

Abstract
Histone acetylation plays an important role in the silencing and activation of genes involved in tumoregenesis. Trichostatin A, originally identified as an anti-fungal drug, is a potent inhibitor of histone deacetylase (HDAC) with potential anti-tumor activity. In this study, we investigated the effect of M344, an amide analogues of trichostatin A, on the growth and differentiation of THP-1 human leukemia cells. We showed that at low doses, (< 0.2 μM), M344 could inhibit the growth of THP-1 cells at G1 phase in vitro with low cytotoxic effect. Low dose of M344 exerted some differentiating effect on THP-1 cells as judged by the expression of c-fms proto-oncogene (M-CSF receptor) and appearance of adherent cells. Growth arrest induced by M344 is associated with increased levels of cyclin–dependent protein kinase inhibitor p21 and cyclin E, in agreement with G1 phase arrest. At higher doses (2 μM), M344 could induce THP-1 cells to undergo apoptosis, which was associated with the cleavage of PARP, cytochrome c release and activation of both caspases-8, -9, followed by the activation of caspase-3. In addition, M344 could increase the levels of pro-apoptotic protein Bax but decreased the levels of anti-apoptotic protein XIAP. M344 is a potent activator of NF-κB transcription factor. RT-PCR assay showed that the M344 could transiently increase IL-1 expression yet markedly decreased TNF-α expression. Our results show that M344 is a potent growth inhibitor and inducer of apoptosis in human leukemia cells and suggest potential therapeutic strategies of HDAC inhibitors for patients with leukemias.
Keywords: apoptosis, cell cycle, differentiation, histone deacetylase inhibitor, leukemia, M344
1. Introduction
The nucleosome core particle is composed of two copies of each histone protein (H2A, H2B, H3 and H4) and 146 base pairs of superhelical DNA. Nuclear processes such as DNA replication, transcription and rearrangements during cell differentiation and/or proliferation are dependent on the chromatin structure and the access of the regulatory proteins to the DNA [1, 2]. The physical and chemical characters of histone proteins play a key role in these processes. One of the regulatory mechanisms involved in the gene expression and transcription is mediated through post-translational modification (acetylation/deacetylation) of the core histone proteins. Modification of histone proteins is established and maintained through two classes of enzymes with opposite functions [3–6]: 1) histone acetyl transferase (HATs) and 2) histone deacetylases (HDACs) [7]. Inhibition of the histone deacetylase will increase the levels of acetylated histone proteins, which allow the easy access of the regulatory proteins and transcriptional factors to the target genes, resulting in increased gene expression.
Recent studies show that histone acetylation/deacetylation may play an important role in the silencing or activation of genes involved in tumoregenesis. Normal cells contain appropriate state of histone acetylation, which is essential for them to undergo normal differentiation and maturation. Tumor cells are characterized by abnormal cell differentiation and proliferation. Inappropriate states of histone acetylation have been found in numerous types of malignant cells [8, 9], implicating potential roles of histone acetylation in tumorigenesis. In recent year, a number of HDAC inhibitors, such as TSA, SAHA, pyroxamde etc., have been identified by various investigators. In vitro studies showed that these inhibitors could induce various tumor cells to undergo growth arrest, differentiation, and/or apoptosis [10–12]. Thus, the use of agents that alter the status of histone acetylation may allow the switching on or off of deregulated genes and induce either differentiation or block uncontrolled proliferation in transformed cells.
Trichostatin A (TSA), a hydroxamic acid, was originally isolated from culture broth of Streptomyces platensis as an anti-fungial antibiotic. Later it was found to be a potent inhibitor of histone deacetylase, which leads to wide investigation as a potential antineoplastic agent [13]. Accumulated evidences suggest that TSA can induce malignancy cells to undergo differentiation or apoptosis and induce cell cycle arrest [14]. TSA has been emerging as a potent novel antineoplastic agent for treating both solid and hematological malignancies. M344 is an amide analogue of trachostatin A developed recently by Jung and co-investigators [15]. Likewise, M344 exhibited an anti-malignance activity, which can induce terminal differentiation of Friend leukemia cells as well as anti-proliferation activity in culture.
In this study, we investigated the effect of M344 on the proliferation and differentiation of human THP-1 leukemia cells. We now report here that M344 is a growth inhibitor at low doses with limited differentiating effect and a potent inducer of apoptotic cell death at high doses in THP-1 leukemia cells. In addition, we showed that G1 growth arrest of THP-1 cells induced by M344 is associated with increased levels of p21 kinase inhibitor and cyclin E.
2. Material and methods
2.1 Cell and Reagents
Human THP-1 monocytic leukemia cell line is obtained from ATCC (Manassas, VA). THP-1 cells were maintained in RPMI 1640 medium containing 10% FBS, 100 unit/ml of penicillin and 100 μg/ml of streptomycin, 100 μM L-glutamine, and 0.1 M HEPES.
M344, an amide analog of Trichostatin A, was purchased from CALBIOCHEM (San Diego, CA, Cat. No. 647925). Anti-c-fms, anti-Bax, anti-p65 Rel A, anti-cytochrome c antibodies were obtained from Santa Cruz Biotechnology Inc (Santa Cruz, CA). Anti-XIAP antibody was obtained from MBL (Nagoya, Japan). Anti-cyclin A, B1, E, p21 and p34, and anti-human caspase-8 antibodies were purchased from PharMingen, Inc. (San Diego, CA). Rabbit anti-human caspase-3 substrate set III were purchase from CALBIOCHEM (San Diego, CA). Monoclonal mouse anti-poly (ADP-ribose) polymerase (PARP) was purchased from Biomol Research Laboratories Inc (Plymouth Meeting, PA). Acridine orange was a product of Sigma (Sigma, St. Louis, MO).
2.2 Acridine Orange Staining
After treatment, 1 × 106 THP-1 cells were harvested and washed once with cold PBS, the cells were then re-suspended in 0.1 ml PBS and added 15 μL of 100 μg/ml acridine orange solution, incubating for 15 min at room temperature. The staining cells were counted under fluorescence microscope. Cells with highly condensed chromatin and fragmented nuclei that are uniformly stained by acridine orange were counted as apoptotic cells. A minimum of 100 cells per sample was counted [16].
2.3 Cellular Fractionation and Caspases Activity Assay
We followed the procedures described previously [16].
2.4 Western Blot [17]
Total cell lysates were boiled in an SDS gel-loading buffer with DTT for 8 min. The total protein concentration was determined using Bio-Rad DC protein assay kit. The samples were subjected to electrophoresis on 10% SDS-polyacrylamide gel. After electrophoresis, proteins were transferred to a 0.2 μm nitrocellulose membrane (Schleicher & Shuell, Keene, NH) at 4°C, 14V overnight. Nonspecific binding sites on the nitrocellulose membrane were blocked by incubating in blocking buffer (5 % nonfat milk in TBS-tween 20) for 2 hours at room temperature. The blots were washed by TBS-tween once and incubated for 2 hours at room temperature with the primary antibody. After removal of excess primary antibody with three washes, the blots were incubated with a secondary antibody (goat anti-mouse or goat anti-rabbit antibodies conjugated with horseradish peroxidase). The membrane was developed with the enhanced chemiluminescence (ECL) reagent and exposed to Hyperfilm-ECL (Amersham Life Science Corp.) for visualization [16].
2.5 Propidium Iodide Staining and Flow Cytometry assay
The THP-1 cells were treated with 2 μM M344 for various time periods. After washing, cells were re-suspended in 0.5 ml PBS and fixed in 4.5 ml cold 70% ethanol. For propidium Iodide staining, cells were washed in cold PBS once and re-suspended in 1 ml of propidium iodide staining solution containing 20 μg/ml propidium iodide, 0.1 % Triton X-100, 200 μg/ml Dnase free Rnase A, for 15 min at room temperature. Cells were then subject to flow cytometric analysis with FACScan (Becton Dickinson, San Jose, CA). Data were analyzed by Modfit Software for cell cycle profiles.
2.6 Electrophoretic mobility-shift assay (EMSA)
EMSA was carried out as described previously [18]. Briefly, 5×106 cells were washed with cold phosphate-buffered saline (PBS) once, and suspended in 400 μl cold lysis buffer (10 mM HEPES-KOH, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, and 1 x Protease Inhibitor Cocktail) with constantly flicking the tube. The cells were allowed to swell on ice for 15 min, and then vortexed vigorously for 10 sec. The homogenate was centrifuged at 6500 rpm for 30 sec, and the supernatant fraction was discarded. The nuclear pellet was resuspended in 50 μl ice-cold nuclear extraction buffer (20 mM HEPES-KOH, pH 7.9, 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, and 1x Protease Inhibitor Cocktail) and incubated on ice for 20 min for high-salt extraction. The mixture was centrifuged for 5 min at 4°C, and the supernatant (nuclear extract) was either used immediately or stored at −70°C.
EMSA was performed by incubating 5 μg of nuclear extract with 32P-labeled NF-κB p50 oligonucleotides, 5′-GCC ATG GGG GGA TCC CCG AAG TCC-3′ (Geneka Biotechnology Inc., Canada) for 25 min at room temperature in a reaction buffer (10 mM Tris, pH 7.5, 50 mM NaCl, 1 mM DTT, 1 mM EDTA, 5% glycerol). The DNA-protein complexes formed were separated from free oligonucleotides on 4% native polyacrylamide gel containing 50 mM Tris pH 7.5, 0.38 M glycine and 2 mM EDTA. The gel was dried and visualized by autoradiography.
2.7 RT-PCR assay
Total RNA was extracted from cultured THP-1 cells using Trizol reagent (Invitrogen Inc. Carlsbad, CA). cDNA was prepared by mixing 1 μg of total cellular RNA with 100 U of M-MLV reverse transcriptase and 20 mM each of dNTP, and oligos d(T)16 (25 μM) in a total volume of 20 μl. The mixtures were incubated at 42° C for 45 min followed by 5 min incubation at 95°C. Seventy-eight μl of PCR master mixtures containing 0.25 U Tag DNA polymerase were then dispensed into each RT product and the corresponding template. Thereafter, 1 μl of “downstream” and “upstream” primers (50 pmol each) were added to relevant tubes. The primers used for PCR were as follows: IL-1α (product size 344bp), forward 5′-CAT CGC CAA TGA CTC AGA GGA AGA-3′, reverse 5′-TGG TTT TGG GTA TCT CAG GCA TCT C-3′, TNF-α (product size 417bp), forward 5′-CCC CAG GGC TCC AGG CGG TGC TTG T-3, reverse 5′-GGA GAC GGC GAT GCG GCT GAT GGT G-3′ and IFN-(product size 324bp), forward 5′-CAG GTC ATT CAG ATG TAG CGG ATA A-3′, reverse 5′-TAG CTG CTG GCG ACA GTT CA-3′). The reaction was carried out in a Perkin Elmer 480 Thermal Cycler under optimal conditions for 30–35 cycles. The PCR reaction products were analyzed by 1.5 % agarose mini-gel electrophoresis after staining with 0.5 μg/ml ethidium bromide. The specificity of the amplified bands was confirmed by the predicted size and by running known PCR templates provided by Clontech for each product.
3. Results
3.1 Effects on THP-1 Cell Proliferation and Differentiation by M344
M344 could induce differentiation and inhibit proliferation of murine erythroleukemia cells at 2 μM [15]. To obtain direct evident that the M344 inhibit THP-1 cell proliferation, we treated the cells with various doses of M344 for 24 hrs or for indicated time, and cells excluding trypan blue dye were counted under a light microscope. We found that, comparing to no treatment group, M344 inhibited THP-1 cell proliferation in a dose-dependent manner (Fig 1A). M344 at 0.2 μM was sufficient to induce a significant inhibition of THP-1 cell growth (Fig 1B). At higher doses (>2 μM), M344 induced cell death as demonstrated by trypan blue staining (data not shown). Western blot analysis showed that M344 induced expression of c-fms (M-CSF receptor), a marker of mononuclear cell differentiation, most evidently at 0.02 μM concentration within 24 hr (Fig 1C). At higher doses (0.2 – 2 μM), M344 is less effective in stimulating the expression of c-fms; and prolonged treatment (48 hr) with M344 at 0.2 μM concentration was required to enhance the expression of c-fms in THP-1 cells (Fig. 1C). Yet, treatment of THP-1 cells with M344 did not significantly increase the number of adherent cells (less than 8%), a functional marker of mature mononuclear cells (Fig. 1D).
Fig 1.

The effects of M344 on cell growth and c-fms expression. A) THP-1 cells were treated with increasing doses of M344. After 24 hrs, cells were stained by trypan blue solution and counted under light microscope. B) THP-1 cells were incubated with 0.2 μM and 2 μM M344, respectively. After indicated time, cells were stained by trypan blue solution and unstained cells were counted under light microscope. C) THP-1 cells were treated with different doses of M344 for 24 hr, or with 0.2 μM of M344 for indicated time periods. Cells were harvested, washed and lysed in SDS-loading buffer. The samples were subjected to western blot assay for c-fms protein level. β-actin level was used as an equal loading control. D) THP-1 cells (105/ml) were treated with various doses of M344 for 24 hr. Adherent cells and nonadherent cells were measured to determine the percentage of adherent cells. E) THP-1 cells were treated with M344 for various time periods. Thereafter, cells were harvested and stained with acridine orange. Apoptotic cells were measured under a fluorescence microscope. F) THP-1 cells were incubated 2 μM M344 for various time periods as indicated. C-fms level was determined by western blot as described in Materials and Methods.
Previous studies showed that HDAC inhibition could induce apoptosis in broad types of carcinoma and leukemia cells [19–21]. We found that treatment with M344 at 2 μM concentration is associated with increasing the number of apoptotic cells as judged by morphologic examination following acridine orange staining under a fluorescence microscope (Fig 1E). Apoptotic cells show highly condensed chromatin and fragmented nuclei that are uniformly stained by acridine orange. Cell death following M344 treatment was confirmed by trypan blue staining in parallel cultures (data not shown). Treatment of THP-1 cells with 0.2 μM M344 slightly increased the number of apoptotic cells within 48 hr (approximately 25%). Yet, treatment with 2 μM M344 for 48 hr induced more than 95% of the THP-1 cells to undergo apoptosis (Fig. 1E). In addition, western blot analysis shows that, concomitant with apoptosis, the levels of c-fms expression decreased in a time-dependent manner when cells were treated with 2 μM M344 (Fig 1F).
3.2 THP-1 Cell Apoptosis Induced with M344
To better understand the nature of apoptosis induced by M344, we examined the expression of a number of pro- and anti-apoptotic regulators. As shown in Fig. 2A and B, along with the apoptotic progress, the anti-apoptotic protein, XIAP, decreased while a pro-apoptotic protein Bax increased, especially the small subunit after prolonged treatment. Since apoptosis of THP-1 cells induced with M344 is associated with the cleavage of PARP protein with the increasing of the 86 kd fragment (Fig. 2B), we conducted a colorimetric assay to evaluate the caspase activity after M344 treatment. Indeed, activation of caspases-3 was confirmed by using cytosolic fraction of THP-1 cells after M344 treatment (Fig. 2C). Compared with no treatment group, M344 at 2 μM concentration significantly increased caspase–3 activity in a time-dependent manner. In addition, cytochrom c in cytosolic fraction of apoptotic THP-1 cells was gradually elevated after M344 treatment (Fig. 2D).
Fig 2.

Apoptosis induction by HDAC inhibitor M344. A) THP-1 cells were treated with various doses of M344 for 24hr. Cells were harvested and lysed in SDS-loading buffer. Total protein concentration was determinate and sample was subjected to western blot assay with anti-XIAP, Bax, and Bcl-2 antibodies. B) THP-1 cells were treated with 2 μM of M344 for various time periods as indicated. Total cell lysates were subjected to western blot assay with anti-XIAP, Bax, Bcl-2 and anti-PARP antibodies. C) THP-1 cells were treated M344 for indicated time periods. The activity of caspase-3 was analyzed as described in Materials and Methods. D) THP-1 cells were treated with 2 μM of M344 for indicated time periods; cytosolic fractions were obtained and subjected to western blot assay for cytochrome c protein. β-actin was used as equal loading control.
3.3 Regulation of Cell Cycle by HDAC Inhibitor M344
Since M344 could prevent THP-1 growth, we investigated the effects of M344 on cell cycle progression. We treated THP-1 cells with M344 for various time periods followed by cell staining with propidium iodide and flow cytometric analysis. As shown in Fig. 3A, treatment of THP-1 cells with M344 (0.2 μM) for 48 hour increased the percentage of cells in G0/G1 phases from 47.11% (control) to 83.32%, while the cells in S phases decreased from 34.33% to 6.15%. Since it has been documented that histone deacetylase inhibitors can induce the expression of cyclin–dependent protein kinase inhibitor p21 [22–24], we asked whether p21 plays a role in M344-induced growth arrest in THP-1 cells. As shown in Fig. 3B, treatment of THP-1 cells with M344 markedly increased the levels of both p21 and cyclin E, reaching peak levels at about 12 hrs following treatment. In contrast, the levels of cyclne A, cycline B1 and cdc2 (p34) were not changed by M344 (unpublished data).
Fig 3.
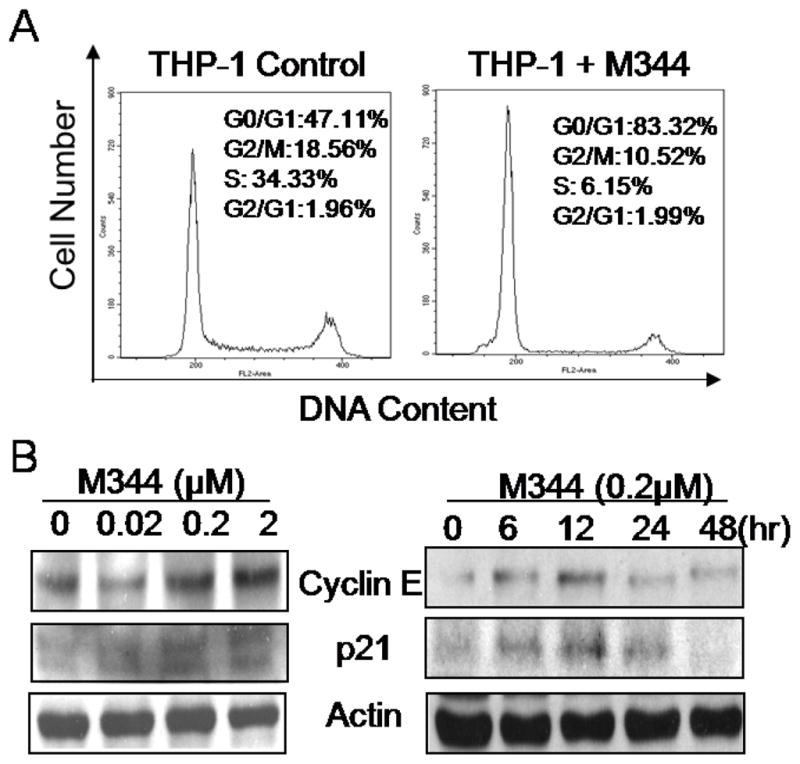
Cell cycle regulation by HDAC inhibitor M344. A) THP-1 cells were treated with 0.2 μM of M344 for 48 hrs. Cells were harvested and stained with propidium Iodide staining solution followed by Flow Cytometry assay to determine the cell cycle stage. B) THP-1 cells were treated with various doses of M344 for 24 hr, or with 2 μM of M344 for indicated time periods. At the end of incubation, cells were harvested and lysed in SDS-loading buffer. Samples were subjected to western blot assay. β-actin level was used as an equal loading control.
3.4 Activation of NF-κB by HDAC Inhibitor M344
Previous studies showed that activation of NF-κB is a key event induced by HDAC inhibitor in various types of cells [25–32]. To determine whether activation of NF-κB was involved in M344-induced THP-1 cell differentiation/apoptosis, electrophoresis mobility-shift assay (EMSA) was conducted to exam the NF-κB specific oligo binding activity of M344 treated nuclear extract. As shown in Fig. 4A, M344 treatment could markedly increase the levels of NF-κB activation in a dose-dependent manner. Activation of NF-κB transcription was detected within 4 hr and reached a peak level 8 hrs after treatment. Western blot analysis using nuclear extract from treated THP-1 cells showed that there is a concomitant increase of p65 RelA subunit of NF-κB (Fig 4B).
Fig 4.
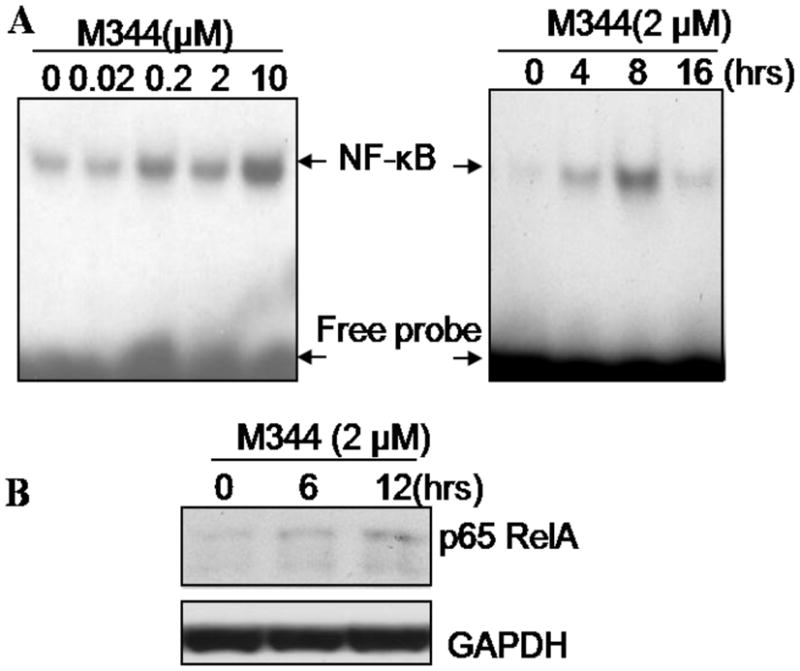
Induction of NF-κB activation by HDAC inhibitor M344. A) THP-1 cells were treated with indicated doses of M344 for 3 hrs, or with 2 μM of M344 for indicated time periods. After incubation, cell nuclear extract was isolated and total protein concentration was determinate. Samples were subjected to EMSA by running through 4% non-denaturing acrylamide gel at 100 v for 30 min. The gel was dried and exposed to X-ray film. B) THP-1 cells were treated with 2 μM of M344 for 0, 6 and 12 hrs and nuclear extractions were obtained; western blot was employed to assay the nuclear p65 Rel A level in equal amount of nuclear extraction. GAPDH was used as equal loading control.
3.5 Expression of Inflammatory Cytokines
A major functional response of mature macrophages is the production of inflammatory cytokines. Since Inhibition of chromatin histone deacetylation could trigger gene expression globally, we asked whether M344 might affect the expression of cytokines in THP-1 cells. We examined the mRNA levels of inflammatory cytokines IL-1α and TNF-α using RT-PCR technique in M344-treated THP-1 cells. Control THP-1 cells expressed basal levels of IL-1α and TNF-α mRNA. When THP-1 cells were induced to undergo apoptosis by treating with 2 μM of M344, the level of IL-1α mRNA increased gradually and reached a peak level at 24 hour. By 48 hour, the level of IL-1α diminished to a minimal level, presumably due to cell death. In contrast, M344 caused a continual decrease in the levels of TNF-α mRNA in THP-1 cells, especially in 2 μM of M344 treated group (Fig 5A).
Fig 5.
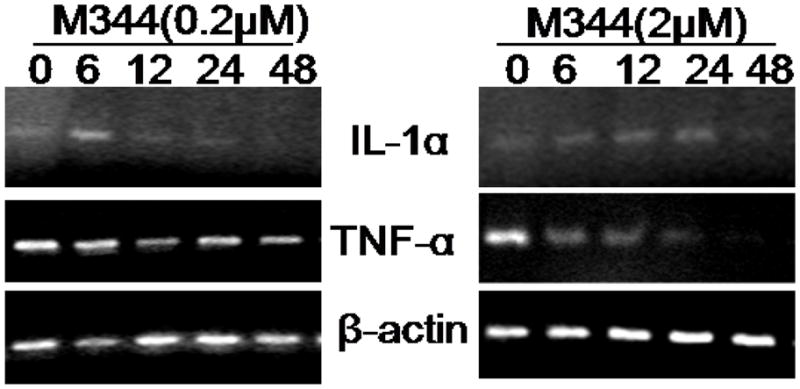
Effect of M344 on the expression of inflammatory cytokines in THP-1 cells. A) THP-1 cells were treated with 0.2 μM and 2 μM of M344, separately for up to 48 hrs. Cells were then harvested at indicated time and total RNA was extracted followed by RT-PCR analysis. One μg of total RNA for each sample was used to generate the cDNA through reverse transcription reaction. PCR assays for individual cytokine were conducted as described in Materials and Methods.
4. Discussion
In this study, we investigated the effect of a newly developed M344 on THP-1 cells, a human myelomonocytic leukemia cell line. We showed that M344 is a potent growth inhibitor [34–37], which arrests cell cycle in G1 phase. At low doses, M344 exhibits some weak differentiating effect on THP-1 cells as judged by enhanced expression of c-fms (M-CSFR) in these cells. Yet, M344 did not fully convert THP-1 cells to become adherent, the most prominent functional marker of mature macrophages. A similar study by Dai et al reported that histone deacetylase inhibitor, sodium butyrate, could arrest U937 human leukemia cells in G1 phase as well as induce U937 cells to undergo maturation as evidenced by the expression of differentiation markers CD11a, CD11b, and CD18 [38]. At higher doses (> 0.2 μM), M344 could induce THP-1 cells to undergo apoptosis following G1 arrest. Treatment of THP-1 cells with 2 μM inhibitor induced more than 95% cells to undergo apoptosis within 48 hr. Apoptosis induced by M344 is associated with the activation of both caspase activation and cytochrome c release from the mitochondria, suggesting that both mitochondria-dependent and receptor-mediated pathways are involved. Other investigators have reported similar results using other types of HDAC inhibitors including MS-275 and FR901228 [20, 39]. These findings indicate that HDAC inhibitors may act through a mechanism distinct from traditional cytotoxic anti-cancer agents, which induce apoptosis primarily through a cytochrome c dependent pathway.
Growth arrest of THP-1 cells induced by M344 is associated with increased levels of both p21 and cyclin E, two major cell cycle regulators of G1 phase. In contrast, the levels of cyclin A, cyclin B1 and cdc 2 (p34) were not substantially changed by M344 in the process. This finding indicates that p21 plays a key role in suppressing the growth of THP-1 cells induced by M344. Given that THP-1 cells are p53 deficiency [40], our results indicate the mechanism by which M344 affect THP-1 growth and survival are probably independent of p53. The importance of p21 in regulating growth inhibition induced by HDAC inhibitors also has been reported previously in other cell types and tissues [23, 36, 37, 41, 42].
Similar to the results reported by other investigators [43, 44], we showed that HDAC inhibitor is a potent activator of NF-κB transcription factor. Activation of NF-κB transcription factor was detected mainly in cells treated with higher dose of HDAC inhibitors (>0.2 μM), suggesting that the activation of NF-κB may contribute primarily to inducing apoptotic cell death rather then cell differentiation.
We hypothesized that inflammatory cytokines such as TNF-α induced by NF-κB activation may play a role in the induction of apoptosis in THP-1 cells. Unexpectedly, we found that instead of up-regulating TNF-α expression, M344 suppressed the expression of TNF-α in THP-1 cells. Inhibition of TNF-α was particularly evident in cultures treated with high doses of the inhibitor (>2 μM). This finding suggests that the activation of apoptotic pathway is not mediated through the production of TNF-α by M344-treated THP-1 cells. In contrast, M344 could rapidly and transiently increase the levels of IL-1 mRNA, another potent inflammatory cytokine, in THP-1 cells. It is not known why HDAC inhibitor could trigger the production of IL-1α but not TNF-α since the production of both cytokines is under the regulation NF-κB transcription factor. Conceivably, HDAC inhibitor may induce additional regulatory proteins such as transcriptional repressors and co-activators, which act in coordination with NF-κB to either promote or suppress the production of these cytokines [46, 47]. The fact that HDAC inhibitor can inhibit TNF-α production by THP-1 cells suggests that HDAC inhibitor could also act as a novel and potential anti-inflammatory agent. In supporting this view, a recent study by Leoni et al. reported that histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits anti-inflammatory properties through the suppression of cytokine production by macrophages [48].
One of the apoptosis regulators affected by M344 is XIAP (X chromosome-linked IAP), a potent anti-apoptotic protein [49, 50]. XIAP is abundantly expressed in THP-1 cells. The levels of XIAP reduced markedly in THP-1 cells that had been treated with high (2 μM) but not low dose of M344. Reduction of endogenous XIAP was detected within 12 hr following HDAC treatment prior to the onset of apoptosis, suggesting that down-regulation of XIAP may play a role in the induction of apoptosis in M344-treated cells. The mechanism by which M344 suppresses the expression of XIAP in THP-1 cells is not known at present. However, of equally likelihood, HDAC inhibitor induced XIAP down regulation may be a direct result of caspase-mediated XIAP degradation during apoptosis as has been reported previously [51].
Advances in the understanding of the acetylation/deacetylation mechanism in cells have prompted investigators to examine the role of HDAC inhibitors as potent anti-cancer agents. Our data showed that M344 could induce THP-1 leukemia cells to undergo growth arrest and promote apoptosis. HDAC inhibitors act with entirely different mechanisms than those with traditional anti-cancer agents. This unique property of HDAC inhibitor allows a combined use of HDAC inhibitors with conventional anti-cancer drugs. Indeed, in a recent report, Denlinger et al [52] showed that treatment of non-small cell lung cancer with combined proteasome inhibitor and HDAC inhibitor greatly promoted apoptosis by 3- to 4-fold compared with HDAC inhibitor alone. More recently, Maggio et al. reported that histone deacetylase inhibitor MS-275 acts synergistically with fludarabine to induce apoptosis in human leukemia cells [53] and Chobanian et al. showed that histone deacetylase inhibitors could enhance paclitaxel-induced cell death in ovarian cancer cell lines independent of p53 status [54]. Still, Coffey et al reported that combined treatment with deacetylase inhibitors and retinoic acids inhibit growth of human neuroblastoma in significantly lower concentrations when used together than when used individually. Thus, combination therapy may improve the ultimate efficacy while reducing the side effects of these agents in clinical use [55].
Acknowledgments
This work was supported by Public Health Service Grant CA 73212 (Chen B) awarded by the National Cancer Institute, Department of Health and Human Services, U.S.A.
Abbreviation
- HDAC
histone deacetylase
- HTA
Histone acetyl transferase
- M-CSF
macrophage colon stimulating factor
- NF-κB
nuclear factor–kappa B
- TNF-α
tumor necrosis factor-alpha
- TSA
trichostatin A
- XIAP
X-linked inhibitor of apoptosis protein
References
- 1.Marmorstein R. Structural and chemical basis of histone acetylation. Novartis Found Symp. 2004;259:78–98. discussion 98–101, 163–9. [PubMed] [Google Scholar]
- 2.Wang S, et al. Transcription regulation by histone deacetylases. Novartis Found Symp. 2004;259:238–45. discussion 245–8, 285–8. [PubMed] [Google Scholar]
- 3.Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. 1998;20(8):615–26. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 4.Ito K, I, Adcock M. Histone acetylation and histone deacetylation. Mol Biotechnol. 2002;20(1):99–106. doi: 10.1385/MB:20:1:099. [DOI] [PubMed] [Google Scholar]
- 5.Chambers AE, et al. Histone acetylation-mediated regulation of genes in leukaemic cells. Eur J Cancer. 2003;39(8):1165–75. doi: 10.1016/s0959-8049(03)00072-8. [DOI] [PubMed] [Google Scholar]
- 6.Cosio BG, et al. Histone acetylase and deacetylase activity in alveolar macrophages and blood mononocytes in asthma. Am J Respir Crit Care Med. 2004;170(2):141–7. doi: 10.1164/rccm.200305-659OC. [DOI] [PubMed] [Google Scholar]
- 7.Tariq M, Paszkowski J. DNA and histone methylation in plants. Trends Genet. 2004;20(6):244–51. doi: 10.1016/j.tig.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Di Gennaro E, et al. Acetylation of proteins as novel target for antitumor therapy: review article. Amino Acids. 2004;26(4):435–41. doi: 10.1007/s00726-004-0087-3. [DOI] [PubMed] [Google Scholar]
- 9.Somech R, Izraeli S, JSA Histone deacetylase inhibitors--a new tool to treat cancer. Cancer Treat Rev. 2004;30(5):461–72. doi: 10.1016/j.ctrv.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 10.Marks PA, et al. Histone deacetylase inhibitors: development as cancer therapy. Novartis Found Symp. 2004;259:269–81. discussion 281–8. [PubMed] [Google Scholar]
- 11.Qu W, et al. Experimental study on inhibitory effects of histone deacetylase inhibitor MS-275 and TSA on bladder cancer cells. Urol Oncol. 2009 doi: 10.1016/j.urolonc.2008.11.018. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe M, et al. Induction of autophagy in malignant rhabdoid tumor cells by the histone deacetylase inhibitor FK228 through AIF translocation. Int J Cancer. 2009;124(1):55–67. doi: 10.1002/ijc.23897. [DOI] [PubMed] [Google Scholar]
- 13.Yoshida M, et al. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265(28):17174–9. [PubMed] [Google Scholar]
- 14.Meinke PT, Liberator P. Histone deacetylase: a target for antiproliferative and antiprotozoal agents. Curr Med Chem. 2001;8(2):211–35. doi: 10.2174/0929867013373787. [DOI] [PubMed] [Google Scholar]
- 15.Jung M, et al. Amide analogues of trichostatin A as inhibitors of histone deacetylase and inducers of terminal cell differentiation. J Med Chem. 1999;42(22):4669–79. doi: 10.1021/jm991091h. [DOI] [PubMed] [Google Scholar]
- 16.Lin H, et al. Mitotic arrest induced by XK469, a novel antitumor agent, is correlated with the inhibition of cyclin B1 ubiquitination. Int J Cancer. 2002;97(1):121–8. doi: 10.1002/ijc.1570. [DOI] [PubMed] [Google Scholar]
- 17.Li X, et al. Maspin augments proteasome inhibitor-induced apoptosis in prostate cancer cells. J Cell Physiol. 2007;212(2):298–306. doi: 10.1002/jcp.21102. [DOI] [PubMed] [Google Scholar]
- 18.Li X, Meng H, Chen BD. Diferentiation-associated expression of conventional protein kinase C isoforms in primary cultures of bone marrow cells induced by M-CSF and G-CSF. Am J Biomed Sci. 2009;1(1):47–55. doi: 10.5099/aj090100047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, et al. Endogenous inhibition of histone deacetylase 1 by tumor-suppressive maspin. Cancer Res. 2006;66(18):9323–9. doi: 10.1158/0008-5472.CAN-06-1578. [DOI] [PubMed] [Google Scholar]
- 20.Lucas DM, et al. The histone deacetylase inhibitor MS-275 induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia cells. Leukemia. 2004;18(7):1207–14. doi: 10.1038/sj.leu.2403388. [DOI] [PubMed] [Google Scholar]
- 21.Bouzar AB, et al. Valproate synergizes with purine nucleoside analogues to induce apoptosis of B-chronic lymphocytic leukaemia cells. Br J Haematol. 2009;144(1):41–52. doi: 10.1111/j.1365-2141.2008.07426.x. [DOI] [PubMed] [Google Scholar]
- 22.Richon VM, et al. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U S A. 2000;97(18):10014–9. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sambucetti LC, et al. Histone deacetylase inhibition selectively alters the activity and expression of cell cycle proteins leading to specific chromatin acetylation and antiproliferative effects. J Biol Chem. 1999;274(49):34940–7. doi: 10.1074/jbc.274.49.34940. [DOI] [PubMed] [Google Scholar]
- 24.Kim YB, et al. Oxamflatin is a novel antitumor compound that inhibits mammalian histone deacetylase. Oncogene. 1999;18(15):2461–70. doi: 10.1038/sj.onc.1202564. [DOI] [PubMed] [Google Scholar]
- 25.Xu RH, et al. Histone acetylation is a checkpoint in FGF-stimulated mesoderm induction. Dev Dyn. 2000;218(4):628–35. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1024>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 26.Rahman I, et al. Oxidative stress and TNF-alpha induce histone acetylation and NF-kappaB/AP-1 activation in alveolar epithelial cells: potential mechanism in gene transcription in lung inflammation. Mol Cell Biochem. 2002;234–235(1–2):239–48. [PubMed] [Google Scholar]
- 27.Zhang W, Kone BC. NF-kappaB inhibits transcription of the H(+)-K(+)-ATPase alpha(2)-subunit gene: role of histone deacetylases. Am J Physiol Renal Physiol. 2002;283(5):F904–11. doi: 10.1152/ajprenal.00156.2002. [DOI] [PubMed] [Google Scholar]
- 28.Miao F, et al. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J Biol Chem. 2004;279(17):18091–7. doi: 10.1074/jbc.M311786200. [DOI] [PubMed] [Google Scholar]
- 29.Graham B, Gibson SB. The two faces of NFkappaB in cell survival responses. Cell Cycle. 2005;4(10):1342–5. doi: 10.4161/cc.4.10.2047. [DOI] [PubMed] [Google Scholar]
- 30.Slevogt H, et al. Moraxella catarrhalis induces inflammatory response of bronchial epithelial cells via MAPK and NF-kappaB activation and histone deacetylase activity reduction. Am J Physiol Lung Cell Mol Physiol. 2006;290(5):L818–26. doi: 10.1152/ajplung.00428.2005. [DOI] [PubMed] [Google Scholar]
- 31.Kim YK, et al. Involvement of HDAC1 and the PI3K/PKC signaling pathways in NF-kappaB activation by the HDAC inhibitor apicidin. Biochem Biophys Res Commun. 2006;347(4):1088–93. doi: 10.1016/j.bbrc.2006.06.196. [DOI] [PubMed] [Google Scholar]
- 32.Kim YK, et al. Activation of NF-kappaB by HDAC inhibitor apicidin through Sp1-dependent de novo protein synthesis: its implication for resistance to apoptosis. Cell Death Differ. 2006;13(12):2033–41. doi: 10.1038/sj.cdd.4401915. [DOI] [PubMed] [Google Scholar]
- 33.Idel S, et al. Branched chain fatty acids induce nitric oxide-dependent apoptosis in vascular smooth muscle cells. J Biol Chem. 2002;277(51):49319–25. doi: 10.1074/jbc.M204639200. [DOI] [PubMed] [Google Scholar]
- 34.Yamashita Y, et al. Histone deacetylase inhibitor trichostatin A induces cell-cycle arrest/apoptosis and hepatocyte differentiation in human hepatoma cells. Int J Cancer. 2003;103(5):572–6. doi: 10.1002/ijc.10699. [DOI] [PubMed] [Google Scholar]
- 35.Greenberg VL, et al. Histone deacetylase inhibitors promote apoptosis and differential cell cycle arrest in anaplastic thyroid cancer cells. Thyroid. 2001;11(4):315–25. doi: 10.1089/10507250152039046. [DOI] [PubMed] [Google Scholar]
- 36.Donadelli M, et al. Trichostatin A, an inhibitor of histone deacetylases, strongly suppresses growth of pancreatic adenocarcinoma cells. Mol Carcinog. 2003;38(2):59–69. doi: 10.1002/mc.10145. [DOI] [PubMed] [Google Scholar]
- 37.Wang ZM, et al. Trichostatin A inhibits proliferation and induces expression of p21WAF and p27 in human brain tumor cell lines. Ai Zheng. 2002;21(10):1100–5. [PubMed] [Google Scholar]
- 38.Dai Y, Rahmani M, Grant S. An intact NF-kappaB pathway is required for histone deacetylase inhibitor-induced G1 arrest and maturation in U937 human myeloid leukemia cells. Cell Cycle. 2003;2(5):467–72. [PubMed] [Google Scholar]
- 39.Imai T, et al. FR901228 induces tumor regression associated with induction of Fas ligand and activation of Fas signaling in human osteosarcoma cells. Oncogene. 2003;22(58):9231–42. doi: 10.1038/sj.onc.1207184. [DOI] [PubMed] [Google Scholar]
- 40.Akashi M, et al. p21WAF1 expression by an activator of protein kinase C is regulated mainly at the post-transcriptional level in cells lacking p53: important role of RNA stabilization. Biochem J. 1999;337( Pt 3):607–16. [PMC free article] [PubMed] [Google Scholar]
- 41.Gray SG, et al. IGF-II enhances trichostatin A-induced TGFbeta1 and p21(Waf1, Cip1, sdi1) expression in Hep3B cells. Exp Cell Res. 1999;253(2):618–28. doi: 10.1006/excr.1999.4661. [DOI] [PubMed] [Google Scholar]
- 42.Gui CY, et al. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci U S A. 2004;101(5):1241–6. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campbell KJ, Perkins ND. Post-translational modification of RelA(p65) NF-kappaB. Biochem Soc Trans. 2004;32(Pt 6):1087–9. doi: 10.1042/BST0321087. [DOI] [PubMed] [Google Scholar]
- 44.Ashburner BP, Westerheide SD, Baldwin AS., Jr The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol. 2001;21(20):7065–77. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Legube G, Trouche D. Regulating histone acetyltransferases and deacetylases. EMBO Rep. 2003;4(10):944–7. doi: 10.1038/sj.embor.embor941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown SA, et al. The Fn14 cytoplasmic tail binds tumour-necrosis-factor-receptor-associated factors 1, 2, 3 and 5 and mediates nuclear factor-kappaB activation. Biochem J. 2003;371(Pt 2):395–403. doi: 10.1042/BJ20021730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leoni F, et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci U S A. 2002;99(5):2995–3000. doi: 10.1073/pnas.052702999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deveraux QL, et al. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388(6639):300–4. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 49.Liston P, et al. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature. 1996;379(6563):349–53. doi: 10.1038/379349a0. [DOI] [PubMed] [Google Scholar]
- 50.Deveraux QL, et al. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. Embo J. 1999;18(19):5242–51. doi: 10.1093/emboj/18.19.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Denlinger CE, et al. Combined proteasome and histone deacetylase inhibition in non-small cell lung cancer. J Thorac Cardiovasc Surg. 2004;127(4):1078–86. doi: 10.1016/s0022-5223(03)01321-7. [DOI] [PubMed] [Google Scholar]
- 52.Maggio SC, et al. The histone deacetylase inhibitor MS-275 interacts synergistically with fludarabine to induce apoptosis in human leukemia cells. Cancer Res. 2004;64(7):2590–600. doi: 10.1158/0008-5472.can-03-2631. [DOI] [PubMed] [Google Scholar]
- 53.Chobanian NH, et al. Histone deacetylase inhibitors enhance paclitaxel-induced cell death in ovarian cancer cell lines independent of p53 status. Anticancer Res. 2004;24(2B):539–45. [PubMed] [Google Scholar]
- 54.Coffey DC, et al. Histone deacetylase inhibitors and retinoic acids inhibit growth of human neuroblastoma in vitro. Med Pediatr Oncol. 2000;35(6):577–81. doi: 10.1002/1096-911x(20001201)35:6<577::aid-mpo18>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]