

Abstract
Nitric oxide (NO) has been considered as an important bio-regulatory molecule in the physiological process. All the existing methods often employed for NO measurement are mainly indirect and not suitable for in vivo conditions. In this paper, we report a systematic study of electrocatalytic NO reduction by comparing the redox properties of NO at carbon microelectrodes functionalized by Fe, Mn and Co protoporphyrins. The mechanisms of electrocatalytic reduction of NO by different metalloporphyrins have been proposed and compared. In addition, by varying the metallic cores of the metalloporphyrins, NO exhibits voltammograms in which the cathodic peak current occur at different potential. A comparative study on the electrochemical behavior of each of these metalloporphyrin (as a result of varying the metallic core) has been performed and a possible mechanism for the observed behavior is proposed. The results confirmed the potential applicability of using metalloporphyrins modified electrodes for voltammetric NO detection.
Keywords: Nitric oxide, biosensor, electrocatalytic reduction, microelectrode, protoporphyrin
1. Introduction
Nitric oxide (NO) is an important tiny molecule responsible for the proper functioning of numerous physiological processes. Although a thorough understanding of NO functions is not possible, its role in the activity of the endothelium-derived relaxing factor [1], as a neurotransmitter [2], preventing platelet aggregation [3], and as a major defense molecule in immune cells against tumor cells and intracellular bacteria [4] has been well studied. The NO molecule has the tendency to form complexes readily with transition metal ions found in metalloproteins and heme-containing proteins. For example, it has been widely accepted that Cytochrome c has the ability to bind with NO and CO, but it is unable to form stable complexes with O2. Crystal structures of Cytochrome c indicates that CO forms a six-coordinate heme-carbonyl (6c-CO) complex on the distal heme face, whereas NO forms an unprecedented five-coordinate heme-nitrosyl (5c-NO) complex on the proximal heme face, replacing the endogenous C-terminal histidine ligand (His 120). This is an important observation that demonstrates steric hindrance is an important factor that influences the distal heme-NO interactions, while the switch from distal to proximal NO binding is governed by the rate of dinitrosyl formation [5]. The biochemical reduction of nitrite to ammonia that occurs in green plants is a six-electron, seven-proton process catalyzed by the nitrite reductase enzymes [6]. The catalytic reduction of nitrite proceeded via the formation of a siroheme-nitrosyl complex, which is the reduced form of ammonia.
Determination of NO has been the subject of interest in many extensive studies. Various methods, such as electron paramagnetic resonance, spectrophotometry and chemiluminescence have been developed for the detection of NO. However, until now, specific determination of NO in a biological sample has been a big challenge as NO is an unstable compound with a half-life time of about five seconds. Furthermore, it reacts rapidly with O2 to form NO2- and NO3-. NO is electrochemically active and can be oxidized at positive potentials at the surface of a metallic, carbon or a carbon modified electrode. Direct NO detections have been attempted using bare or unmodified electrodes, but these platforms showed poor selectivity due to the interference of other electroactive species at a similar or closer potential. Functionalization of electrodes is a routine way to improve the sensitivity and selectivity of the electrocatalytic detection of NO. The electro oxidation of NO on micro-membranes such as bromophenol blue film, Nafion, cellulose acetate modified carbon, platinum, and gold electrodes have been reported [7-10]. These functional thin layers allow selective permeation of NO towards the working electrode and discriminate the other interfering compounds. In addition, various interesting novel molecular materials are being extensively studied with the aim of investigating their role in the electrocatalytic reduction of nitrite, nitrosyl and NO in aqueous conditions.
NO reduction can be electrocatalyzed in solution, and under anaerobic conditions by numerous transition metal complexes, such as iron phenanthroline, pyridine-EDTA complexes [11] and metal-substituted heteropolyanions [12]. In the past, several reports have described the use of electropolymerized metalloporphyrins as new electrode materials with the aim of investigating the electrocatalytic reduction or oxidation of nitrite, nitrosyl and nitric oxide in aqueous solutions [13-15]. Furthermore, the hydrogen atom present in the meso position can be replaced by other functional groups, thereby modifying the electron donor-acceptor capability [16]. These materials constitute potential molecular materials suitable for electrode surface modification to design new electrochemical micro-sensors for the detection and quantitative determination of NO. Kitajama et al. [17] transferred the reactivity of an iron porphyrin to the electrode-solution interface by incorporating the porphyrin into a thin polymeric film by oxidative electropolymerization. Younathan et al. [18] reported a pioneering study that involved the application of electropolymerized iron protoporphyrin films to model the activity of nitrite reductase enzymes. It was reported that the electropolymerized films of iron protoporphyrin (IX) dimethyl ester are effective catalysts for the electroreduction of HONO/NO2- or NO to N2O, N2, N2OH and NH3. Water-soluble cobalt tetrakis (N-methyl-2-pyridyl) porphine [Co(2-TMPyP)] has been investigated for its catalytic behavior towards nitric oxide reduction [19]. The proximity of the N-methyl pyridinium group to the porphine ring has strong effects on the axial ligand coordination properties and the redox potential of metalloporphyrins [8, 9]. The presence of strong electron withdrawing substituents makes cobalt porphyrin, a robust catalyst for NO catalytic reduction. In many cases, the NO sensitive electrodes are made out of a thin carbon fiber coated with a polymeric nickel porphyrin and Nafion. In the present work, calculated NO concentration exhibited a linear relationship in therange of 1 nM-100 μM in physiological phosphate buffer solution (pH 7.4) [20-22].
Most recently, we reported a study of electrochemical properties on a series of engineered metallo protoporphyrins and their reconstituted myoglobin, including Co, Mn and Ru substituted porphyrin [23-25]. In this paper, we will discuss a functional electrochemical biosensor using metalloporphyrins (Fe, Mn, Co protoporphyrin IX) modified microelectrodes for the measurement of NO. Results indicated that the electrocatalytic response of NO varies by varying the metal substituted protoporphyrin modified electrode. The electrochemical behaviors were compared with each other and a suitable mechanism has been proposed. In the present work, we specifically employ microelectrodes to study the NO redox reaction, because they offer extremely high sensitivity due to the high signal noise ratio, enhanced mass transport and reduced ohmic drop. In addition, the use of microelectrodes opens up the possibility of the electrochemical detection of NO in living cells and tissue cultures.
2. Experimental section
2.1 Chemicals
All solutions were prepared from analyticalgrade chemicals and triply distilled water. Phosphate-buffered saline solutions (PBS, 0.1M) of pH 4.5 and 7.2 were prepared with high purity salts without further purification. Mn protoporphyrin IX and Co protoporphyrin IX were synthesized according to a procedure described previously [23-25]. Hemin was purchased form Aldrich. Dimethylsulfoxide (DMSO), tetrabutylammonium tetrafluoroborate (TBABF4) were of reagent grade and used as received. Nafion solution (4%) was purchased from Aldrich.
2.2 Microelectrode preparation
Briefly, a single carbon fiber of diameter 5 μm was attached to the stripped end of an insulated copper wire (diameter, 0.2 mm) using a silver conductive paste. The silver paste was allowed to dry, after which the free end of the copper wire was cannulated into a borosilicate glass capillary. The copper wire was fixed to the capillary using a fast drying epoxy resin. The capillary was then pulled using a micropipette puller. This pulling procedure resulted in two glass pipettes, one of which was discarded, and the other which holds the carbon fiber protruding from its tip will be employed for further experiments. The junction between the carbon fiber and capillary is sealed with a drop of the sealguard silicone and allowed to cure until it became firm. The protruding fiber was trimmed
2.3 Experimental set-up
Cyclic voltammetric experiments were carried out in a three-electrode system at room temperature. The counter electrode was a platinum wire and an AgǀAgCl (saturated KCl) was used as the reference electrode. The electrochemical cell was covered with a Teflon covering and all the electrodes were inserted into the cell through the holes drilled on the Teflon cover. All electrochemical measurements were conducted inside a glove box designed for complete exclusion of oxygen.
The microelectrode designed as mentioned above was immersed in 1.0 M sulfuric acid for 5 min to pre-activate the electrode surface and thoroughly rinsed prior to use. Cyclic voltammetry was then performed using this microelectrode in DMSO + 0.1 M TBABF4/0.1 M metalloporphyrin solution. By performing the foretold cyclic voltammetry for ten consecutive cycles (cycling between −1.0 V to 0.35 V with a scan rate of 0.1 V/s) a uniform polymeric film of metalloporohyrin was casted onto the microelectrode surface. The electrode casted with the polymeric film was then taken out of the solution and rinsed with triply distilled water. Following this, the electrode was immersed in a cuvette containing 19μl of 0.5% (w/v) nafion/ethanol for a minute and then the electrode was taken out and allowed to dry in air.
Saturated NO solution was prepared as follows: About 10 ml of phosphate buffer (pH 7.0) was degassed by sonication under vacuum for 30 minutes. Following this, the headspace is replaced using nitrogen for 30 minutes to remove oxygen and transferred to a sample bottle with a silicon rubber stopper. NO was continuously purged into the deoxygenated buffer (approximately for an hour) until the buffer became saturated. NO saturated in the aqueous phase had an approximate concentration of 2.0 mM at 20 °C. This saturated solution is stable for 48 hours at 20 °C and was kept under NO atmosphere until use.
3. Results and discussion
3.1 Preparation and characterization of electrode films containing metalloporphyr
Figure 2a represents the repetitive cyclic voltammetric response of a carbon fiber microelectrode in 0.1 M of the iron protoporphyrin IX chloride (hemin) in DMSO+0.1 M TBABF4 solution (scan range from −1.0 V to 0.35 V). A pair of well-defined peaks that appears at Eeq = −0.28 V vs. AgǀAgCl during the first scan corresponds to the oxidation of Fe(II) to Fe(III) present in the porphyrin. A continuous increase in the amplitude of these cyclic voltammetric peaks during repetitive scans (within the reported potential range) confirmed the formation of a film on the electrode surface as a consequence of the anodic electropolymerization of the hemin. The effect of hemin concentration on the cyclic voltammetric response was studied after an open circuit delay of 2 min; the dependence of cathodic peak height (Ipc) with hemin concentration is shown in Figure 2b. From Figure 2b, it is apparent that the peak current value (Ipc) increases with increase in hemin concentration; however, no linear dependence is observed in the range (0-0.2 mM) investigated, owing to adsorption phenomena. After the completion of coating process, the electrode is rinsed using triply distilled water. Later, cyclic voltammetry was performed using this electrode in 0.1 M PBS buffer. In the resulting voltammogram, we noticed a pair of peaks at Epa = − 0.27 V and Epc = − 0.34 V that can be assigned oxidation of Fe(II) to Fe(III) and the reduction of Fe(III) to Fe(II) respectively (present in the polymeric porphyrin). Following this, the stability of the hemin filmadsorbed on the microelectrode surface was examined. Next, the electrode was thoroughly rinsed with triply distilled water to remove the excess of adsorbed monomer (if any had been present). An intense colored layer was clearly visible on the electrode surface, indicating the strong adsorption of hemin on the microelectrode surface to impart a very compact and stable film.
Figure 2.

(a) Cyclic voltammograms of a carbon fiber microelectrode in DMSO+0.1 M TBAF4 solution (scan rate: 100 mV/s) containing 150 μM hemin; (b) Calibration plot showing the dependence of cathodic peak current with varying hemin concentrations (after an open circuit delay of 2 min) (c) Cyclic voltammetry of 200 μM Mn-protoporphyrin IX in DMSO+0.1 M TBAF4 solution (scan rate: 100 mV/s).
Figure 2c is the representative repetitive voltammograms of 0.1 mM Mn-PP in DMSO + 0.1 M TBABF4 solution at a PG electrode (Scan range: from −1.0 V to 0.5 V). In this voltammogram, we noticed a pair of well-defined peaks appearing at Epa = −0.31 V and Epc = −0.40 V vs. AgǀAgCl. This corresponded to the reduction of Mn(III) to Mn(II) and its oxidation back to Mn(III) on the polymerized Mn-PP surface. Upon continuous cycling, a growth pattern is observed for Mn-PP (noticed only in basic solution) which clearly indicated the formation of a film on the surface of the electrode surface.
However, the surface modification of the electrode was not successful in Co-PP, when the same technique was employed. The cyclic voltammogram obtained using Co(III)-PP/Co(II) PP in DMSO+ 0.1 M TBABF4 solution at a carbon fiber electrode (not shown) with repetitive potential scans (from −1.5 V to 0.5 V) exhibited a formal potential at E0’= −0.10 V vs. AgǀAgCl corresponding to the reduction of Co (III)-PP/Co(II)-PP. The peak-to-peak separation of Co(III)-PP/Co(II)-PP redox reaction is approximately 550 mV, indicating a slow heterogeneous electron transfer rate as reported previously [25]. In addition, another reversible redox peak was witnessed (not shown) corresponding to the reduction of Co(II)-PP/Co(I) PP at E0’= −0.95 V vs. AgǀAgCl. However, the Co PP film was not stable and washed off when rinsed using triply distilled water. When cyclic voltammetry was performed repetitively using this electrode in PBS, the voltammogram exhibited a marked decrease in the observed current magnitude. After several scans, the redox peak totally disappeared indicating the inability of Co PP films to adhere strongly to the electrode surface.
3. 2. Cyclic voltammetry of NO at a bare carbon fiber microelectrode
NO can be easily oxidized to nitrite (NO2−) and nitrate (NO3−). When cyclic voltammetry was performed in 100 μM NO in phosphate buffer (pH 7.0), we observed a voltammogram with two sharp electrochemically irreversible peaks at +0.85 V and +1.20 V respectively (Figure 3a). The redox potentials of other species such as nitrite, nitrate and ascorbate are very similar to that of NO and this makes the NO detection a challenging task. In addition, not only can nitrite be oxidized at a similar potential as that of NO, but also NO oxidation can yield nitrite. As a result, the voltammetric technique (or the electrode) employed should not only be sensitive to the oxidation of both NO and the oxidation of nitrite. Further, to confirm their interference, cyclic voltammetry was performed using the microelectrode in sodium nitrite in pH 7.0 phosphate buffer. In this experiment, we witnessed a totally irreversible oxidation peak (near +1.15 V) corresponding to the oxidation of nitrite.
Figure 3.

Cyclic voltammetry of 100 μM NO in 0.1 M PBS (pH 7.4) (scan rate = 100mV/s) on (a) a bare electrode and (b) a Nafion film modified electrode.
Following this, the electrode was coated with a thin film of Nafion in order to stabilize nitrite and prevent its further oxidation to NO3−. Next, cyclic voltammetry was performed using this nafion-coated electrode in the same NO solution. The resulting voltammograms shown in Figure 3a and Figure 3b exhibited a voltammetric peak at +0.85 V corresponding to NO oxidation and indicating the tendency of nafion film to completely mask the oxidation of nitrite. As a result, NO easily diffuse throughout the entire nafion film. In addition, the peak current that corresponded to NO oxidation increased with an increase in the solution concentration.
3.3. Measurement of NO using metalloporphyrins modified carbon fiber microelectrodes
As shown in Figure 4A (a), the cyclic voltammogram of iron protoporphyrin IX film in pH 7.4 PBS buffer solution exhibited a pair of peaks corresponding to the metal center pair Fe(III)/Fe(II). Furthermore, the peaks of the metal center pair disappeared upon the addition of saturated NO solution. A new cathodic and irreversible peak appeared at Epc = −0.82 V vs. AgǀAgCl (Figure 4A (b)). Earlier, Barley et al. [26] reported that the disappearance of the Fe (III)/Fe(II) redox peaks can be attributed to the complex formation of [Fe(III)] porphyrin and NO (for both dissolved iron porphyrins and surface polymerized porphyrins) [27]. NO can react directly with the polymeric Fe(III) form of porphyrin to give the porphyrin-nitrosyl complex. The formation of Fe-porphyrin-nitrosyl complex was suggested to be governed by axial complex of the hemin. The appearance of the cathodic peak at Epc = −0.82 V can be attributed to the reduction of iron porphyrin-nitrosyl complex formed within the iron porphyrin film as depicted in Scheme A.
Figure 4.

(A) Cyclic voltammetry of the Nafion-hemin modified electrode in 0.1 M PBS (pH = 7.4) (a) without NO (b) in the presence of 200 μM NO. (Scan rate: 100 mV/s). (B) Cyclic voltammetry of Mn-PP modified PG electrode in 0.1 M PBS (pH 7.4) (a) without NO; (b) after addition of NO (100 μM). (C) Reduction peak current (at −0.82 V) vs. [NO] on the Mn-PP modified electrode.
Scheme (A).

Proposed electrocatalytic reduction mechanism of NO by Hemin
The formation of the iron nitrosyl-porphyrin complex, [Fe(III)(NO)]+ is supported by absorption spectral changes observed at ITO coated electrodes [27]. A visible shift observed in the Soret band from R= 419 to 422 nm confirmed the formation of iron-nitrosyl-porphyrin complex.
Furthermore, as an extension, the electrocatalytic reduction of nitric oxide and the activities of other metalloporphyrins became a topic of interest. Manganese porphyrins are ideal candidates for NO reduction and the redox chemistry of manganese and iron porphyrins were known to exhibit parallel properties. Diab et al. [28] and Su et al. [29] reported that the electrocatalytic reduction of NO by water-soluble manganese porphyrins (Mn(III)TMPyP) proceeds via a pathway different from that of iron porphyrins. The electrocatalytic reduction of NO by water-soluble manganese porphyrin proceeds through the pathway as shown in Scheme (B). The pathway involves an ECE mechanism, which initially involves the reduction of Mn(III)P, followed by NO coordination and finally the electro reduction of (NO)Mn(II)P. In the case of iron protoporphyrin IX, the iron(III) porphyrin is coordinated initially with NO and (NO)Fe(III)-PP is subsequently reduced at moderate potential to (NO)Fe(II)-PP.
Scheme (B).

Proposed electrocatalytic reduction mechanism of NO by Mn protoporphyrin
Figure 4B represents the cyclic voltammogram of a PG electrode modified by Mn(III) protoporphyrin IX in a pH 5.0 phosphate buffer with (voltammogram a) and without (voltammogram b) the addition of saturated NO solution. A pair of peaks that appears at E0’= −0.46 V corresponds to the redox reaction of Mn(III)-PP to Mn(II)-PP. In the presence of NO, the peak at −0.51 V disappeared while a new cathodic peak appeared at Epc = −0.85 V, which may be attributed to the reduction of (NO)Mn(II)PP. A calibration plot (as shown in Figure 4C) is obtained by plotting the reduction peak current (observed by means of constant potential amperometry at a potential of −0.85 V) following successive additions of aliquots of saturated NO solution. From Figure 4C, it is evident that the peak current observed at −0.85 V increases linearly with increase in NO concentration (0.5 μM–50 μM).
In this work, we utilized cobalt protoporphyrin IX as an electrocatalyst for the reduction of nitrite. The Co-PP films modified on the microelectrode surfaces by means of electrochemical deposition could not adhere strongly on to the electrode surface. The films are not stable when they are rinsed with aqueous or organic solvents. As a result, the electrocatalytic reduction of NO by cobalt protoporphyrin was carried out in DMSO+0.1 M TBABF4 solution containing Co-PP and saturated NO aqueous solution.
A pair of redox peaks corresponding to the redox reaction of Co(III)-PP/Co(II)-PP and Co(II)-PP/Co(I)-PP were observed in DMSO + 0.1 M TBABF4 solution [25]. Upon the addition of saturated NO aqueous solution, a new irreversible reduction wave at Epc = −1.05 V grows up (not shown). This observed additional catalytic current is attributed to the reduction of NO or NO2−. However, in the present study, it is impossible to account for a detailed mechanism describing the NO electrocatalytic reduction by cobalt proptoporphyrin IX, as the electro reduction mechanism in aqueous solution is totally different from that in organic solvent.
4. Conclusion
NO can be electrochemically detected either by its oxidation or reduction reaction. Our results indicated that the functional groups of metal reconstituted protoporphyrins are more efficient towards the catalytic reduction of NO. Our results have also confirmed the possibility of using metalloporphyrins as functional groups for the electrochemical biosensing of NO. Furthermore, in this paper, the mechanism corresponding to the electrocatalytic reduction of NO on Fe, Mn and Co protoporphyrin IX modified electrodes has been proposed and compared with one another. Results indicated that the electrodes modified with different metalloporphyrins exhibited different electrocatalytic reduction properties, which can be used for the development of highly sensitive and selective NO biosensors.
Figure 1.
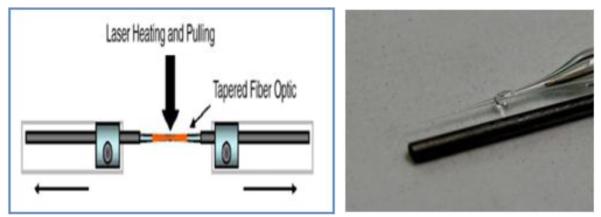
Laser microelectrode pulling system and image of carbon fiber microelectrode with a 0.5 mm (dia.) alongside a pencil lead as a contrast.
Acknowledgements
This current work is partially supported under grant FA9550- 07-1-0344 of Department of Defense/ Air Force Office of Scientific Research, FIU Faculty Research Award, Kauffman Foundation and NSF MRI 0821582 grant.
References
- 1.Bassenge E. Clinical relevance of endothelium-derived relaxing factor (EDRF) Br J Clin Pharmacol. 1992;34(Suppl 1):37S–42S. doi: 10.1111/j.1365-2125.1992.tb04147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuriyama K, Ohkuma S. Role of nitric oxide in central synaptic transmission: effects of neurotransmitter release. Jpn J Pharmacol. 1995;69:1–8. doi: 10.1254/jjp.69.1. [DOI] [PubMed] [Google Scholar]
- 3.Nong Z, Hoylaerts M, Van Pelt N, Collen D, Janssens S. Nitric Oxide Inhalation Inhibits Platelet Aggregation and Platelet-Mediated Pulmonary Thrombosis in Rats. Circulation Research. 1997;81:865–869. doi: 10.1161/01.res.81.5.865. [DOI] [PubMed] [Google Scholar]
- 4.Chakravortty D, Hensel M. Inducible nitric oxide synthase and control of intracellular bacterial pathogens. Microbes and Infection. 2003;5:621–627. doi: 10.1016/s1286-4579(03)00096-0. [DOI] [PubMed] [Google Scholar]
- 5.Pixton DA, Petersen CA, Rudi van Eldik AJ, Garton EM, Andrew CR. Activation Parameters for Heme: NO Binding in Alcaligenes xylosoxidans Cytochrome c: The Putative Dinitrosyl Intermediate Forms via a Dissociative Mechanism. J. Am. Chem. Soc. 2009;131:4846–4853. doi: 10.1021/ja809587q. [DOI] [PubMed] [Google Scholar]
- 6.Swamy U, Wang M, Tripathy JN, Kim SK, Hirasawa M, Knaff DB, Allen JP. Structure of Spinach Nitrite Reductase: Implications for Multi-electron Reactions by the Iron–Sulfur: Siroheme Cofactor. Biochemistry. 2005;44:16054–16063. doi: 10.1021/bi050981y. [DOI] [PubMed] [Google Scholar]
- 7.Hakim TS, Sugimori K, Camporesi EM, Anderson G. Half-life of nitric oxide in aqueous solutions with and without haemoglobin. Physiol Meas. 1996;17:267–77. doi: 10.1088/0967-3334/17/4/004. [DOI] [PubMed] [Google Scholar]
- 8.Peng YF, Hu CH, Zheng DY, Hu SS. A sensitive nitric oxide microsensor based on PBPB composite film modified carbon fiber microelectrode. Sensors and Actuators B: chemical. 2008;133:571–576. [Google Scholar]
- 9.Chen X, Xie P, Tian Q, Hu S. Amperometric nitric oxide sensor based on poly(thionine)/nafion-modified electrode and its application in monitoring nitric oxide release from rat kidney, Anal. Lett. 2006;39:1321–1332. [Google Scholar]
- 10.Katrlík J, Zále áková P. Nitric oxide determination by amperometric carbon fiber microelectrode. Bioelectrochemistry. 2002;56:73–76. doi: 10.1016/s1567-5394(02)00024-5. [DOI] [PubMed] [Google Scholar]; 11 Kleifges K-H, Juzeli nas E, Jüttner K. Electrochemical study of direct and indirect NO reduction with complexing agents and redox mediator. Electrochimica Acta. 1997;42:2947–2953. [Google Scholar]
- 11.Kleifges K-H, Juzeli nas E, Jüttner K. Electrochemical study of direct and indirect NO reduction with complexing agents and redox mediator. Electrochimica Acta. 1997;42:2947–2953. [Google Scholar]
- 12.Kuznetsova LI, Yurchenko ÉN, Paukshtis EA, Detusheva LG, Litvak GS. Reaction of nitrogen oxides with salts of metal-substituted heteropolyanions. Russian Chemical Bulletin. 1992;41:1526–1530. [Google Scholar]
- 13.Diab N, Oni J, Schulte A, I. Blfchl RA, Schuhmann W. Pyrrole functionalized metalloporphyrins as electrocatalysts for the oxidation of nitric oxide. Talanta. 2003;61:43–51. doi: 10.1016/S0039-9140(03)00358-8. [DOI] [PubMed] [Google Scholar]
- 14.Lin R, Bayachou M, Greaves J, Farmer PJ. Nitrite Reduction by Myoglobin in Surfactant Films. J. Am. Chem. Soc. 1997;119:12689–12690. [Google Scholar]
- 15.Burg A, Lozinsky H, Cohen H, Meyerstein D. Mechanism of Reduction of the Nitrite Ion by CuI Complexes. Eur. J. Inorg. Chem. 2004;18:3675–3680. [Google Scholar]
- 16.Chen J, Ikeda O. Redox Mechanism of NO in Water-Soluble Iron Porphyrin. Electroanalysis. 2001;13:1076–1081. [Google Scholar]
- 17.Kitajama A, Miyake M, Koyama T, Ikada O, Kijima K, Komura T, Uno A, Yamatodani A. Detection of Nitric Oxide with the Iron (III) Porphyrin Doped NafionǀGlassy Carbon Electrode. Denki Kagaku oyobi Kogyo Butsuri Kagaku. 1999:784–788. [Google Scholar]
- 18.Younathan JN, Wood KS, Meyer TJ. Electrocatalytic reduction of nitrite and nitrosyl by iron(III) protoporphyrin IX dimethyl ester immobilized in an electropolymerized film. Inorg. Chem. 1992;31:3280–3285. [Google Scholar]
- 19.Cheng SH, Su YO. Electrocatalysis of Nitric Oxide Reduction by Water-Soluble Cobalt Porphyrin. Spectral and Electrochemical Studies. Inorg. Chem. 1994;33:5847–5854. [Google Scholar]
- 20.Sun J, Zhang X, Broderick M, Fein H. Measurement of Nitric Oxide Production in Biological Systems by Using Griess Reaction Assay. Sensors. 2003;3:276–284. [Google Scholar]
- 21.Nizam Diab N, Oni J, Schuhmann F. Electrochemical nitric oxide sensor preparation: a comparison of two electrochemical methods of electrode surface modification. Bioelectrochemistry. 2005;66:105–110. doi: 10.1016/j.bioelechem.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 22.Lee Y, Oh BK, Meyerhoff ME. Improved Planar Amperometric Nitric Oxide Sensor Based on Platinized Platinum Anode. 1. Experimental Results and Theory When Applied for Monitoring NO Release from Diazeniumdiolate-Doped Polymeric Films. Anal. Chem. 2004;76:536–544. doi: 10.1021/ac035064h. [DOI] [PubMed] [Google Scholar]
- 23.Li C-Z, Taniguchi I, Mulchandani A. Redox properties of engineered ruthenium myoglobin. Bioelectrochemistry. 2009 doi: 10.1016/j.bioelechem.2009.04.003. in press. [DOI] [PubMed] [Google Scholar]
- 24.Taniguchi I, Li C-Z, Ishida M, Yao Q. Electrochemical and spectroelectrochemical properties of manganese(III) reconstituted myoglobin. J. Electroanal. Chem. 1999;460:245–250. [Google Scholar]
- 25.Li C-Z, Nishiyama K, Taniguchi I. Electrochemical and spectroelectrochemical studies on cobalt myoglobin. Electrochim. Acta. 2000;45:2883–2888. [Google Scholar]
- 26.Barley MH, Rhodes MR, Meyer TJ. Electrocatalytic Reduction of Nitrite to Nitrous Oxide and Ammonia Based on the N-Methylated, Cationic Iron Porphyrin Complexes [FeIII(H2O)(TMPyP)]5+ Inorg. Chem. 1987;26:1746–1750. [Google Scholar]
- 27.Bedioui F, Trevin S, Albin V, Villegas MGG, Devynck J. Design and characterization of chemically modified electrodes with iron(III) porphyrinic-based polymers: Study of their reactivity toward nitrites and nitric oxide in aqueous solution. Anal. Chim. Acta. 1997;341:177–185. [Google Scholar]
- 28.Diab N, Schuhmann W. Electropolymerized manganese porphyrin/polypyrrole films as catalytic surfaces for the oxidation of nitric oxide. Electrochim. Acta. 2001;47:265–273. [Google Scholar]
- 29.Yu CH, Su YO. Electrocatalytic reduction of nitric oxide by water-soluble manganese porphyrins. J. Electroanal. Chem. 1994;368:323–327. [Google Scholar]