

Summary
Dicer is a specialized ribonuclease that initiates RNA interference (RNAi) by cleaving double-stranded RNA (dsRNA) into small RNA fragments about 22 nucleotides in length. Here, we present the three dimensional structure of human Dicer bound to the protein TRBP at ~20 angstrom resolution determined by negative stain electron microscopy (EM) and single particle analysis. Our analysis reveals that the Dicer-TRBP complex is an L-shaped molecule with a long edge of 150 angstroms and a 100-angstrom extension on one end. A surface trench runs the length of the long edge of the molecule, defining a putative dsRNA-binding site. Docking the crystal structure of Giardia Dicer, which represents the nuclease core of human Dicer, into the EM map suggests two possible overall molecular architectures for human Dicer. These results offer insights into the structure of Dicer proteins found in multi-cellular organisms and provide a conceptual framework for understanding the initiation of RNAi.
Introduction
RNAi is a broad spread mechanism of gene silencing involved in many aspects of eukaryotic biology (Ghildiyal and Zamore, 2009). RNAi is triggered in the cytosol of most eukaryotic cells by the presence of dsRNA (Fire et al., 1998). During the initiation of RNAi the trigger dsRNA is cleaved into small RNA duplexes, typically about 22 nt. in length, by a specialized ribonuclease named Dicer (Bernstein et al., 2001). Dicer products are called small interfering RNAs (siRNA) when generated by sequential cleavages of long dsRNAs (Elbashir et al., 2001). In humans, and many other eukaryotes, Dicer also produces microRNAs (miRNA), which are endogenous small RNAs that regulate the translation of about a third of all human genes (Lewis et al., 2005). Dicer generates miRNAs by cleaving the terminal loop off of endogenous hairpin-structured pre-miRNAs (Grishok et al., 2001; Hutvagner et al., 2001).
Insight into the structural mechanism used by Dicer to generate small RNAs came from the crystal structure of a Dicer protein derived from the protozoan Giardia intestinalis (MacRae et al., 2006). This structure suggested that Dicer functions as a molecular ruler, which recognizes the open end of dsRNA helices and cleaves a set distance away. Giardia Dicer contains a PAZ domain, which binds the end of dsRNA, that is separated from two catalytic ribonuclease III (RNase III) domains by a flat, positively charged surface. The distance between the PAZ and RNase III domains matches the length spanned by 25 base pairs of RNA, which is the typical size of the small RNA products generated by Giardia Dicer.
Although the structure and mechanism of Giardia Dicer has been studied in detail (MacRae and Doudna, 2007; Macrae et al., 2006; MacRae et al., 2007; MacRae et al., 2006), much less is known about Dicer proteins from higher eukaryotes. Moreover, the Giardia enzyme is an atypical Dicer protein in that it is much smaller and simpler than any Dicer protein found in any other organism to date. For example, human Dicer (219 kDa) is nearly 3-times larger than Giardia Dcier (82 kDa). The difference in molecular mass is accounted for by at least five additional protein domains found in most Dicer proteins. These include an N-terminal DExD helicase-like domain (Ma et al., 2008), a TRBP-binding domain (Lee et al., 2006), a putative dsRNA binding domain named DUF283 (Dlakic, 2006), an Ago-binding domain (Sasaki and Shimizu, 2007) and a C-terminal dsRNA binding domain (Provost et al., 2002). These additional domains participate in dsRNA processing, regulate dicing activity and serve as molecular scaffolds for consolidating protein factors involved in the initiation of RNAi (Chendrimada et al., 2005; Gregory et al., 2005; Ma et al., 2008; Maniataki and Mourelatos, 2005; Soifer et al., 2008). Therefore, in order to gain insight into molecular structure of the large Dicer proteins found in multi-cellular organisms we generated a 3D reconstruction of human Dicer bound to the protein TRBP using electron microscopy and single particle analysis.
Results
Reconstitution of Dicer-TRBP complex
We recently established a protocol for in vitro reconstitution of the human RISC-loading complex, a trimeric assembly containing the proteins Dicer, Argonaute2 (Ago2) and TRBP (MacRae et al., 2008). Here, we used a similar protocol to assemble the binary Dicer-TRBP complex from purified recombinant proteins. The assembled Dicer-TRBP complex is stable in solution and can be separated from excess free TRBP by size exclusion chromatography. The Dicer-TRBP complex is catalytically active on pre-miRNA substrates (data not shown). We do not know the stoichiometry of Dicer:TRBP in the reconstituted complex. However, based on previous work with Dicer-TRBP-Ago2 we suspect a ration of 1:1 (MacRae et al., 2008).
We initially hoped that this simple preparation would be suitable for visualization of single particles by EM. However, even after purification by size exclusion these samples often resulted in poor negatively stained images when applied to a carbon grid—many particles appeared aggregated and heterogeneous in size (data not shown). To further purify the Dicer-TRBP complex we therefore included an additional purification step in which applied the complex to a glycerol gradient by ultracentrifugation. The resulting preparation consistently yielded singular, homogenous particles on uranyl acetate stained grids suitable for single particle analysis (Fig. 1).
Figure 1. Reconstitution of Dicer-TRBP complex.

(a) Schematic illustrating the relative size of Human Dicer, Giardia Dicer and TRBP molecules, including major protein domains. The scale bar indicates the number of amino acids. (b) An example of a negatively-stained micrograph of Dicer-TRBP. Particles are mostly mono-dispersed and adopt a large variety of orientations.
Initial model generation of the Dicer-TRBP complex
Once we established a reproducible method for imaging Dicer-TRBP particles in negative stain we endeavored to generate a 3D reconstruction of the complex. The first step towards this goal was to establish a reliable initial model, which is essential to any successful single particle reconstruction. Currently, the only crystal structure of a Dicer protein available is the homolog from Giardia intestinalis. Unfortunately, Giardia Dicer is a poor initial model of Dicer-TRBP because it lacks many domains in human Dicer and is less than a third of the size of the human Dicer-TRBP complex (MacRae et al., 2006). We therefore began our analysis ab initio by generating an initial model using the random conical tilt (RCT) method (Radermacher et al., 1986).
We used a recently developed automated RCT data collection module of the Leginon software package to collect a series of tilt pairs (Yoshioka et al., 2007). After classifying the particles in the untilted micrographs, we used the corresponding particles in the tilted micrographs for volume back-projection, constrained by the geometrical relationship between the particle pairs. Using 840 particles, we obtained an RCT model of the Dicer-TRBP complex (Fig. 2). The model revealed that the Dicer-TRBP complex is an “L”-shaped molecule. The long edge of the model, or back of the L, is approximately 150 Å and the base of the L protrudes out approximately 100 Å.
Figure 2. Random conical tilt (RCT) and initial model generation of Dicer-TRBP complex.
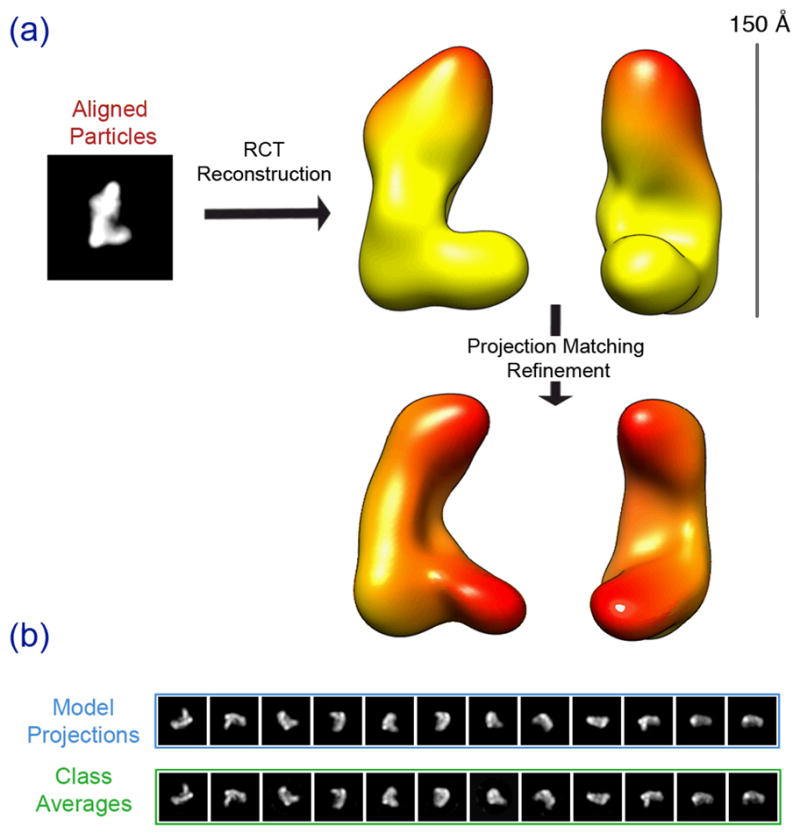
Untilted particles of the RCT datasets are aligned and classified, resulting in a class average shown in (a). RCT reconstruction using the tilted particles provides a 3D volume of Dicer-TRBP, which essentially adopts an “L”-shape. Further refinement of the RCT model against untilted dataset results in a more refined volume (a:bottom). Scale bar indicates the size of the volume. (b) Model projections are in good agreement with the corresponding class averages at the final iteration of projection matching refinement.
We further improved the RCT model by performing a projection-matching based iterative refinement. We collected an untilted 35,000-particle dataset and refined the RCT model by projection-matching using the EMAN software package (Ludtke et al., 1999). The refined model retained the L shape and overall dimensions of the RCT model (Fig. 2). New features also began to emerge during refinement. Most notably, the refined initial model hinted at the presence of a surface grove on the inner face along the long edge of the model.
Validation of the initial model
To further validate and test the robustness of our initial model, we performed a control refinement experiment. From our raw micrographs, the class averages and the RCT model, it is clear that the Dicer-TRBP complex has an overall L-shaped architecture. However, we wanted to verify low-resolution details such as the cleft in the center of the model as well as the shape of the base. Therefore, we generated a synthetic model using two cylinders placed adjacent to one another to mimic the L-shaped structure (Fig. 3). Our rationale was that this model has roughly the same overall shape as the RCT model, but lacks all of the surface details derived from the RCT reconstruction and subsequent projection matching refinement.
Figure 3. Validation of the Dicer-TRBP initial model.

Using cylinders, an “L” shape 3D synthetic model is generated to mimic the dimensions of Dicer-TRBP complex. This synthetic model (a: left) was used as the starting model for projection matching refinement against the same dataset used in Figure 2. The result of the refinement is shown in (a: right). Model projections are compared to their corresponding class averages for the final iteration of projection matching refinement. (b) The refined RCT model (solid) and the refined synthetic cylinder model (transparent) overlayed. The bottom panel compares the corresponding 2D projections between the two models.
The synthetic model was refined using the same protocol and 35,000-particle stack as was used in refinement of the RCT model. The refined synthetic cylinder-model looks very similar to the refined RCT initial model, apart from a mirror symmetry due to the ambiguity of the handedness of the particles in projection (Fig. 3a). Although RCT is in principle capable of determining the correct handedness of the reconstructed volume, we have not seen a compelling feature in the RCT model that can be used as a determinant for handedness. Therefore, we have arbitrarily chosen one as a reference. Overlaying the two models revealed that the two are nearly identical (Fig 3b). This is further illustrated by comparing the 2D model projections of the RCT-refined with the corresponding projections of the synthetic cylinder-refined model. Thus, we have generated an initial model of Dicer-TRBP that converged from two independent starting models. This model reveals the overall shape of human Dicer-TRBP with a high degree of confidence and serves as a robust initial model for obtaining a higher resolution reconstruction of the complex.
Refinement of the Dicer-TRBP complex reconstruction
Having established a reliable initial model of the Dicer-TRBP complex, we next sought to generate a more detailed, higher resolution 3D reconstruction by refining the structure against a large dataset of particles. We collected an untilted dataset in negative stain consisting of about 130,000 Dicer-TRBP particles. We then employed a combination of routines from the EMAN and SPIDER reconstruction packages (Frank et al., 1996; Ludtke et al., 1999) for projection-matching refinement. Briefly, projections of the 3D model and subsequent classification of particles was performed by EMAN, after which a SPIDER script was employed to perform a reference-free hierarchical clustering analysis of the particles in each class. The resulting SPIDER class that exhibited the highest cross-correlation value to the corresponding model projection was used in the creation of the 3D density for the following iteration. The final refined model was reconstructed to an estimated resolution of ~18 Å, as judged by Fourier shell correlation (FSC) at a cutoff of 0.5 (van Heel and Harauz, 1986), and the rmeasure criterion (Sousa and Grigorieff, 2007).
The refined structure of Dicer-TRBP has the L-shape and overall dimensions similar those of the initial model, but also possesses many more fine surface details (Fig. 4). First, there is a clear surface trench that runs along the inner face of the long edge of the L. The trench is most pronounced at the bottom of the L, where it forms a deep cleft between the back and base of the L. There is also a distinct ridge that runs down the backside of the L. The refined structure also suggests that the complex is composed of multiple interconnected globular structures. The base of the L is formed by a globular domain that extends over the deep cleft in the surface trench, and the top of the L appears to be composed of a large globular structure as well.
Figure 4. Iterative Refinement of the Dicer-TRBP initial model.

Using the refined initial model, projection matching iterative refinement was carried out against a 130,000-particle dataset. Panel (a) shows four views of the resulting Dicer-TRBP model. Panel (b) shows the model projections in comparison with the class averages at the final iteration of the refinement protocol. Panel (c) shows the FSC curve of the final iteration of the refinement. At a 0.5 cutoff, the resolution is determined to be approximately 15 Å. Panel (d) shows the particle distribution across all possible euler angles at the final refinement iteration.
Interpretation of the model
Although our reconstruction of the Dicer-TRBP complex is limited to a moderate resolution (about 18 Å), the structure contains sufficient detail to allow us to begin to interpret the model and gain biological insight into the architecture and mechanism of human Dicer. First, we are intrigued by the size and shape of the surface trench that runs along the inner surface of the long edge of the L. The trench is roughly 20 Å in diameter and over 100 Å long. These measurements are nearly identical to the dimensions of the dsRNA substrates that are processed by Dicer. We therefore hypothesize that the surface trench is an extended binding site used by the enzyme to recruit and position dsRNA substrates for catalysis.
Previous biochemical experiments indicate that human Dicer uses a measuring mechanism similar to that of Giardia Dicer to generate small RNAs (Ye et al., 2007; Zhang et al., 2004). We therefore proposed that Giardia Dicer, which is about one third the size of human Dicer, represents the conserved nuclease core structure within the human enzyme (Macrae et al., 2006). To explore this possibility we attempted to locate the position of the Dicer nuclease core in our reconstruction by manually docking the crystal structure of Giardia Dicer into the human Dicer-TRBP model. This exercise immediately revealed that the base of the L is not long enough to accommodate the Giardia Dicer crystal structure (not shown). In contrast, the back portion of the L is both wide enough and long enough to contain Giardia Dicer, and hence, is the likely candidate for nuclease core portion of the complex. We next identified two ways of reasonably docking Giardia Dicer into the Dicer-TRBP volume into the back of the L. We named these two possible positions for the Dicer nuclease core Model A and Model B (Fig. 5). In both Model A and B the distinct ridge on the back of the L aligns with a similarly shaped ridge on the backside of Giardia Dicer. The difference between Model A and Model B is that the position of Giardia Dicer is essentially flipped between the two models—Giardia Dicer in Model A is related to Model B by an approximate 180° rotation perpendicular to the long axis of the L.
Figure 5. Modeling the Giardia Dicer crystal structure into the Dicer-TRBP reconstruction.

Two possible models are proposed based on the docking of the Giardia Dicer crystal structure into the Dicer-TRBP volume. In Model A, the PAZ domain (pink) of Giardia Dicer resides near the top of the “L”, with the modeled dsRNA extending through the base of the “L”, where the RNase III domains (orange) are located. In Model B, the Giardia Dicer is 1800 rotated, with the PAZ domain (pink) now oriented towards the base of the “L” and the RNase III domains (orange) at the tip of the “L”. The views at the bottom of both panels are directed along the long axis of the modeled dsRNA, as indicated by the arrows.
We previously found that the front face of Giardia Dicer binds and processes dsRNA substrates (MacRae et al., 2007; MacRae et al., 2006). Intriguingly, in both Model A and Model B the front face of Giardia Dicer is coincident with the surface trench in Dicer-TRBP that we had hypothesized binds to dsRNA. To examine how Model A and Model B might accommodate a dsRNA substrate we docked a 40-basepair RNA hairpin onto the Giardia Dicer crystal structure in each model. In both Model A and Model B the RNA hairpin is easily accommodated in the surface trench with almost no steric clashes (Fig. 5). In Model A the open end of the hairpin, which is essential for recognition by Dicer (Lund and Dahlberg, 2006; MacRae et al., 2007), lies adjacent to the globular structure at the top of the L. The rest of the modeled RNA extends down along the surface trench and passes under the base of the L where the RNase III domains are positioned. In Model B the open end of the dsRNA is buried in the deepest cleft of the trench, between the back and base of the L. The RNA extends up from this point to the top of the L, where the RNase III domains are set to cleave it. Thus, we have identified a probable dsRNA-binding trench in the Dicer-TRBP complex and proposed two reasonable mechanisms by which this trench may be used by the enzyme to generate small RNAs.
The base of the L, which is too small to be the nuclease core of the enzyme, is a good candidate for the location of N-terminal helicase-like domains of Dicer. Inspection of our model suggests that the base of the L is not an integral part of the nuclease core structure. This brings to mind previous biochemical studies that found that removal of the helicase by mutagenesis does not disrupt stability or activity of the enzyme (Lee et al., 2006; Ma et al., 2008; Macrae et al., 2006). We note, however, that the location of the helicase in our reconstruction has yet to be examined experimentally.
Discussion
One of the major challenges in solving an ab initio structure by single particle electron microscopy is obtaining a reliable initial model. This is especially true for small asymmetric samples that are potentially also conformationally flexible. Here, we report development of an initial model for Dicer-TRBP by RCT and the subsequent refinement of the structure by projection-matching. We are confident in our reconstruction because independent refinements of the RCT model and a test synthetic model converged to a single 3D volume.
The 3D reconstruction of Dicer-TRBP reveals an L-shaped molecule with a surface trench that we believe is likely the dsRNA substrate-binding pocket of the enzyme. We were also able to dock Giardia Dicer into two distinct positions in the Dicer-TRBP model. This is consistent with the proposal that human Dicer contains a nuclease core structure similar to the crystal structure of Giardia Dicer (MacRae et al., 2006). Because Giardia Dicer fits into the Dicer-TRBP model reasonably well in both Model A and B it is not possible for us to distinguish with certainty which is the true orientation of the nuclease core at this point. However, given the present data, we favor Model A over Model B for two reasons. First, the RNase III portion of Giardia Dicer fits into the reconstruction in Model A slightly better than it does in Model B (Fig. 5). Second, the deep cleft in the surface trench of our reconstruction, between the back and base of the L, is highly reminiscent of the “catalytic valley” observed in active site of all RNase III enzymes (Akey and Berger, 2005; Du et al., 2008; Gan et al., 2006; Takeshita et al., 2007). In Model A the catalytic valley of the Giardia Dicer crystal structure falls exactly coincident with the deep cleft in our reconstruction.
Prior to this work all structural information describing Dicer enzymes has been restricted to either the minimal Dicer from Giardia or truncated individual domains of the human enzyme (Du et al., 2008; Takeshita et al., 2007). The reconstructions presented here are the first established structures of an intact full-sized Dicer enzyme from a multi-cellular organism. These results provide a solid foundation for further exploring the structure of human Dicer and understanding the initiation of RNAi on a deeper mechanistic level. The refined structure contains several interesting surface features that remain to be correlated with many of the discrete domains apparent in the amino acid sequence of Dicer. Therefore, future efforts will be directed towards identifying individual domains of Dicer and TRBP in the EM maps and improving the quality and resolution of the structures by employing cryo-EM.
RNAi is a mechanism of gene regulation that touches nearly every aspect of human biology. A comprehensive understanding of RNAi will ultimately require establishing detailed structures of the complex protein assemblies that carry out this essential cellular process. Because Dicer is the main scaffold for the initiation of RNAi the structure presented here represent an important first step towards this goal.
Experimental Procedures
Protein sample preparation
Human Dicer and TRBP proteins were produced and purified separately using a recombinant baculovirus expression system as described (MacRae et al., 2008). To assemble the Dicer-TRPB complex, purified Dicer and TRBP samples were mixed in a 1:2 ratio, ensuring that TRBP was in excess of Dicer. The concentrated sample was then applied to a Superose 6 10/300 GL column (GE Healthcare Life Sciences) equilibrated in 0.1 M NaCl, 0.5 mM TCEP, 40 mM Tris, pH 8, to separate Dicer-TRBP complex from free TRBP.
To further increase the purity of the assembled protein complex, a final purification step using a glycerol gradient was employed. 10 to 30% glycerol gradients containing 150 mM KCl and 20mM HEPES at pH 7.5 were prepared using a Gradient Master 107 (BioComp Instruments, Canada). Typically, 20 uL of Dicer-TRBP, at approximately 1 mg/mL, was layered on top of a 5 ml gradient. Loaded gradients were centrifuged for 24 hours at 45,000 rpm in a Beckman SW60 swinging-bucket rotor. After ultracentrifugation, gradients were separated into 0.4 ml fractions and proteins were visualized by SDS-PAGE using silver stain (Silver Stain Plus Kit, BioRad, USA). The best fractions, judged by protein purity, were pooled and used directly for negative stain microscopy.
Negative staining
Purified Dicer-TRBP samples were negatively stained using the carbon sandwich method (Ohi et al., 2004). Approximately 4 μL of protein sample was deposited on plasma-cleaned, regular C-flat grids (with thin carbon over the holes), and allowed to absorb for 30 to 60 seconds. Excess sample was blotted from the side of the grid and replaced with 2% uranyl acetate (UA) solution. The UA stain was blotted off and replaced with fresh stain. This process was repeated 3 to 5 times. After the last iteration, the grid set in stain for another 30 to 60 sec. While waiting, a piece of thin carbon was floated over a well of UA stain. The grid was then used to pick up the piece of thin carbon. With the top layer of carbon, the grid was blotted from the bottom to remove excess stain and allowed to dry.
Electron microscopy
Data were acquired using a Tecnai F20 Twin transmission electron microscope operating at 120 keV, using a dose of ~20 e-/Å2 and a nominal average defocus of −1.5 μm; 947 and 2023 images were automatically collected during two different sessions at a nominal magnification of between 50,000× and 62,000x at a pixel size of 0.151 nm and 0.131 nm at the specimen level respectively. All images were recorded with a Tietz F415 4 × 4 K pixel CCD camera (15 μm pixel) utilizing the Leginon data collection software (Suloway et al., 2005). Random Conical Tilt (RCT) experiments were carried out using the RCT node of Leginon (Yoshioka et al., 2007), with image pairs taken at 0 and 50 degrees. A total of 335 image pairs were recorded.
Image processing and reconstruction (initial model)
All experimental data were processed by the Appion software package, which interfaces with the Leginon database infrastructure (Lander et al., 2009). For the RCT dataset, particle pairs were picked using tiltpicker (Voss et al., 2009). Contrast transfer function (CTF) corrections for the untilted images were estimated using Automated CTF Estimation (ACE) package (Mallick et al., 2005). Only images whose CTF estimation had an ACE confidence of 0.8 or better were extracted. 11,804 particles from untilted images were then aligned and classified. RCT volumes were generated within Appion from select class average using 840 aligned particle pairs. The synthetic two-cylinder model was generated using the MO 3 method in SPIDER (Frank et al., 1996).
For initial model generation, a 2023-micrograph dataset was processed. Particles were picked using two different methods available in Appion – DoG picker (Voss et al., 2009) and template-based particle picker (Roseman, 2004). CTF for the images were estimated using Automated CTF Estimation (ACE) and corrected at the whole image level. 41,176 particles were extracted from the dataset at a box size of 240 pixels using the 80% confidence CTF cut-off. A crude reference-free classification was run to assess quality of particles as well as to remove bad particles. The final stack contained 35,453 particles.
All 3D reconstructions were carried out using the EMAN reconstruction package (Ludtke et al., 1999). The RCT volume and the two-cylinder model were used as initial models in two separate reconstructions. The resolution of the final volumes were assessed by calculating the FSC at a cutoff of 0.5, resulting in a value of 19 Å for both the RCT model and two-cylinder model (van Heel and Harauz, 1986). Calculation of resolution by rmeasure (Sousa and Grigorieff, 2007) at a 0.5 cutoff yielded a resolution of 27 Å and 28 Å for the RCT model and two-cylinder model, respectively.
Image processing and reconstruction (refined model)
For refinement of the initial model, 146,531 particles were extracted from a dataset of 947 images. Processing was done similarly to the previous untilted dataset. After filtering out the bad particles, the final stack for reconstruction contained 129,554 particles.
For 3D reconstruction, a combination of SPIDER and EMAN reconstruction packages was used (Frank et al., 1996; Ludtke et al., 1999). The starting model was obtained from the initial model generated from the previous step. Creation of projections of the 3D model and subsequent classification of the particles was performed by EMAN, after which a SPIDER script was employed to perform a reference-free hierarchical clustering analysis of the particles in each class. This procedure usually generates approximately 4 – 5 SPIDER classes. The class that exhibited the highest cross-correlation value to the corresponding model projection was used in the creation of the 3D density for the following iteration. The refinement consisted of 25 iterations with angular increments of 100 (180 projections) for the first 10 iterations, 80 (280 projections) for the next 10 iterations and 50 (711 projections) for the final 5 iterations. The resolution of the final reconstruction was assessed by calculating the FSC at a cutoff of 0.5, which provided a value of 15 Å (van Heel and Harauz, 1986), and by rmeasure (Sousa and Grigorieff, 2007) at a 0.5 cutoff, which yielded a value of 21 Å.
Acknowledgments
We are grateful to Kristi E. Kass for technical support during sample preparation. Electron microscopic imaging and reconstruction were conducted at the National Resource for Automated Molecular Microscopy, which is supported by the National Institutes of Health (NIH) through the P41 program of the National Center for Research Resources (RR17573). This work was also supported by the NIH grant R01 GM086701 to I.J.M. P.W.L is a pre-doctoral fellow of the American Heart Association. I.J.M. is a Pew Scholar in the Biomedical Sciences.
Footnotes
Accession Numbers
The refined reconstruction of Dicer-TRBP has been deposited at the Electron Microscopy Data Bank (EMDB) and assigned accession code EMD-1646.
References
- Akey DL, Berger JM. Structure of the nuclease domain of ribonuclease III from M. tuberculosis at 2.1 A. Protein Sci. 2005;14:2744–2750. doi: 10.1110/ps.051665905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, Shiekhattar R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740–744. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlakic M. DUF283 domain of Dicer proteins has a double-stranded RNA-binding fold. Bioinformatics. 2006;22:2711–2714. doi: 10.1093/bioinformatics/btl468. [DOI] [PubMed] [Google Scholar]
- Du Z, Lee JK, Tjhen R, Stroud RM, James TL. Structural and biochemical insights into the dicing mechanism of mouse Dicer: a conserved lysine is critical for dsRNA cleavage. Proc Natl Acad Sci U S A. 2008;105:2391–2396. doi: 10.1073/pnas.0711506105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Gan J, Tropea JE, Austin BP, Court DL, Waugh DS, Ji X. Structural insight into the mechanism of double-stranded RNA processing by ribonuclease III. Cell. 2006;124:355–366. doi: 10.1016/j.cell.2005.11.034. [DOI] [PubMed] [Google Scholar]
- Ghildiyal M, Zamore PD. Small silencing RNAs: an expanding universe. Nat Rev Genet. 2009;10:94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005;123:631–640. doi: 10.1016/j.cell.2005.10.022. [DOI] [PubMed] [Google Scholar]
- Grishok A, Pasquinelli AE, Conte D, Li N, Parrish S, Ha I, Baillie DL, Fire A, Ruvkun G, Mello CC. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell. 2001;106:23–34. doi: 10.1016/s0092-8674(01)00431-7. [DOI] [PubMed] [Google Scholar]
- Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau PW, et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol. 2009;166:95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Hur I, Park SY, Kim YK, Suh MR, Kim VN. The role of PACT in the RNA silencing pathway. Embo J. 2006;25:522–532. doi: 10.1038/sj.emboj.7600942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- Lund E, Dahlberg JE. Substrate selectivity of exportin 5 and Dicer in the biogenesis of microRNAs. Cold Spring Harb Symp Quant Biol. 2006;71:59–66. doi: 10.1101/sqb.2006.71.050. [DOI] [PubMed] [Google Scholar]
- Ma E, MacRae IJ, Kirsch JF, Doudna JA. Autoinhibition of human dicer by its internal helicase domain. J Mol Biol. 2008;380:237–243. doi: 10.1016/j.jmb.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacRae IJ, Doudna JA. An unusual case of pseudo-merohedral twinning in orthorhombic crystals of Dicer. Acta Crystallogr D Biol Crystallogr. 2007;63:993–999. doi: 10.1107/S0907444907036128. [DOI] [PubMed] [Google Scholar]
- Macrae IJ, Li F, Zhou K, Cande WZ, Doudna JA. Structure of Dicer and mechanistic implications for RNAi. Cold Spring Harb Symp Quant Biol. 2006;71:73–80. doi: 10.1101/sqb.2006.71.042. [DOI] [PubMed] [Google Scholar]
- MacRae IJ, Ma E, Zhou M, Robinson CV, Doudna JA. In vitro reconstitution of the human RISC-loading complex. Proc Natl Acad Sci U S A. 2008;105:512–517. doi: 10.1073/pnas.0710869105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacRae IJ, Zhou K, Doudna JA. Structural determinants of RNA recognition and cleavage by Dicer. Nat Struct Mol Biol. 2007;14:934–940. doi: 10.1038/nsmb1293. [DOI] [PubMed] [Google Scholar]
- MacRae IJ, Zhou K, Li F, Repic A, Brooks AN, Cande WZ, Adams PD, Doudna JA. Structural basis for double-stranded RNA processing by Dicer. Science. 2006;311:195–198. doi: 10.1126/science.1121638. [DOI] [PubMed] [Google Scholar]
- Mallick SP, Carragher B, Potter CS, Kriegman DJ. ACE: automated CTF estimation. Ultramicroscopy. 2005;104:8–29. doi: 10.1016/j.ultramic.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Maniataki E, Mourelatos Z. A human, ATP-independent, RISC assembly machine fueled by pre-miRNA. Genes Dev. 2005;19:2979–2990. doi: 10.1101/gad.1384005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohi M, Li Y, Cheng Y, Walz T. Negative Staining and Image Classification - Powerful Tools in Modern Electron Microscopy. Biol Proced Online. 2004;6:23–34. doi: 10.1251/bpo70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provost P, Dishart D, Doucet J, Frendewey D, Samuelsson B, Radmark O. Ribonuclease activity and RNA binding of recombinant human Dicer. EMBO J. 2002;21:5864–5874. doi: 10.1093/emboj/cdf578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radermacher M, Wagenknecht T, Verschoor A, Frank J. A new 3-D reconstruction scheme applied to the 50S ribosomal subunit of E. coli. J Microsc. 1986;141:RP1–2. doi: 10.1111/j.1365-2818.1986.tb02693.x. [DOI] [PubMed] [Google Scholar]
- Roseman AM. FindEM--a fast, efficient program for automatic selection of particles from electron micrographs. J Struct Biol. 2004;145:91–99. doi: 10.1016/j.jsb.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Shimizu N. Evolutionary conservation of a unique amino acid sequence in human DICER protein essential for binding to Argonaute family proteins. Gene. 2007;396:312–320. doi: 10.1016/j.gene.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Soifer HS, Sano M, Sakurai K, Chomchan P, Saetrom P, Sherman MA, Collingwood MA, Behlke MA, Rossi JJ. A role for the Dicer helicase domain in the processing of thermodynamically unstable hairpin RNAs. Nucleic Acids Res. 2008;36:6511–6522. doi: 10.1093/nar/gkn687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa D, Grigorieff N. Ab initio resolution measurement for single particle structures. J Struct Biol. 2007;157:201–210. doi: 10.1016/j.jsb.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. Automated molecular microscopy: the new Leginon system. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Takeshita D, Zenno S, Lee WC, Nagata K, Saigo K, Tanokura M. Homodimeric structure and double-stranded RNA cleavage activity of the C-terminal RNase III domain of human dicer. J Mol Biol. 2007;374:106–120. doi: 10.1016/j.jmb.2007.08.069. [DOI] [PubMed] [Google Scholar]
- van Heel M, Harauz G. Resolution criteria for three dimensional reconstruction. Optik. 1986;73:119–122. [Google Scholar]
- Voss NR, Yoshioka CK, Radermacher M, Potter CS, Carragher B. DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J Struct Biol. 2009;166:205–213. doi: 10.1016/j.jsb.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Paroo Z, Liu Q. Functional anatomy of the Drosophila microRNA-generating enzyme. J Biol Chem. 2007;282:28373–28378. doi: 10.1074/jbc.M705208200. [DOI] [PubMed] [Google Scholar]
- Yoshioka C, Pulokas J, Fellmann D, Potter CS, Milligan RA, Carragher B. Automation of random conical tilt and orthogonal tilt data collection using feature-based correlation. J Struct Biol. 2007;159:335–346. doi: 10.1016/j.jsb.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Kolb FA, Jaskiewicz L, Westhof E, Filipowicz W. Single processing center models for human Dicer and bacterial RNase III. Cell. 2004;118:57–68. doi: 10.1016/j.cell.2004.06.017. [DOI] [PubMed] [Google Scholar]