

Abstract
The first organocatalytic enantioselective radical polycyclization has been accomplished using singly occupied molecular orbital (SOMO) catalysis. The presented strategy relies on a selective single-electron oxidation of chiral enamines formed by condensation of polyenals with an imidazolidinone catalyst employing a suitable copper(II) oxidant. The reaction proceeds under mildly acidic conditions at room temperature and shows compatibility with an array of electron-poor as well as electron-rich functional groups. Upon termination by radical arylation, followed by subsequent oxidation and rearomatization, a range of polycyclic aldehydes has been accessed (12 examples, 54-77% yield, 85-93% ee). The enantioselective formation of up to six new carbocycles in a single catalyst-controlled cascade is described. Evidence for a radical-based cascade mechanism is indicated by a series of experimental results.
Inspired by the biosynthetic origins of steroids and terpenoidal natural products, synthetic organic chemistry has witnessed considerable efforts in the field of biomimetic polyene cyclization.1 Most importantly, cationic polycyclizations, originally pioneered by van Tamelen, Johnson, and Goldsmith, have emerged as a valuable tool for the generation of diverse polycylic skeleta.2,3 While substrate- or auxiliary-mediated induction has been traditionally employed to render such cascade reactions stereoselective, recent advances by Yamamoto, Ishihara, and Gagné have shown that catalytic enantiocontrol can also be exploited in these cation-driven bond constructions.4 In contrast, radical-mediated polyene cyclizations, as originally proposed (and dismissed) by Breslow as a biosynthetic pathway,5 have received considerably less attention.1,6 Moreover, while auxiliary-based cascade radical cyclizations have been reported,6 enantioselective approaches via catalysis remain largely elusive.7,8 With the objective to develop a general method for asymmetric catalytic polyene cyclizations, we reasoned that the application of our previously reported SOMO-activation strategy9 should allow us to establish a powerful cascade reaction10 via a SET-induced open-shell pathway.5,6e-f,11 In this communication, we report the successful execution of these ideals and a new application of SOMO catalysis to the enantioselective construction of multiple C–C bonds and contiguous stereocenters in the context of steroidal and terpenoidal architecture.
Design Plan
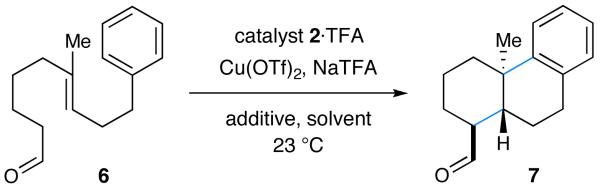
We hypothesized that a suitably functionalized aldehyde (e.g., 1) bearing the requisite degree of tethered unsaturation should condense with imidazolidinone catalyst 2 to give α-imino radical intermediate 3 upon oxidation with an appropriate metal oxidant (Scheme 1). At this stage, we expected radical cation 3 to engage in a series of 6-endo-trig radical cyclizations terminated by a suitable arene to give cyclohexadienyl radical 4. A second oxidation step would then furnish the corresponding cyclohexadienyl cation which, upon rearomatization and liberation of the catalyst, would deliver the requisite pentacycle 5.12 In analogy to our previous SOMO-catalysis studies,9g we presumed that catalyst 2 would favor the α-imino radical geometry as shown for 3 in which the polyene chain is oriented away from the bulky tert-butyl substituent. At the same time the aryl moiety on the catalyst framework should effectively shield the Si-face leaving the Re-face exposed to addition across the proximal trisubstituted alkene. As a key design element, the catalyst-induced enantio- and diastereocontrol that arises in the production of the first carbocycle should be structurally relayed in subsequent ring formations. Moreover, we recognized that the electronic properties of the tethered polyene would play a pivotal role in partitioning the putative single-electron pathway towards cascade ring construction, in lieu of non-productive mechanisms such as (i) radical oxidation or (ii) non-regioselective alkene addition. To this end, polyolefins (such as 1), that incorporate an alternating sequence of polarity-inverted C=C bonds (acrylonitrile and isobutene moieties) were chosen as suitable substrates that would electronically match nucleophilic olefins with electron-deficient radical intermediates (and vice versa). Moreover, the selection of trisubstituted nucleophilic olefins was expected to favor the desired 6-endo-trig mechanism over a competing 5-exo-trig mode, as radical additions to fully substituted carbons are strongly disfavored.13 Similarly, the use of unsaturated nitriles should enforce 6-endo-regiocontrol while functional conversion of C≡N to a range of common steroidal substituents (H, CHO, Me) is known to be operationally trivial.6f,14
Scheme 1.

Enantioselective Polycyclization via SOMO Catalysis.
Our proposed polycyclization strategy was first evaluated using bicyclization substrate 6, imidazolidinone 2a, and a series of metal oxidants. Surprisingly, our initial survey revealed that strong oxidants which had been successful in previous SOMO-activation studies (e.g., cerium(IV) ammonium nitrate (CAN)9a-d,f or [Fe(phen)3](PF6)39g) did not generate any of the desired tricyclic product 7. In both cases, products derived from premature oxidation of the tertiary alkyl radical intermediate or capture of the radical by external nucleophiles were predominant.9d In contrast, the use of Cu(OTf)2 in acetonitrile with sodium trifluoro-acetate (NaTFA) as a base furnished a small amount of the desired tricycle 7 albeit in low enantioselectivity (Table 1, entry 1).15,16 Subsequent optimization studies revealed that and slow addition of the oxidant (via syringe pump) greatly improved the yield of desired tricyclic product 7 presumably due to the kinetic preference for a unimolecular cascade process versus an interrupted pathway that involves bimolecular radical oxidation. While it is possible that the carbocation intermediates from an interrupted step may still participate in a productive cyclization pathway, it is clear that β-H elimination remains the dominant mechanism in this case.17 A subsequent evaluation of catalyst architecture revealed markedly improved enantioselection with catalyst 2b which replaces the benzyl group of catalyst 2a with the extended shielding of a (1-naphthalene)methyl moiety (74% ee, entry 4). Moreover, a survey of reaction media revealed a pronounced effect on enantiomeric excess in that a mixture of isobutyronitrile with 1,2-dimethoxyethane (3:2) gave rise to synthetically useful results (70% yield, 87% ee, entry 6).
Table 1.
Reaction Optimization for Enantioselective Bicyclization.a
 |
|||||
|---|---|---|---|---|---|
| entry | catalyst | additiveb | solvent | yield (%)c | ee (%)d |
| 1 | 2a, Ar = Ph (20 mol%) | none | MeCN | 11 | 34 |
| 2 | 2a, Ar = Ph (20 mol%) | TFA | MeCH | 16 | 35 |
| 3e | 2a, Ar = Ph (20 mol%) | TFA | MeCH | 42 | 42 |
| 4e | 2b, Ar = 1-Np (20 mol%) | TFA | MeCH | 56 | 74 |
| 5e | 2b, Ar = 1-Np (20 mol%) | TFA | i-PrCN/DMEf | 54 | 87 |
| 6e | 2b, Ar = 1-Np (30 mol%) | TFA | i-PrCN/DMEf | 70 | 87 |
Reactions peformed on a 0.20 mmol scale using 2.5 equiv of Cu(OTf)2 and 2.0 equiv of NaTFA.
3.0 equiv of TFA used.
Isolated yield.
ee determined by chiral HPLC analysis.
Slow addition of oxidant and base as a solution in MeCN or i-PrCN.
3:2 i-PrCN/DME (0.08 M).
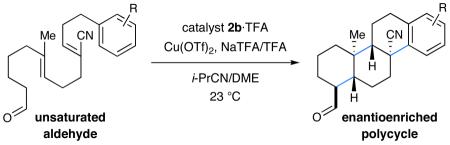
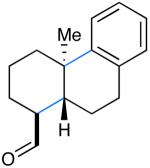
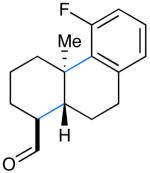
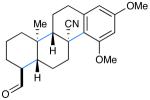
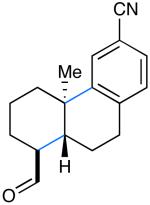
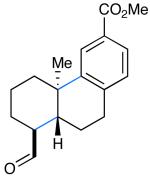
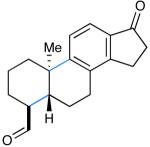
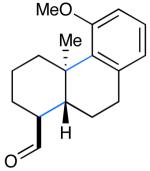
We next evaluated the generality of our newly developed organocatalytic protocol in a series of cascade bicyclizations (Table 2). A wide array of arenes readily act as terminating groups; in addition to electron-neutral phenyl rings (7, 70% yield 87% ee), electron-rich and poor aryl aldehydes also participate to furnish 8-12 in good yields and comparable enantioselectivty (65-77% yield, 85-90% ee).18 While the reaction yields for all aryl-terminators examined were similar, we observed that electron-deficient arenes were successful over a larger range of reaction concentrations, a result that supports the matched interaction of a nucleophilic trialkyl radical with an electron-poor terminator in the final SRAr cyclization.19 In such cases, we presume that electron-withdrawing substituents increase the substitution rate and stabilize the newly formed cyclohexadienyl radical intermediate. Notably, when meta-substituted arenes were subjected to these cascade conditions, cyclization at the aryl-2-position was predominant (to render products 10 and 12), again emphasizing the radical nature of this process (2-4:1 ortho:para selectivity, 65-75% yield, 88-90% ee).9g,19 Indeed, the prevalent formation of ortho-substituted product 12 is in sharp contrast to the literature precedent for para-selectivity when cationic conditions were employed with similar polyene substrates.2d It is important to note that all products of this survey were obtained as single diastereomers.20
Table 2.
 | ||
|---|---|---|
|
|
|
| 7: 70% yield | 10d: 65% yield | 13: 61% yield |
| 87% ee | 90% eee | 91% ee |
|
|
|
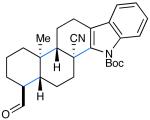
| 8: 74% yield | 11: 77% yield | 14: 71% yield |
| 88% ee | 87% ee | 92% ee |
|
|
|
| 9: 76% yield | 12d: 75% yield | 15: 54% yield |
| 85% ee | 88% eee | 86% ee |
Conditions: Slow addition (7 h) of Cu(OTf)2 (2.5 equiv), NaTFA (2.0 equiv), and TFA (3.0 equiv) in i-PrCN (2 parts) to aldehyde and catalyst (30 mol%) in i-PrCN/DME (1:2) to give 0.08 M solution and subsequent stirring for 17 h at room temp.
Isolated yield.
ee determined by chiral HPLC analysis.
Mixture of ortho/para (4:1 for 10; 2:1 for 12).
ee of ortho product; 91% ee for para regioisomer of 12.
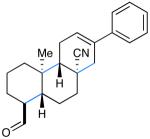
We next tested the capacity of our SOMO catalysis strategy to accomplish polyene tricyclization. Based on our design principles (outlined above), we assumed that aldehydic substrates that incorporate an alternating sequence of electron-rich/electron-poor olefinic acceptors (isobutene and acrylonitrile) would be suitably matched to allow intramolecular radical chain propagation prior to coupling and termination with a suitable π-rich aryl ring.9g,19 To our delight, a range of tricyclization substrates readily furnished the desired products 13-15 in moderate to good yields and useful enantioselectivities (Table 2, 54-71% yield, 86-92% ee).21 It should be noted that electron-rich benzene (13) and indole (14) terminators can be tolerated using these mild oxidative conditions. Moreover, the successful generation of tricyclic aldehyde 15 reveals that π-nucleophilic olefins such as 1,1-disubstituted styrenes can also serve as suitable terminating groups.
As outlined in Scheme 2, we finally sought to probe the limitations of this radical cyclization strategy via application to extended ring systems such as tetra-, penta- and hexacycles. Remarkably, for the SOMO-catalyzed tetracyclization of trienal 1 we observed similar levels of reaction efficiency and enantiocontrol to those obtained in bicycle and tricycle formation (pentacycle 5, 56% yield, 92% ee, single diastereomer). Moreover, extension of this multi-bond forming reaction to tetraene cascade ring construction cleanly afforded the pentacyclization product 16 in almost identical yield and enantiomeric excess (63% yield, 93% ee). Perhaps most notably, this new SOMO-polyene cyclization concept has been successfully extended to the enantioselective22 production of the hexacyclization adduct 17 as single diastereomer and in 62% yield (corresponding to an average yield of 92% per bond formation). In the course of this cascade bond construction, a total of eleven contiguous stereocenters, of which five are all-carbon quaternary centers, were formed from a simple acyclic starting material under the influence of imidazolidinone 2b. It is instructive to consider that the last stereogenic center formed in the course of this cascade resides 11.7 Å from the catalyst binding point23 thus showcasing the stereoinduction efficiency of this new SOMO cyclization sequence.
Scheme 2.

Extended Ring Systems by Organo-SOMO Catalysis.a
In summary, we have developed the first catalytic enantioselective cyclization strategy to access steroidal and terpenoidal frameworks using organocatalysis. This strategy represents an ambient temperature protocol, which is previously unprecendented in SOMO-activation catalysis with respect to carbon-carbon bond formation. Future work will be devoted to the application of this new technology to the synthesis of complex natural products and pharmaceutically relevant entities.
Supplementary Material
Acknowledgement
Financial support was provided by NIHGMS (R01 01 GM093213-01) and kind gifts from Merck and Amgen. S. R. thanks the Deutsche Forschungsgemeinschaft (DFG) for a postdoctoral fellowship (Re 2882/1-1).
Footnotes
Supporting Information Available. Experimental procedures, syntheses of starting materials, X-ray crystallographic proof of absolute configuration, and spectral data for all new compounds are provided (162 pages) (PDF).
References
- (1) (a).Yoder RA, Johnston JN. Chem. Rev. 2005;105:4730–4756. doi: 10.1021/cr040623l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2) (a).van Tamelen EE. Acc. Chem. Res. 1968;1:111. [Google Scholar]; (b) van Tamelen EE, McCormick JP. J. Am. Chem. Soc. 1969;91:1847. doi: 10.1021/ja00706a071. [DOI] [PubMed] [Google Scholar]; (c) Johnson WS, Kinnel RB. J. Am. Chem. Soc. 1966;88:3861. doi: 10.1021/ja00968a035. [DOI] [PubMed] [Google Scholar]; (d) Johnson WS, Semmelhack MF, Sultanbawa MUS, Dolak LA. J. Am. Chem. Soc. 1968;90:2994. [Google Scholar]; (e) Goldsmith DJ, Phillips CF. J. Am. Chem. Soc. 1969;91:5862. [Google Scholar]
- (3) (a).For an early review, see: Bartlett PA. In: Asymmetric Synthesis. Morrison JD, editor. Vol. 3. Academic Press; Orlando: 1984. p. 341. For selected examples of substrate- or auxiliary-controlled enantioselective synthesis, see: Bartlett PA, Johnson WS. J. Am. Chem. Soc. 1973;95:7501. doi: 10.1021/ja00803a047. Johnson WS, Brinkmeyer RS, Kapoor VM, Yarnell TM. J. Am. Chem. Soc. 1977;99:8341. doi: 10.1021/ja00467a048. Johnson WS, Fletcher VR, Chenera B, Bartlett WR, Tham FS, Kullnig RK. J. Am. Chem. Soc. 1993;115:497. Corey EJ, Lin S. J. Am. Chem. Soc. 1996;118:8765. Bogenstätter M, Limberg A, Overman LE, Tomasi AL. J. Am. Chem. Soc. 1999;121:12206. For a reagent-controlled enantioselective example, see: Zhao Y-J, Loh T-P. J. Am. Chem. Soc. 2008;130:10024. doi: 10.1021/ja802896n.
- (4) (a).Ishihara K, Nakamura S, Yamamoto H. J. Am. Chem. Soc. 1999;121:4906. [Google Scholar]; (b) Ishihara K, Ishibashi H, Yamamoto H. J. Am. Chem. Soc. 2001;123:1505. [Google Scholar]; (c) Ishibashi H, Ishihara K, Yamamoto H. J. Am. Chem. Soc. 2004;126:11122. doi: 10.1021/ja0472026. [DOI] [PubMed] [Google Scholar]; (d) Sakakura A, Ukai A, Ishihara K. Nature. 2007;445:900. doi: 10.1038/nature05553. [DOI] [PubMed] [Google Scholar]; (e) Mullen CA, Campbell MA, Gagné MR. Angew. Chem., Int. Ed. 2008;147:6011. doi: 10.1002/anie.200801423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5) (a).Breslow R, Barrett E, Mohaosi E. Tetrahedron Lett. 1962:1207. Breslow R, Olin SS, Groves JT. Tetrahedron Lett. 1968:1837. For a related example, see: Lallemand JY, Julia M, Mansuy D. Tetrahedron Lett. 1973:4461.
- (6) (a).For a review, see: Dhimane A-L, Fensterbank L, Malacria M. In: Radicals in Organic Synthesis. Renaud P, Sibi MP, editors. Vol. 2. Wiley-VCH; Weinheim: 2001. p. 350. For selected non-enantioselective examples, see: Chen L, Gill GB, Pattenden G. Tetrahedron Lett. 1994;35:2593. Handa S, Nair PS, Pattenden G. Helv. Chim. Acta. 2000;83:2629. Dombroski MA, Kates SA, Snider BB. J. Am. Chem. Soc. 1990;112:2759. Zoretic PA, Weng X, Caspar ML, Davis DG. Tetrahedron Lett. 1991;32:4819. Zoretic PA, Fang H, Ribeiro AA. J. Org. Chem. 1998;63:7213. doi: 10.1021/jo980518+. For selected substrateor auxiliary-controlled examples, see: Heinemann C, Demuth M. J. Am. Chem. Soc. 1997;119:1129. Heinemann C, Demuth M. J. Am. Chem. Soc. 1999;121:4894. Barrero AF, Herrador MM, Quílez del Moral JF, Arteaga P, Arteaga JF, Piedra M, Sánchez EA. Org. Lett. 2005;7:2301. doi: 10.1021/ol050335r.
- (7) (a).For examples of enantioselective atom-transfer radical bicyclizations employing chiral Lewis acids, see: Yang D, Gu S, Yan Y-L, Zhao H-W, Zhu N-Y. Angew. Chem., Int. Ed. 2002;41:3014. doi: 10.1002/1521-3773(20020816)41:16<3014::AID-ANIE3014>3.0.CO;2-J. Yang D, Zheng B-F, Gao Q, Shen G, Zhu N-Y. Angew. Chem., Int. Ed. 2006;45:255. doi: 10.1002/anie.200503056.
- (8) (a).For reviews on catalytic enantioselective radical (cascade) reactions, see: Godineau E, Landais Y. Chem.—Eur. J. 2009;15:3044. doi: 10.1002/chem.200802415. Miyabe H, Takemoto Y. Chem.—Eur. J. 2007;13:7280. doi: 10.1002/chem.200700864. Sibi MP, Manyem S, Zimmermann J. Chem. Rev. 2003;103:3263. doi: 10.1021/cr020044l.
- (9) (a).Beeson TD, Mastracchio A, Hong J-B, Ashton K, MacMillan DWC. Science. 2007;316:582. [PubMed] [Google Scholar]; (b) Yang H-Y, Hong J-B, MacMillan DWC. J. Am. Chem. Soc. 2007;129:7004. doi: 10.1021/ja0719428. [DOI] [PubMed] [Google Scholar]; (c) Kim H, MacMillan DWC. J. Am. Chem. Soc. 2008;130:398. doi: 10.1021/ja077212h. [DOI] [PubMed] [Google Scholar]; (d) Graham TH, Jones CM, Jui NT, MacMillan DWC. J. Am. Chem. Soc. 2008;130:16494. doi: 10.1021/ja8075633. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Amatore M, Beeson TD, Brown SP, MacMillan DWC. Angew. Chem., Int. Ed. 2009;48:5121. doi: 10.1002/anie.200901855. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wilson JE, Casarez AD, MacMillan DWC. J. Am. Chem. Soc. 2009;131:11332. doi: 10.1021/ja904504j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Conrad JC, Kong J, Laforteza BN, MacMillan DWC. J. Am. Chem. Soc. 2009;131:11640. doi: 10.1021/ja9026902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10) (a).Walji AM, MacMillan DWC. Synlett. 2007:1477. [Google Scholar]; (b) Enders D, Grondal C, Hüttl MRM. Angew. Chem., Int. Ed. 2007;46:1570. doi: 10.1002/anie.200603129. [DOI] [PubMed] [Google Scholar]; (c) Tietze LF. Chem. Rev. 1996;96:115. doi: 10.1021/cr950027e. [DOI] [PubMed] [Google Scholar]
- (11).Snider BB. Chem. Rev. 1996;96:339. doi: 10.1021/cr950026m. [DOI] [PubMed] [Google Scholar]
- (12).This mechanism is in agreement with our previously described intramolecular α-arylation of aldehydes; see ref. 9g.
- (13) (a).Curran DP, Giese B, Porter NA. Stereochemistry of Radical Reactions. VCH; Weinheim: 1996. p. 71. [Google Scholar]; (b) Spellmeyer DC, Houk KN. J. Org. Chem. 1987;52:959. doi: 10.1021/jo950884i. [DOI] [PubMed] [Google Scholar]; (c) Beckwith AJL. Tetrahedron. 1981;37:3073. [Google Scholar]
- (14).Tobisu M, Nakamura R, Kita Y, Chatani N. J. Am. Chem. Soc. 2009;131:3714. doi: 10.1021/ja810142v. [DOI] [PubMed] [Google Scholar]
- (15) (a).Jenkins CL, Kochi JK. J. Am. Chem. Soc. 1972;94:843. [Google Scholar]; (b) Jenkins CL, Kochi JK. J. Am. Chem. Soc. 1972;94:856. [Google Scholar]
- (16).Additon of both base (NaTFA or NaHCO3) and TFA was found to be necessary for high conversion. Replacement of Cu(OTf)2/NaTFA by Cu(TFA)2·H2O/NaOTf showed inferior reactivity presumably due to the kinetic inaccessibility of the active oxidant [(i-PrCN)4Cu]X2 (outer sphere complex, X = OTf, TFA) from inner sphere complex Cu(TFA)2; ref. 15a.
- (17).Under the reaction conditions alkene protonation followed by subsequent cationic cyclization is not a feasible process. Using typical cationic cyclization conditions (see ref. 4d), a cationic cyclization could be achieved in a separate step.
- (18).Structural assignments of 8 and 13 as well as absolute configuration of 8 (upon chemical derivatization) were secured by X-ray crystallographic analysis; see Supporting Information for details.
- (19).Tiecco M, Testaferri L. In: Reactive Intermediates. Abramovitch RA, editor. Vol. 3. Plenum Press; New York: 1983. p. 61. [Google Scholar]
- (20).General Procedure for the Enantioselective Polyene Cyclization: An oven-dried vial equipped with a magnetic stir bar and a rubber septum was charged with a solution of catalyst 2b·TFA (0.060 mmol, 0.30 equiv) and the polyenal (0.200 mmol, 1.00 equiv) in i-PrCN/DME (1:2, 1.5 mL) under argon atmosphere. To this mixture, a solution of Cu(OTf)2 (0.500 mmol, 2.50 equiv), NaTFA (0.400 mmol, 2.00 equiv), and TFA (0.400 mmol, 2.00 equiv) in i-PrCN (1.0 mL) was added slowly using a syringe pump within 7 h at room temp. After stirring had been continued for further 17 h, the light-green solution was subjected to aqueous work-up and purified by flash chromatography to afford the cyclization product.
- (21).Further evidence against a radical polar crossover reaction mechanism was obtained by the examination of a mismatched tricyclization substrate consisting of two trisubstituted electron-rich alkene moieties: Significantly lower reaction efficiency (25% yield of desired tetracycle) and only partial conversion were observed.
- (22).We assume similar levels of enantiocontrol as observed for 5 and 16.
- (23).Estimated by semi-empirical PM3 calculation of 17 (Gaussian 03).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.