage in the absence of treatment (Pb) is estimated to 5.39 × 10−10 (Fig. 3). In the high-transmission setting, immunity suppresses the level of parasites below 108 preventing any emerged resistance from being transmitted through gametocytes.
Figure 2.

The baseline scenarios for the low (black) and high (red) transmission intensity setting respectively. In the absence of treatment, the parasite biomass of a nonimmune person would increase lethally over time. For a host with some immunity, the parasite biomass could be suppressed around the detectable or pyrogenic levels. Parameter estimates are given in Appendix D.
Figure 3.

The probability of artemisinin-resistance emerging at different stages. The dots show the probability of resistance emerging from the sporozoite (Ps), the hepatic merozoite (Ph) and blood stage (Pb). The dotted line represents the range for the probability of resistance emerging in the hepatic merozoite stage (Ph) in the presence of some residual drugs. For the blood stage, the probability of resistance emerging is calculated at cycle 10 when total parasite biomass is equal or above 108.
An additional effect of immunity is that it decreases a patient's chance of becoming symptomatic (i.e. there is a greater per-infection probability of infection resolution below the density threshold at which illness develops). In low-transmission settings where there is less immunity, hosts present with symptoms more often. In general, the effect of this on the risk of resistance emergence resistance is difficult to predict [but see Figure 3 of Boni et al. (2008b)]. For model comparison, and simplicity, the pyrogenic threshold (py) or the parasite density at which the infection becomes symptomatic is assumed to be the same for all infections.
The impact of deployment of ACT on the emergence of arteminsinin resistance is considered with two possible scenarios: (i) when parasites are sensitive to the artemisinin's partner drug and (ii) there is already resistance to the artemisinin's partner drug. If there is still no resistance to the partner drug, it is assumed that ACTs kill parasites effectively and this results in a 99.999% reduction in the artemisinin-sensitive parasite population and a 99.9% reduction in the artemisinin-resistant parasite population (as the partner drug is still efficacious). In reality, the treatment efficacy could be smaller due to poor adherence. If the partner drug is failing because of pre-existing resistance, then the antimalarial effect is more dependent on the artemisinin. To illustrate this, we assume that in this case, the ACT can still kill 40% of the parasites if the parasites are resistant to both artemisinin and the partner drug. If the parasites are artemisinin-sensitive but partner-drug resistant then the ACT is assumed to kill 80% of the parasite population.
From Fig. 3, we conclude that artemisinin resistance is most likely to emerge in an individual from a low-transmission intensity area when it is used in combination with a partner drug to which resistance already exists (Pb = 9.71 × 10−7). An ineffective partner drug (due to pre-existing resistance) not only reduces the efficacy of the ACT treatment, but also increases the chance of artemisinin-resistance emerging. In the high-transmission setting, both specific and nonspecific immunity act to reduce the parasite population, decreasing the chance of resistance emerging and producing resistant gametocytes. The chance of resistance emergence is relatively small in high-transmission settings and situations when the partner drug is still working.
As mentioned earlier, residual partner drug effects on the emergence of resistance are more likely in high transmission settings where presumptive drug use is common (Hastings and Watkins 2005). When there are some residual drugs, we estimate the chance of resistance emerging from the mutant merozoites leaving the liver (Ph) in the range of 10−10–10−7 (equation 4). To prevent such an event, it is important to minimize the chance of parasites seeing residual drug level that fall in the selective window by choosing appropriate combinations to which no resistance exists and/or combinations whose elimination half-lives are matched. In addition, the presumptive use of some long elimination half-life antimalarials should also be restricted.
The effect of hyperparasitaemia on the emergence of resistance was also explored. Hyperparasitaemia is defined here as an infection in which the proportion of infected erythrocytes varies from 5% to 70%. This is most likely to result from unrestricted multiplication. We also modelled two possible alternative causes: (i) a large number of parasites released from the liver or (ii) more rapid increase of parasites over the erythrocytic cycles (high multiplication rate). These two parameters were modified simultaneously to characterize the hyperparasitaemia infection in the low-transmission setting: the parasite multiplication rate and the initial number of hepatic merozoites. Hyperparasitaemia in the blood-stage model is established through either an increase in the per-generation multiplication rate from 10 to 13 or an increase in the initial number of parasites entering the blood stage from 105 to 107.
Figure 3 shows the effect of hyperparasitaemia on the probability of resistance emerging when the resistance to the partner drug is present (Pb = 4.84 × 10−6). Resistance is more likely to emerge from hyperparasitaemic infections when treating with an ineffective ACT. The probability of resistance emergence increases more under the hyperparasitaemic scenario where 107 parasites enter the blood from the liver. When hyperparasitaemia is created by an increase in the parasite multiplication rate, the probability of resistance emergence does not differ from nonhyperparasitaemia (Pb = 9.60 × 10−7) as increasing the multiplication rate causes an increase in both the sensitive and resistant parasites equally.
Discussion
We investigated the origins of antimalarial resistance by considering the likelihood of resistance emergence at three different stages of the parasite life cycle: the sporozoite stage, the hepatic merozoite stage and the asexual blood stage. We concentrated particularly on artemisinin resistance in the context of current use. Our estimates of absolute probabilities of resistance emergence are not evidence-based and may well be inaccurate. They make a number of simplifying assumptions which may not be justified, but they do provide a framework for comparing probabilities at different stages of the parasite life cycle. This comparative analysis of the de novo resistance in different stages of life cycle shows that in the absence of antimalarials, the emergence of resistance is rare and similar among the different stages of infection (estimated range between 10−10 < P < 10−9). Although parasite numbers vary hugely among the different stages of development, there is a series of bottlenecks which tend to dominate the overall dynamics and evolution. Exposure of the asexual cycle to antimalarial drugs, and subsequent recrudescence allows selection of the resistant subpopulation. Thus, in the specific case of artemisinin combination treatment, artemisinin resistance is most likely to emerge at the blood stage when there is already resistance to the partner drug (10−6 < Pb < 10−5) or when liver-stage parasites first enter the blood in small numbers and encounter residual drug levels (10−10 < Ph < 10−7).
The chance of resistance emergence increases in hyperparasitaemic infections (Pb = 10−5). Hyperparasitaemia reflects a failure of host defence, and carries a greater risk of recrudescence with concomitant gametocytaemia. This is the essential amplification step that leads to spread (White and Pongtavornpinyo 2003). When we compared different transmission intensity settings, the emergence of resistance was more likely to occur from the blood stage in a low-transmission area where the human population is mostly nonimmunes and therefore treated. The chance of resistance emerging from mutation at the hepatic merozoite stage is much lower for several reasons (Stepniewska and White 2008) but could contribute if the hosts are frequently exposed to antimalarials and tend to have residual drug in their blood.
Understanding the driving force of resistance is necessary when choosing an appropriate treatment strategy. If resistance is driven by treatment failure, i.e. by the selection of mutant parasites in the blood stage at the time of drug treatment, effective ACTs (in combination with a still-effective partner drug) are beneficial for delaying the emergence of artemisinin resistance. However, if resistance is driven by the selection of resistant mutant hepatic merozoites encountering a subtherapeutic drug concentration then the control of presumptive treatments and the choice of combination therapies become more important.
A possible solution to the problem of increasing antimalarial resistance, is to use a combination of three drugs with one being an artemisinin derivative and the two other drugs having longer, matching half-lives, but different mechanisms of action, thereby protecting each other after the rapid elimination of the artemisinin derivative. This could be beneficial in both delaying the emergence and spread of resistance. The use of triple combination drugs is not new: it is routinely used to treat other infectious diseases such as HIV and tuberculosis.
It is important to mention some caveats and limitations to the equations presented in this study. Like all models, the one presented here has its limitations as it does not take into account many important features of malaria biology such as characteristics of the host-immune response, var gene switching rates, gametocytocidal effects of ACTs, complex variations in parasite population size, different gametocyte switching rates for resistant and sensitive parasites (Buckling et al. 1997), patient adherence to drug regimen, mechanisms of resistance and cross-resistance, factors affecting cure rates, fitness costs of resistance for resistant phenotypes and many more.
In particular, the conclusion concerning resistance emergence during the blood stage depends entirely on how we quantify the various impediments that restrict successful transmission of resistant forms from the treated blood stage. These impediments (such as var switching, low survival probabilities) appear substantial to explain the genetic field data that resistance to chloroquine and sulphadoxine–pyrimthamine arises rarely (Hastings 2004). They are formally present as ρR in equation 10 but are absent from the dynamic model equation 8 used to produce Fig. 3. Setting ρR to a low value in equation 10 shows that these impediments can potentially make treated blood stages an unlikely source of mutation.
The assumption that a single point mutation was sufficient for the parasite to become resistant to the treatment may raise some concerns. Some resistance arises from a single point mutation (e.g. atovaquone resistance), but other types of resistance arise through a series of mutations or a primary mutation that provides an essential step, augmented by secondary mutations that progressively increase the level of resistance. A more comprehensive model involving more complex genetic events may be required (Sibley et al. 2001; Hastings et al. 2002a,b; Hyde 2002). When multiple unlinked events are required for encoding resistance and when more than one parasite genotype is considered, there is the possibility of outbreeding of multigenic resistance mechanisms through recombination breakdown during meiosis.
While more complex models will no doubt shed light on the quantitative details of resistance emergence, simpler models provide insights into the basic evolutionary principles at work as the parasite moves through the different stages of its life cycle (Boni et al. 2008a). The simple model presented here gives some clues as to the origins of antimalarial resistance, suggesting that blood-stage replication and small parasite populations encountering residual drug are the most likely scenarios for the emergence of drug resistance. This type of understanding can help us monitor resistance and patient drug levels, and hopefully, design treatments that minimize the probability of resistance emerging. The dynamics of antimalarial treatment and resistance evolution bring together the most basic elements in the fields of medicine and evolution: treatment of a diseased patient with an effective drug and the adaptation of an organism to a novel, unfriendly environment. Knowledge from each field sheds light on the other and will hopefully lead to new developments that improve the long-term efficacy of antimalarial drugs and help relieve the overall human burden of malaria.
Acknowledgments
This study was part of the Wellcome Trust Mahidol University Oxford Tropical Medicine Research Programme (077166/Z/05/F) funded by the Wellcome Trust of Great Britain. IMH is supported by the Liverpool School of Tropical Medicine and Bill & Melinda Gates Foundation (project #39777), and MFB is supported by the MRC Centre for Genomics and Global Health.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Appendix S1. Additional assumptions. All model assumptions based on all three stages where resistance emerges are shown below.
Appendix S2. Details of the dynamic model for calculating the probability of resistance emergence in the blood stage. The model is implemented in the Microsoft Excel file given in the online supporting material.
Appendix S3. Calculation of the probability of resistance emerging at the blood stage when the parasite is at equilibrium.
Appendix S4. The parameter estimates for baseline scenarios when deriving the probability of resistance emerges in the blood stage
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Literature cited
- Adjuik M, Babiker A, Garner P, Olliaro P, Taylor W, White N. Artesunate combinations for treatment of malaria: meta-analysis. Lancet. 2004;363:9–17. doi: 10.1016/s0140-6736(03)15162-8. [DOI] [PubMed] [Google Scholar]
- Ashley EA, White NJ. Artemisinin-based combinations. Current Opinion in Infectious Diseases. 2005;18:531–536. doi: 10.1097/01.qco.0000186848.46417.6c. [DOI] [PubMed] [Google Scholar]
- Barnes KI, White NJ. Population biology and antimalarial resistance: the transmission of antimalarial drug resistance in Plasmodium falciparum. Acta Tropica. 2005;94:230–240. doi: 10.1016/j.actatropica.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Boni MF, Buckee CO, White NJ. Mathematical models for a new era of malaria eradication. PLoS Medicine. 2008a;5:e231. doi: 10.1371/journal.pmed.0050231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni MF, Smith DL, Laxminarayan R. Benefits of using multiple first-line therapies against malaria. Proceedings of the National Academy of Sciences of the United States of America. 2008b;105:14216–14221. doi: 10.1073/pnas.0804628105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckling AG, Taylor LH, Carlton JM, Read AF. Adaptive changes in Plasmodium transmission strategies following chloroquine chemotherapy. Proceedings. Biological Sciences. 1997;264:553–559. doi: 10.1098/rspb.1997.0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross AP, Singer B. Modelling the development of resistance of Plasmodium falciparum to anti-malarial drugs. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1991;85:349–355. doi: 10.1016/0035-9203(91)90286-8. [DOI] [PubMed] [Google Scholar]
- Curtis CF, Otoo LN. A simple model of the build-up of resistance to mixtures of anti-malarial drugs. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1986;80:889–892. doi: 10.1016/0035-9203(86)90248-8. [DOI] [PubMed] [Google Scholar]
- Dondorp A, Nosten F, Stepniewska K, Day N, White N. Artesunate versus quinine for treatment of severe falciparum malaria: a randomised trial. Lancet. 2005;366:717–725. doi: 10.1016/S0140-6736(05)67176-0. [DOI] [PubMed] [Google Scholar]
- Dye C, Williams BG. Multigenic drug resistance among inbred malaria parasites. Proceedings of the Royal Society of London. Series B: Biological Sciences. 1997;264:61–67. doi: 10.1098/rspb.1997.0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatton ML, Hogarth W, Saul A. Time of treatment influences the appearance of drug-resistant parasites in Plasmodium falciparum infections. Parasitology. 2001;123:537–546. doi: 10.1017/s0031182001008824. [DOI] [PubMed] [Google Scholar]
- Hastings IM. A model for the origins and spread of drug-resistant malaria. Parasitology. 1997;115:133–141. doi: 10.1017/s0031182097001261. [DOI] [PubMed] [Google Scholar]
- Hastings IM. The origins of antimalarial drug resistance. Trends in Parasitology. 2004;20:512–518. doi: 10.1016/j.pt.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Hastings IM, Watkins WM. Intensity of malaria transmission and the evolution of drug resistance. Acta Tropica. 2005;94:218–229. doi: 10.1016/j.actatropica.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Hastings IM, Bray PG, Ward SA. Parasitology. A requiem for chloroquine. Science. 2002a;298:74–75. doi: 10.1126/science.1077573. [DOI] [PubMed] [Google Scholar]
- Hastings IM, Watkins WM, White NJ. The evolution of drug-resistant malaria: the role of drug elimination half-life. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences. 2002b;357:505–519. doi: 10.1098/rstb.2001.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutagalung R, Htoo H, Nwee P, Arunkamomkiri J, Zwang J, Carrara VI, Ashley E, et al. A case-control auditory evaluation of patients treated with artemether-lumefantrine. American Journal of Tropical Medicine and Hygiene. 2006;74:211–214. [PubMed] [Google Scholar]
- Hyde JE. Mechanisms of resistance of Plasmodium falciparum to antimalarial drugs. Microbes and Infection. 2002;4:165–174. doi: 10.1016/s1286-4579(01)01524-6. [DOI] [PubMed] [Google Scholar]
- Institute of Medicine. Saving Lives, Buying Time: Economics of Malaria Drugs in an Age of Resistance. Washington, DC: The National Academies Press; 2004. [PubMed] [Google Scholar]
- Koella JC, Antia R. Epidemiological models for the spread of anti-malarial resistance. Malaria Journal. 2003;2:3. doi: 10.1186/1475-2875-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin BR. Models for the spread of resistant pathogens. Netherlands Journal of Medicine. 2002;60:58–64. [PubMed] [Google Scholar]
- Newton PN, Angus BJ, Chierakul W, Dondorp A, Ruangveerayuth R, Silamut K, Teerapong P, et al. Randomized comparison of artesunate and quinine in the treatment of severe falciparum malaria. Clinical Infectious Diseases. 2003;37:7–16. doi: 10.1086/375059. [DOI] [PubMed] [Google Scholar]
- Paget-McNicol S, Saul A. Mutation rates in the dihydrofolate reductase gene of Plasmodium falciparum. Parasitology. 2001;122:497–505. doi: 10.1017/s0031182001007739. [DOI] [PubMed] [Google Scholar]
- Pongtavornpinyo W. Liverpool: Liverpool School of Tropical Medicine; 2006. Mathematical modelling of antimalarial drug resistance. PhD Dissertation. [Google Scholar]
- Price RN, Nosten F, Simpson JA, Luxemburger C, Phaipun L, Ter Kuile F, Van Vugt M, et al. Risk factors for gametocyte carriage in uncomplicated falciparum malaria. American Journal of Tropical Medicine and Hygiene. 1999;60:1019–1023. doi: 10.4269/ajtmh.1999.60.1019. [DOI] [PubMed] [Google Scholar]
- Sibley CH, Hyde JE, Sims PF, Plowe CV, Kublin JG, Mberu EK, Cowman AF, et al. Pyrimethamine-sulfadoxine resistance in Plasmodium falciparum: what next? Trends in Parasitology. 2001;17:582–588. doi: 10.1016/s1471-4922(01)02085-2. [DOI] [PubMed] [Google Scholar]
- Smithuis F, Kyaw MK, Phe O, Aye KZ, Htet L, Barends M, Lindegardh N, et al. Efficacy and effectiveness of dihydroartemisinin-piperaquine versus artesunate-mefloquine in falciparum malaria: an open-label randomised comparison. Lancet. 2006;367:2075–2085. doi: 10.1016/S0140-6736(06)68931-9. [DOI] [PubMed] [Google Scholar]
- Stepniewska K, White NJ. Pharmacokinetic determinants of the window of selection for antimalarial drug resistance. Antimicrobial Agents and Chemotherapy. 2008;52:1589–1596. doi: 10.1128/AAC.00903-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins WM, Mosobo M. Treatment of Plasmodium falciparum malaria with pyrimethamine-sulfadoxine: selective pressure for resistance is a function of long elimination half-life. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1993;87:75–78. doi: 10.1016/0035-9203(93)90431-o. [DOI] [PubMed] [Google Scholar]
- White NJ. Qinghaosu (artemisinin): the price of success. Science. 2008;320:330–334. doi: 10.1126/science.1155165. [DOI] [PubMed] [Google Scholar]
- White NJ, Pongtavornpinyo W. The de novo selection of drug-resistant malaria parasites. Proceedings of the Royal Society of London. Series B: Biological Sciences. 2003;270:545–554. doi: 10.1098/rspb.2002.2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White NJ, Chapman D, Watt G. The effects of multiplication and synchronicity on the vascular distribution of parasites in falciparum malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1992;86:590–597. doi: 10.1016/0035-9203(92)90141-x. [DOI] [PubMed] [Google Scholar]
- World Health Organisation (WHO) Guidelines for the Treatment of Malaria. Geneva, Switzerland: WHO; 2006. [Google Scholar]
- Yeung S, Pongtavornpinyo W, Hastings IM, Mills AJ, White NJ. Antimalarial drug resistance, artemisinin-based combination therapy, and the contribution of modeling to elucidating policy choices. American Journal of Tropical Medicine and Hygiene. 2004;71:179–186. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.