

Abstract
A wide array of readily prepared pivalates of ketoximes can be converted to the corresponding ketones in good yields by treatment with iron powder in THF containing catalytic amounts of both trimethylsilyl chloride and glacial acetic acid at room temperature for 30 minutes, followed by a brief aqueous workup.
Keywords: Oximes, Reduction
We have recently been actively involved in developing methodology for effecting both inter- and intramolecular Michael-type conjugate additions of carbon nucleophiles to in situ-produced vinylnitroso compounds.1,2 Thus, we have found that nitrosoalkene species 2 can easily be generated from α-chloro-O-TBS-oximes 1 using a fluoride source in the presence of a carbon nucleophile to form α-alkylated oximes 3 (Scheme 1). For many of our intended purposes, however, it was necessary to regenerate the carbonyl compounds 4 from the resulting oximes 3.
Scheme 1.

Numerous methods have previously been described for converting oximes and their O-acyl derivatives to carbonyl compounds.3 These procedures generally involve either hydrolytic, oxidative or reductive conditions. After some disappointing results using a few of the more common literature cleavage procedures (e.g. TiCl3,4 Dess-Martin periodinane,5 etc.), we decided to explore new methodology for this transformation. In particular, we were interested in developing a mild general procedure which would utilize inexpensive, commercially available reagents having long shelf lives. Moreover, since hydrolytic methods usually involve stringent reaction conditions, and oxidative procedures are often incompatible with functionality in some of our systems (e.g. amines, indoles, etc.), we primarily focussed on devising a reductive protocol.
In 1998, Burk and coworkers discovered that N-acetyl enamides can be prepared in moderate to good yields directly from ketoximes by heating at 70 °C in toluene/acetic anhydride in the presence of iron powder.6a In an improvement of this methodology, Zhang et al. found that these reactions can be effected at room temperature if DMF is used as solvent, and also that the reaction is initiated by the addition of a catalytic amount of trimethylchlorosilane.6b More recently, we reported that other acylating reagents can be used in this process.6c We considered the possibility of using a variation of this methodology for converting oximes to carbonyl compounds by omitting the acylating reagent in order to form the NH imine, which would be susceptible to rapid hydrolysis. It should also be noted that an iron(0)-promoted cleavage of oximes has been reported but which requires forcing conditions (Fe powder/conc. HCl/MeOH, reflux).7,8
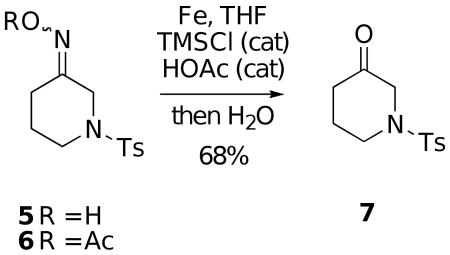
In initial exploratory experiments, the oxime 5 (12:1 E/Z mixture) was used as a test system (Eq. 1). However, exposure of this compound to iron powder and a catalytic amount of trimethylsilyl chloride in THF, MeOH or toluene at either room temperature or at reflux led to no observable cleavage reaction but rather only oxime E/Z isomerization occurred. On the other hand, we were pleased to find that the corresponding oxime acetate 6, when treated with iron powder in THF containing a catalytic amount of TMSCl at room temperature for about one hour, followed by stirring with water for an additional hour, afforded the desired ketone 7 in reasonable yield (>60%).
Although this transformation worked as planned, we observed that the reaction was highly irreproducible. Surprisingly, in some runs ketone 7 was formed cleanly and in others no reaction occurred. After some detective work, it was discovered that the successful reactions had been run in round-bottomed flasks that were previously cleaned in a concentrated nitric acid bath, followed by washing with water several times before drying. Reactions run in flasks which had not been treated in this manner only led to recovery of starting material. Thus, it became evident that the oxime acetate reduction by iron(0) is apparently catalyzed by a trace of acid remaining in the flask. In fact, addition of a catalytic amount of either nitric acid, p-toluenesulfonic acid or trifluoroacetic acid led to successful reactions, but the best yields of ketone 7 (68%) were obtained by addition of a small amount of glacial acetic acid to the mixture. It should also be noted that omission of the TMSCl shuts down the cleavage. However, during the course of these studies it was also observed that some deacetylation of 6 was occurring under these conditions, leading to unreactive oxime 5. Therefore, in an attempt to avoid this problem, we decided to explore the cleavage of other O-acyl oxime derivatives.
 |
(1) |
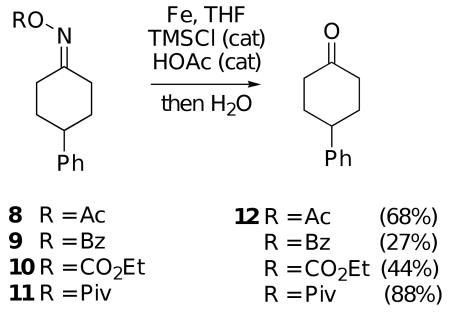
These studies were conducted on acyl derivatives 8-11 prepared from the oxime of 4-phenylcyclohexanone, and the results are outlined in Eq. 2. Thus, it was found that the pivalate ester9 of this oxime gave the best yield of ketone 12. The acetate 8, benzoate 9 and ethyl carbonate 10 all produced 12 in significantly lower yields. Using pivalate 11, further optimization studies showed that the cleavage works best if the reduction step is carried out at room temperature for 30 minutes, followed by addition of water and stirring for 15 minutes to effect imine hydrolysis. Longer reaction times for the reduction step generally led to lower yields of the ketone and to formation of significantly larger amounts of by-products. Also, using DMF as the solvent gave lower isolated yields of product 12 than reactions run in THF.
 |
(2) |
Using what was learned above, a general procedure has been developed for the cleavage of a broad range of ketoxime pivalates9 to the corresponding ketones under the reaction conditions shown in Eq. 3. This methodology has been applied to the representative examples listed in Table 1. It should be noted, however, that aldehyde oxime pivalates gave mixtures of the aldehyde and the corresponding nitrile under these reaction conditions. Thus, the work described here provides a very mild, general method for converting readily prepared pivalates of ketoximes to the corresponding ketones.
Table 1.
Conversion of oxime pivalates to the corresponding carbonyl compounds
| entry | oxime pivalate | ketone product | isolated yield |
|---|---|---|---|
| A | 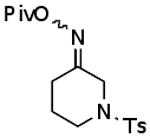 |
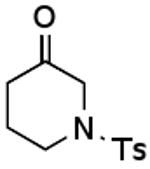 |
81% |
| B | 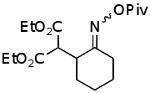 |
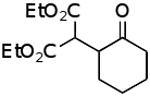 |
54% |
| C | 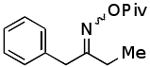 |
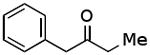 |
71% |
| D | 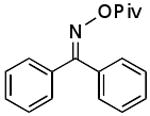 |
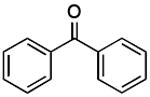 |
95% |
| E | 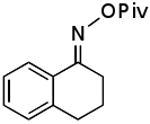 |
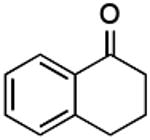 |
91% |
| F | 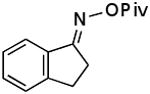 |
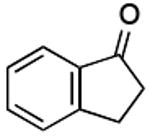 |
75% |
| G | 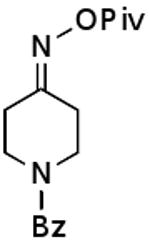 |
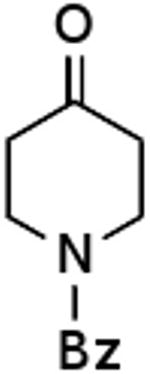 |
79% |
| H | 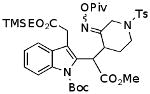 |
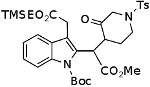 |
84%a,b,c |
This reaction was run at 0.03 M concentration.
Under the same condtions the corresponding oxime acetate gave 57% of the ketone.
These compounds are mixtures of diasteromers. Full spectral data will be reported in a subsequent paper.
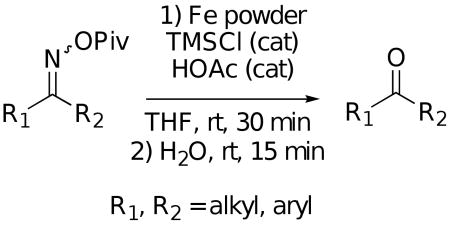 |
(3) |
General procedure for cleavage of oxime pivalates
To a solution of the oxime pivalate9 (0.10 mmol) in THF (1 mL) was added iron powder (55.8 mg, 1.0 mmol) followed by glacial AcOH (1 drop) and TMSCl (1 drop). After stirring for 30 min at rt, the reaction mixture was diluted with H2O (1 mL) and stirred for an additional 15 min. The liquid phase was separated from the remaining Fe powder using a pipette and transferred to a separatory funnel. The Fe powder was then washed with EtOAc (3 × 2 mL) which was added to the separatory funnel. The organic layer was separated and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give a residue, which was purified by flash column chromatography on silica gel eluting with a mixture of ethyl acetate and hexanes. Isolated yields of carbonyl products formed from a series of oxime pivalates are shown in Table 1.
Acknowledgments
We are grateful to the National Institutes of Health (9R56GM-087733) for financial support of this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.For reviews of vinylnitroso compounds see: Gilchrist TL. Chem Soc Rev. 1983;11:53.Lyapkalo IM, Ioffe SL. Russ Chem Rev. 1998;67:467.
- 2.(a) Korboukh I, Kumar P, Weinreb SM. J Am Chem Soc. 2007;129:10342. doi: 10.1021/ja074108r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li P, Majireck MM, Witek JA, Weinreb SM. Tetrahedron Lett. 2010;51:2032. doi: 10.1016/j.tetlet.2010.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For reviews see: Corsaro A, Chiacchio U, Pistara V. Synthesis. 2001:1903.Corsaro A, Chiacchio U, Pistara V. Curr Org Chem. 2009;13:482.Sahu S, Sahu S, Patel S, Dash S, Mishra BK. Ind J Chem. 2008;47B:259.
- 4.Wildsmith E, Timms GH. Tetrahedron Lett. 1971;12:195. [Google Scholar]
- 5.Chaudhari SS, Akamanchi KG. Synthesis. 1999:760. [Google Scholar]
- 6.(a) Burk MJ, Casey G, Johnson NB. J Org Chem. 1998;63:6084. doi: 10.1021/jo9809332. [DOI] [PubMed] [Google Scholar]; (b) Zhu G, Casalnuovo AL, Zhang X. J Org Chem. 1998;63:8100. [Google Scholar]; (c) Sun C, Weinreb SM. Synthesis. 2006:3585. [Google Scholar]
- 7.Pradhan PK, Dey S, Jaisankar P, Giri VS. Synth Commun. 2005;35:913. [Google Scholar]
- 8.N-O bonds of oxazolines have been cleaved with iron: Jiang D, Peng J, Chen Y. Org Lett. 2008;10:1695. doi: 10.1021/ol8002173.
- 9.For synthesis of oxime pivalates see: Davidson JP, Corey EJ. J Am Chem Soc. 2003;125:13486. doi: 10.1021/ja0378916.