

Abstract
Plasmid expression vectors carrying either the entire engrailed coding region or a subfragment including the homoeo box, produce protein fusions having sequence-specific DNA binding activity.
Mutations in Drosophila have identified genes that control major steps in development1–3. Some of these mutants, the segmentation mutants, are defective in the processes that subdivide the embryo into the segmented body plan4–7 while others, the homoeotic mutants, improperly specify the developmental fate of particular regions of the fly1. Garcia-Bellido8 suggested that these mutations affect ‘selector genes’ that act, in those cells in which they are expressed, to select the developmental pathway. It was proposed that they function by controlling ‘cytodifferentiation genes’8.
A segmentation gene
Each morphologically obvious segment is composed of cells from two distinct lineages termed anterior and posterior compartments9,10. Genetic analysis suggests that the engrailed gene product is required to specify cells as members of posterior compartments11–14. As anticipated by the selector gene hypothesis8, by 3.5 h of development engrailed gene product accumulates in narrow bands corresponding to the primordia of the posterior part of each segment15–17. Apparently, the engrailed regulatory activity acts wherever it is expressed to direct cells along a pathway of development suited to cells of posterior compartments8. In engrailed mutants, the segmental fusions and the failure to specify cells of posterior compartments are thought to result from the absence of this regulator or from alterations in its expression.
Selector genes interact
In addition to engrailed, several other Drosophila developmental genes are expressed in spatially restricted patterns consistent with their apparent roles in directing particular portions of the developmental programme15–19. These studies have focused interest on two related issues. How are these genes regulated to achieve the appropriate pattern of expression, and how do they regulate subsequent development? Recent studies suggest that the selector genes interact in a complex regulatory network. Based on phenotypes of double mutant combinations, Struhl20 argued that the Ubx gene product represses Scr expression in the mesothorax. Molecular studies21 have offered further suggestions of interactions. Regulatory interactions among six different homoeotic loci appear to coordinate their spatial patterns of expression (ref. 22 and C. Wedeen and M. Levine, personal communication).
Recent molecular analyses suggest that the segmentation genes interact to control the expression of one another. Immunofluorescent staining revealed that engrailed protein appears first in alternate segments and only later in every segment17. This led to the suggestion that engrailed expression is regulated by the products of another class of segmentation genes, the pair-rule genes17,23, which are expressed in alternate segments19,24. Alterations in the pattern of engrailed expression in pair-rule mutants have confirmed this prediction (S. DiNardo and P.H.O’F., unpublished observations; K. Howard and P. Ingham, personal communication). Similar analyses of fushi tarazu expression suggest that its expression is also influenced by several other segmentation genes (S. Carroll and M. Scott, personal communication).
These observations suggest an extensive network of regulatory interactions among the Drosophila developmental genes, and imply that selector genes are themselves targets for the regulators they encode17,23.
The homoeo box
The products of a number of the developmental genes include a conserved protein domain of 60 amino acids, the homoeo domain. Related sequences have been identified in species from human to annelids25–30. This remarkable conservation suggests that the homoeo domain has common physical interactions in all these organisms. A portion of this domain is found in two yeast proteins, a1 and α2, that determine the cell fate (mating type) via transcriptional regulation31,32. Because of this homology it has been proposed that the developmental genes of Drosophila might function similarly, by interacting with DNA27,33. Further, it has been noted that sequences within the homoeo domains are compatible with a protein structural motif that characterizes bacterial sequence-specific DNA binding proteins27,34.
Here we address three issues. Does a homoeo-domain-containing protein bind to DNA? Is the binding specific? Is the homoeo domain responsible for binding?
Construction of engrailed fusion proteins
To study how the engrailed protein product might function as a regulator, we constructed bacterial expression vectors that encoded the engrailed protein as carboxy-terminal extensions of β-galactosidase. In order to test for possible autonomous functions of the homoeo domain, we constructed three fusion proteins (Fig. 1). The full-length fusion contained the sequence encoding the entire 552 amino acids of the engrailed protein. This engrailed protein sequence has been derived from an open reading frame in the engrailed cDNA sequence35. The pattern of evolutionary sequence conservation of this open reading frame suggests that it encodes protein (J. Kassis, D. K. Wright, and P.H.O’F., unpublished observations). We think that this predicted protein is made in vivo because two antisera directed against different domains of this predicted sequence detect expression of these domains in the posterior compartments of segments17. The ‘homoeo domain fusion’ includes only the terminal quarter of the protein coding sequence, encompassing the 60-amino-acid homoeo domain and an additional 44 amino acids on the N-terminal side plus 39 amino acids on the C-terminal side. The ‘non-homoeo domain fusion’ is deleted for a 196-amino-acid region and lacks the homoeo domain (Fig. 1).
Fig. 1.

Construction of lacZ-engrailed fusions and expression in Escherichia coli. a, Gene fusions; b, polyacrylamide gel electrophoresis of bacterial extracts; FL, full-length fusion; HD, homoeo domain fusion; NHD, non-homoeo domain fusion; lacZ, β-galactosidase.
Methods: a, DNA fragments derived from the engrailed locus17,35,38 were inserted in the polylinker of expression vector plasmids pUR 290, 291 or 292 (ref. 47). These different plasmids allow inserted DNA to be expressed as a C-terminal extension of β-galactosidase. In the full-length fusion protein (FL) the extension includes 7 amino acids that precede the first methionine of the engrailed protein as well as the entire engrailed protein. In the homoeo domain construct (HD), a BamHI fragment was spliced out of the FL construction, keeping only the last quarter of the engrailed coding sequence from amino acid 409 to the end. This fusion protein contains the entire homoeo domain (amino acids 453–513). Amino acids 347–542, containing the homoeo domain, have been deleted in the non-homoeo domain protein (NHD) by removing a XhoI fragment from the FL construct. In the absence of insertion the lacZ gene is expressed as the complete β-galactosidase. b, Polyacrylamide gel electrophoresis of bacterial extracts. The expression of the fusion proteins from chimaeric plasmids is under the control of the lac promoter in a lacI overproducing strain DG101 (a gift from D. Gelfand, Cetus Corporation). Bacteria in exponential growth were induced with isopropyl β-thiogalactoside at an absorbance of 0.5 (at 600 nm) and collected 2 h later. Cells were resuspended in about 0.005 culture volume of 25% sucrose, 0.2 mM EDTA, 40 mM Tris-HCI pH 7.5 and 1 mM dithiothreitol (DTT). Lysis and protein solubilization were achieved by lysozyme treatment (0.4 mg ml−1 for 1 h at 0 °C followed by addition of urea to 4 M and further incubation at 0 °C for 1 h). After centrifugation at 20,000 r.p.m. for 1 h the urea was removed from the supernatant by dialysis first against 10 mM Tris-HCI pH 7.5, 25 mM NaCl, 1 mM EDTA, 0.1% Triton X-100, 1 mM DTT, 10% glycerol, 1 mM phenylmethylsulphonyl fluoride, and 0.1 mM benzamidine containing 2 M urea and subsequently against the same buffer without urea. Glycerol was added to the extracts to a final concentration of 50% and they were stored at −20 °C. When analysed by SDS-gel electrophoresis the fusion proteins are seen as abundant high relative molecular mass proteins (arrowheads). The larger fusion protein is present in lower amounts and we detect numerous minor bands that are presumed to result from degradation of the fusion proteins.
Nonspecific DNA binding
We first tested whether the fusion proteins would bind DNA nonspecifically. We mixed the fusion proteins with labelled restriction fragments of DNA and then, using an antibody directed against bacterial β-galactosidase and fixed Staphylococcus aureus as an immunoadsorbent, we precipitated the fusion protein along with bound DNA fragments31,36. The bound DNA fragments were then separated by electrophoresis and detected by autoradiography. At low salt concentration (50 mM NaCl) and in the absence of carrier DNA, the full-length fusion protein and the homoeo domain fusion protein bound all of the HaeIII restriction fragments of bacteriophage Φ×174 DNA, whereas neither β-galactosidase alone, nor the non-homoeo domain fusion protein bound significant amounts of DNA (Fig. 2). Though these observations are consistent with the hypothesis that the homoeo domain would function in DNA binding, the results could potentially be due to simple ionic interactions. Sequence-specific binding would suggest that the fusion protein has a DNA binding domain.
Fig. 2.
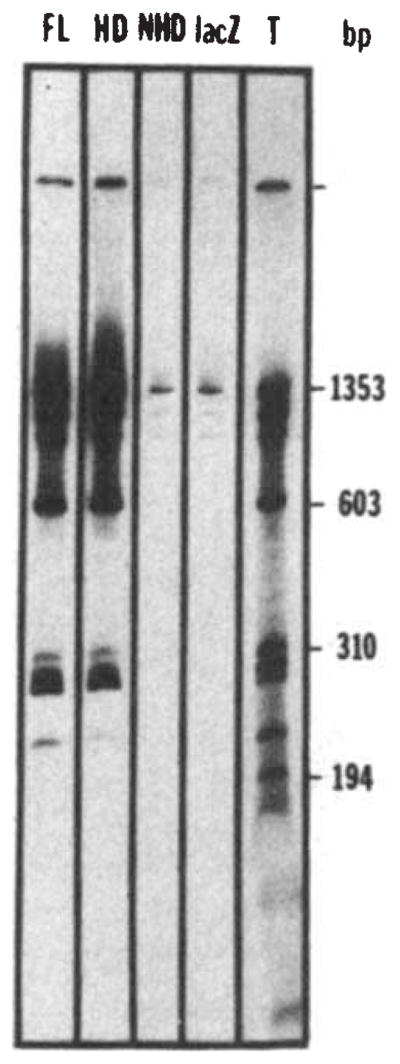
Nonspecific binding of the fusion proteins to DNA. Bacteriophage Φ×174 DNA was cleaved by HaeIII and end-labelled using T4 polymerase48. The labelled DNA (about 30 ng) was incubated for 30 min at 0 °C in 25 μ1 of binding buffer (BB) (50 mM NaCl, 20 mM Tris pH 7.6, 0.25 mM EDTA, 1 mM DTT, 10% glycerol) in the presence of a bacterial extract containing the fusion protein (about 10 μg of total protein extract, see Fig. 1 legend). The complexes formed between DNA and the fusion proteins were then immunoprecipitated by 30 min further incubation at 0 °C with an anti-β-galactosidase monoclonal antibody (provided by Tom Mason and Judy Partaledis, courtesy of Mike Hall) preadsorbed on cross-linked Staphylococcus (Pansorbin, Calbiochem). The pellet was washed twice in BB, phenol extracted and the DNA ethanol precipitated before polyacrylamide gel electrophoresis and autoradiography. The autoradiogram shows the results of precipitations performed in the presence of extracts containing the full-length fusion (FL), the homoeo domain fusion (HD), the non-homoeo domain fusion (NHD) or β-galactosidase (lacZ). Lane T shows the labelled digest before immunoprecipitation, representing 25% of the counts added to the incubation mixture in the immunoprecipitation experiments.
Sequence-specific DNA binding
Since we had no knowledge of what the target sequences for binding of engrailed protein might be, we sought a generalized approach to detect sequence specificity. Sequence-specific DNA binding proteins recognize degenerate versions of a consensus binding site. Sufficiently complex DNA should contain, by chance, sequences recognized by the protein. For example, Ross and Landy37 identified the sequence of several binding sites for λ integrase (Int protein) in pBR322 DNA.
To pursue this approach, we digested λ-phage DNA to produce more than 100 fragments and labelled their 3′ ends. This DNA was used in the assay described above, except that we added increasing amounts of salt or carrier DNA so that only fragments that bound to the fusion proteins with higher affinities would appear in the precipitate. Figure 3 shows that at low concentrations of carrier DNA all the λ DNA fragments are bound nonselectively (lanes 2, 5), but as the stringency of the binding conditions is increased the binding becomes selective. For example, at high concentration of carrier DNA (Fig. 3, lanes 4, 7), only 4 fragments out of 115 DNA fragments are retained. These experiments demonstrate the specificity of engrailed fusion protein binding to DNA.
Fig. 3.
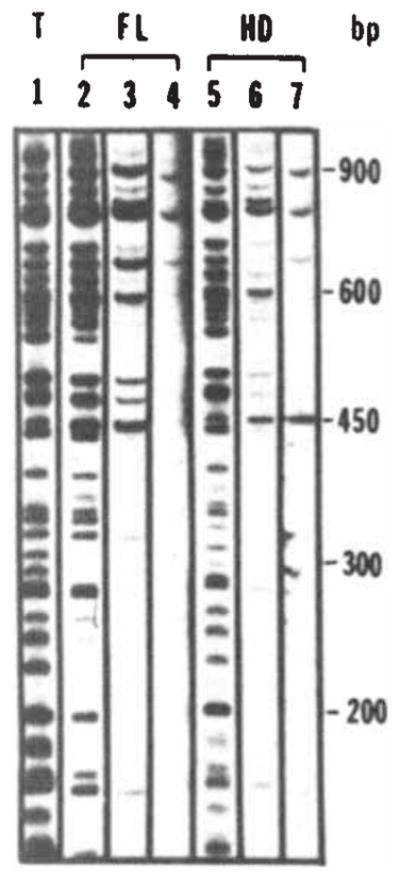
Sequence-specific DNA interaction of the fusion protein with bacteriophage λ DNA fragments. Bacteriophage λ DNA was restricted with Sau3A and labelled using T4 polymerase48. Binding assays were performed as described in Fig. 2 legend with the addition of carrier DNA as indicated. Fragments were separated on a 5% polyacrylamide gel. Lane 1 shows the total digest. The amount of DNA loaded in lane 1 is one-quarter of the input amount used in the binding assays (10ng; 0.4 μg ml−1). Lanes 2, 3 and 4 show immunoprecipitates obtained following addition of the full-length fusion extract in the presence of 0, 4 and 40 μg ml−1 of carrier DNA (fragmented calf thymus DNA), respectively. Lanes 5, 6 and 7 show the results of a similar experiment using the homoeo domain fusion extract. Lanes 4 and 7 were exposed four times longer because of the reduced recovery of bands at high levels of carrier.
Whether the homoeo domain fusion or the full-length fusion is used in binding assays, the same fragments are recovered in immunoprecipitates at comparable efficiencies (Fig. 3). Thus, the two fusions bind with the same specificity and generally exhibit similar relative affinities for these fragments. The 143 amino acids of the engrailed sequence present in the homoeo domain fusion must include a domain competent in specific binding. At least under the conditions of our assay, the additional 409 amino acids of the full-length fusion protein make little or no contribution to the specificity of binding.
To estimate the minimal binding constant of the binding interaction, we assume that all of the fusion protein is active31,36. When our binding assay contains less than 10−8 M fusion protein and less than 10−11 M DNA fragments, recovery of specific DNA fragments in the immunoprecipitate exceeds 50% (for example Fig. 4A, engrailed fragments f and k). Accordingly, the binding constant must exceed 5 × 107 mol−1 (at 170 mM NaCl). Further more, preliminary evidence indicating that only a fraction of the fusion protein is active (J.T., unpublished observations) suggests that the binding constant must be higher, and may well be comparable to the binding constants of other sequence-specific DNA binding proteins34.
Fig. 4.

Sequence-specific interaction of the homoeo domain fusion protein with restriction fragments of cloned fushi tarazu and engrailed sequences. The fushi tarazu clone (p6-3, derived from clone pDmA439, a gift from Matt Scott)49 contains 900 bp of upstream sequence, the entire coding sequence and 800 bp of downstream sequence. The engrailed clone (p615) contains 2.3 kb of upstream sequence, the complete first exon and most of the first intron35,38 (panel B). Restriction fragments of p6-3, p615 or λ DNA were end-labelled and tested for fusion protein binding as described in the legend for Fig. 2, except that a higher salt concentration (170 mM NaCl) was used to diminish nonspecific interactions. Fragments were separated on a 5% acrylamide gel. In the example shown in A, the fushi tarazu plasmid (ftz) DNA was digested with RsaI and the engrailed plasmid (en) was digested with HinfI and ClaI. Lanes marked T show the total fragment pattern and lanes marked I show the fragments recovered in the immunoprecipitate. For both plasmids the amount of DNA loaded in T is one-quarter of the amount subjected to immunoprecipitation and displayed in I. Bands derived totally from plasmid sequences are identified with a ‘p’. Bands derived at least in part from insert sequences are designated as a to j (ftz) or a to o (en) and their positions within the cloned sequence are indicated in B. Similar experiments using AluI, HaeIII, BstNI, DdeI or HpaII to digest these plasmids gave comparable results (not shown). B shows a map of the inserts in p6-3 (ftz) and p615 (en). The orientation of the coding region within the insert is indicated. Except for the dashed portion of the engrailed insert, the sequence is known (refs 27, 35, and J. Kassis, D. K. Wright and C.D., unpublished). The positions of restriction enzyme sites (RsaI sites in ftz and HinfI and ClaI sites in en) are indicated and all fragments detectable in the separation shown in panel A are lettered. Those fragments detected as bound by the homoeo domain fusion protein are indicated as +. Because they were not detected, a few small restriction fragments (unlettered) could not be scored in the binding experiment (A) but analysis of these DNAs cleaved with other restriction enzymes failed to detect additional binding sites. Exons of the transcribed region are indicated with a bold line and introns with sloping lines. The hatched region indicates the position of a subfragment that was purified and used for additional binding studies (Fig. 5). Note that the region responsible for binding of ftz, fragment a, was further localized by analysis of AluI, HaeIII, BstNI, DdeI, HpaII and HinfI digests that defined only one site; this site corresponds to that mapped in more detail by analysis of the purified subfragment (Fig. 5). C Compares the efficiency of homoeo domain fusion protein binding to engrailed or λ sites. The binding assays were performed as described in A except that increasing amounts of unlabelled DNA (HinfI-restricted p615) was added as competitor. Lanes T represent 10% of the counts of the total digest submitted to immunoprecipitation in lanes 1, 2, 3 and 4. In lane 1, no unlabelled DNA was added while in lanes 2, 3 and 4, 30 ng, 250 ng and 4 μg, respectively, were added, en lanes: plasmid p615 was restricted with ClaI and HinfI labelled and submitted to immunoprecipitation. Lanes 1, 2, 3 and 4 represent immunoprecipitated DNA from ~4 ng of labelled DNA (~ 1 fmol). Obviously the four bands retained in A behave differently when unlabelled competitor DNA is added. Three bands (arrows) corresponding to fragments f, k and h in A, are detected in the immunoprecipitate in the presence of 1,000-fold excess of unlabelled DNA. λ lanes: λ DNA was restricted with ClaI and HinfI, labelled and processed under the same conditions as for en DNA. An equimolar amount of DNA (1 fmol) was used in this experiment (20 ng). Two bands were retained at the highest stringency (arrowheads). en + λ lanes: T is an equimolar mixture of en and λ DNA restricted with ClaI and HinfI. The bands that are retained are the same as those seen in the experiments testing en and λ DNAs individually. Five bands are retained in the presence of the highest concentration of competitor (arrows and arrowheads).
The binding behaviour of the fusion protein described here may not accurately reproduce the behaviour and specificities of the natural engrailed protein. The binding specificity might be influenced by interactions that the fusion protein cannot reproduce or by modifications that would be missing in a protein produced in Escherichia coli. Furthermore, our simple in vitro binding assay may lack accessory factors that influence in vivo binding of the engrailed protein to DNA. Nonetheless, because other work using fusion proteins or proteolytic fragments31,34 suggests that DNA binding domains can function relatively autonomously, we believe that the results reported here are likely to reflect at least a subset of the activities of the normal engrailed protein.
Specific binding to Drosophila DNA
If engrailed and other selector genes act as pleiotropic regulators of transcription, we might expect their protein products to interact with DNA near the promoters of a number of target genes. Can we identify any plausible candidates for target genes? There is considerable evidence that selector genes regulate each other’s expression (summarized above). Thus, we envision that the developmental genes will include regulatory sites that are targets for interaction with the products of other developmental genes. Because of the high degree of relatedness of the developmental genes, the various target sites might also be homologous. Because it is a member of this group of related proteins, perhaps the engrailed fusion protein will exhibit site-specific interaction with all or a subset of the related regulatory sites.
Following this line of logic, we decided to look for engrailed fusion protein binding adjacent to cloned selector genes. Because a detailed analysis would require DNA sequence information, we chose to examine the engrailed locus itself, for which we have 1.2 kilobases (kb) of upstream sequence (unpublished data), and the fushi tarazu locus that had been sequenced by Laughon and Scott27.
We looked for engrailed fusion protein binding to a 4.9-kb EcoRI fragment that includes 2.6 kb of engrailed coding sequence and 2.3 kb of upstream sequences38 and to a 3.2-kb fragment that includes the fushi tarazu coding sequence and flanking sequences49. Figure 4A shows that both the cloned engrailed sequences and cloned fushi tarazu sequences contain fragments that bind to the engrailed fusion protein under stringent assay conditions. In fact, a number of binding fragments are detected (Fig. 4A, C). The positions of binding fragments are indicated in Fig. 4B.
We purified the subfragments indicated by the hatched lines in Fig. 4B and used these to map more precisely the binding interactions upstream of the engrailed and fushi tarazu coding regions. Secondary digests of these fragments were tested for interactions with the engrailed fusion protein (Fig. 5). These analyses localized three binding sites within the 900-base-pair (bp) region 5′ to the engrailed cDNA. The higher resolution and sensitivity of these experiments showed that the binding fragment k (Fig. 4A, B) actually contained two binding sites (sites a and b in Fig. 5) and that fragment d, though not detected as a binding fragment in experiments using the whole plasmid, contains a weak binding site (site c in Fig. 5). The analysis of the fushi tarazu subfragment did not reveal any new binding sites but did contribute to more accurate localization of the upstream site (Fig. 5). At present the accuracy of localization of the sites does not allow us to identify a consensus binding site unambiguously.
Fig. 5.

Localization of binding sites in the 5′ regions of engrailed and fushi tarazu. In digests of whole plasmid DNA we identified fragments that were bound by the homoeo domain fusion protein (Fig. 4). To localize more precisely the binding sites immediately upstream of the engrailed and fushi tarazu coding regions, we purified a 1,180-bp ClaI-PstI fragment from the engrailed clone and a 939-bp HinfI-TaqI fragment from the fushi tarazu clone (Fig. 4B). These purified fragments were digested with additional enzymes as indicated and the binding of subfragments assayed as before (Fig. 4). In each of the restriction digests shown here the binding fragments are indicated as solid lines. Although the overlap of binding sites: a ClaI-AluI fragment (a, the binding regions, this could be misleading if the binding sites were complex. Therefore, in the summary diagrams above the restriction patterns, we have indicated the precision of the localization of the binding sites based on the minimal fragment showing binding (bold line). For engrailed, three fragments were identified as containing binding sites: a, ClaI-AluI fragment (a, 67 bp), a HaeIII-AluI fragment (b, 100 bp) and a HhaI fragment (c, 88 bp). For ftz a single site was localized to a 165-bp HinfI-RsaI fragment(*).
Binding sites
Without a functional assay we cannot directly assess the importance of the binding sites detected in cloned Drosophila sequences. However, we can test whether affinities, frequency of occurrence, clustering and location of binding sites differ from fortuitous sites.
If the frequency of fortuitous sites were extremely low (less than 1 per 1,000 kb), the presence of a cluster of binding sites within a few kilobases of engrailed DNA would be highly significant. This is not the case. Although the frequency of binding sites on a 4.9-kb fragment of engrailed DNA is higher than the density of λ DNA, the difference was not very large—about 10-fold (Fig. 4C).
Fortuitous binding sites should have a wide range of affinities depending on their similarity to an optimal site. Higher-affinity fortuitous sites should be less frequent (chance might produce an optimal binding site but should do so less frequently than imprecise approximations of this sequence). We tested the relative affinity of the engrailed fusion protein for various binding sites. We used conditions where a number of labelled restriction fragments are bound selectively. Addition of cold competitor DNA displaced bacteriophage λ DNA fragments with different efficiencies (Fig. 4C). Thus, as expected, the binding sites in λ DNA have a range of affinities and there are few high-affinity sites. Assuming that the binding sites in λ DNA occur by chance, the specific binding of 14 restriction fragments at intermediate stringencies suggests that the sequence recognized by the engrailed fusion protein is relatively short (about 6 bp) or substantially degenerate.
For some sequence-specific DNA binding proteins (such as lac repressor), fortuitous occcurrence of high-affinity binding sites is extremely unlikely. For these a site of functional interaction (the lac operator39) has a distinctively high affinity. For other sequence-specific interactions (for example λ integrase37,40) the affinities of fortuitous and functional sites overlap. Depending on the characteristics of the engrailed fusion protein, functionally relevant sites might have distinctively high affinities. We therefore examined the relative binding affinity of engrailed and bacteriophage λ DNA fragments. We observe differing affinities for the engrailed fusion protein interaction with various sites on engrailed DNA (Fig. 4C). Using our present assay, the ranges of affinities seen for λ and engrailed sites overlap (Fig. 4C). Three engrailed fragments bind with particularly high affinity (arrows) compared with two λ fragments (arrowheads).
The binding data provide no support for suggestions that the binding sites in Drosophila DNA are functional. It should, however, be made clear that the opposing conclusion also cannot be reached from these data; that is, the binding sites in engrailed DNA cannot be dismissed as nonfunctional because they have properties similar to fortuitous binding sites. Thus, the issue of function remains open.
Although the functional importance of the binding sites still requires experimental test, we propose that the binding sites we have detected in Drosophila DNA function in vivo as targets for interaction with either the engrailed protein or closely related gene products (that is, other selector genes). We make this suggestion on the basis of the location, clustering and conservation of the sites. The positions of binding sites in relation to the fushi tarazu and the engrailed coding regions are reminiscent of the positions of enhancer elements in other systems41–43. The clustering of binding sites is unlikely to be coincidental. Such clustering of binding sites for regulatory proteins is fairly common (for example, refs 44,45). Finally, if functional, the binding sites will be conserved in evolution. In the absence of a functional test, we believe that the best way to distinguish fortuitous and functional binding sites is to see whether protein binding occurs at analogous positions in distantly related genomes. We have cloned the engrailed gene of a distantly related Drosophila species, D. virilis46. A preliminary analysis indicates that the fusion protein binds to fragments upstream of the D. virilis engrailed gene (D. Wright, unpublished data).
Binding specificity
Together with previous arguments27, our results predict that the homoeo domain imparts a sequence-specific DNA binding activity to the protein. Accordingly, other homoeo domain-containing proteins should also bind DNA in a sequence-specific manner and such proteins having closely homologous homoeo domains should have similar sequence specificity. We presently recognize two classes of homoeo domain sequences. Class I is comprised of seven genes which have highly homologous homoeo domains and are located in two clusters of developmental genes (the bithorax complex and the Antennapedia complex)22,25. Class II is comprised of the engrailed homoeo domain and the highly homologous homoeo domain of the engrailed related gene35. The homoeo domains of different classes have lower homology (Fig. 6). As noted previously, the regions of sequence identity suggest that class I homoeo domains might specify binding to the same sequence27. The differences between class I and class II homoeo domains might include an alteration of the sequence specificity.
Fig. 6.

Family tree of relatedness of homoeo domains. Pairwise comparisons of the protein sequences of the homoeo domains encoded by Drosophila genes (Antp, Ubx, ftz, en and er), yeast genes (a1 and α2) and sequences isolated by homology from humans (Hu1 and Hu2) and frogs (AC1) were used to score divergence. The comparison was confined to residues 29-58 because this region is well conserved among all of these sequences. To produce homology scores we gave one point for amino-acid identity and half a point for similarity. The data are assembled into a family tree by indicating the divergence of each sequence that approximates all the pairwise comparisons. The sequence data are taken from Levine et al28 (Hu1 and Hu2), Scott and Weiner26 (Ubx and ftz), McGinnis et a125 (Antp), Poole et al.35 (en and er), Carraso et al.30 (AC1), and Tatchell et al.32 (α2 and a1). Note that the discrepancies in the published sequence25,26 for Ubx are significant in comparison to the divergence among class I homoeo domains.
Evolution of proteins
Duplication and divergence of a primordial gene encoding a DNA binding protein might lead to a family of interacting regulators. If the primordial protein included sequences for dimerization and for DNA binding, newly duplicated coding regions would have common binding specificities and could form heterotypic dimers. The interactions between products of duplicated genes would persist if the dimerization function and DNA binding function were conserved. We suggest that continued duplication and divergence can result in a family of DNA binding proteins that interact physically by forming heterotypic associations and interact functionally by competition for binding to related DNA binding sites. Such interactions would link the various genes in a regulatory network. The evolution of the members of the family would be coupled because of the importance of maintaining the interactions among the members of this regulatory network. Since coordinate change of many genes is an extraordinarily unlikely event, such a network of interaction may contribute to the extraordinary conservation of homoeo domain sequences. It should be possible to test the predictions of this rationale using approaches similar to those used here to show that the engrailed gene encodes a sequence-specific DNA binding activity.
Acknowledgments
We thank our colleagues for discussions and experimental assistance, particularly Steve DiNardo, Mike Hall, Sandy Johnson, Judy Kassis, Jerry Kuner, Roger Miesfield, Sandro Rusconi, Elizabeth Sher and Deann Wright. We thank Steve Poole for the gift of en cDNA and sequence information before publication, Matt Scott for the fushi tarazu clone and for encouragement, and Sandy Johnson and Keith Yamamoto for their comments on the manuscript. This work was funded by NSF grant PCM-8418263 and by NIH grant GM 31286. C.D. was supported by a Fogarty fellowship and by ARC, and J.T. by an NIH training grant.
References
- 1.Lewis EB. Nature. 1978;276:565–570. doi: 10.1038/276565a0. [DOI] [PubMed] [Google Scholar]
- 2.Kaufman TC, Lewis R, Wakimoto B. Genetics. 1980;94:115–133. doi: 10.1093/genetics/94.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia-Bellido A, Santamaria P. Genetics. 1972;72:87–104. doi: 10.1093/genetics/72.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nusslein-Volhard C, Wieschaus E. Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- 5.Nusslein-Volhard C, Wieschaus E, Kluding H. Wilhelm Roux Arch dev Biol. 1984;193:267–282. doi: 10.1007/BF00848156. [DOI] [PubMed] [Google Scholar]
- 6.Weischaus E, Nusslein-Volhard C, Jurgens G. William Roux Arch dev Biol. 1984;193:296–307. doi: 10.1007/BF00848158. [DOI] [PubMed] [Google Scholar]
- 7.Jurgens G, Wieschaus E, Nusslein-Volhard C, Kluding H. Willhelm Roux Arch dev Biol. 1984;193:283–295. doi: 10.1007/BF00848157. [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Bellido A. CIBA Fdn Symp. 1975;29:161–182. doi: 10.1002/9780470720110.ch8. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Bellido A, Ripoll P, Morata G. Nature new Biol. 1973;245:251–253. doi: 10.1038/newbio245251a0. [DOI] [PubMed] [Google Scholar]; Devl Biol. 1976;48:132–147. [Google Scholar]
- 10.Crick FHC, Lawrence PA. Science. 1975;189:340–347. doi: 10.1126/science.806966. [DOI] [PubMed] [Google Scholar]
- 11.Morala G, Lawrence PA. Nature. 1975;255:608–617. doi: 10.1038/255614a0. [DOI] [PubMed] [Google Scholar]
- 12.Lawrence PA, Morata G. Wilhelm Roux Arch dev Biol. 1979;187:375–379. doi: 10.1007/BF00848471. [DOI] [PubMed] [Google Scholar]
- 13.Struhl G. Devl Biol. 1981;84:372–385. doi: 10.1016/0012-1606(81)90406-1. [DOI] [PubMed] [Google Scholar]
- 14.Kornberg T. Proc natn Acad Sci USA. 78:1095–1099. doi: 10.1073/pnas.78.2.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]; Devl Biol. 1981;86:363–372. [Google Scholar]
- 15.Kornberg T, Siden I, O’Farrell P, Simon M. Cell. 1985;40:45–53. doi: 10.1016/0092-8674(85)90307-1. [DOI] [PubMed] [Google Scholar]
- 16.Fjose A, McGinnis WJ, Gehring WJ. Nature. 1985;313:284–289. doi: 10.1038/313284a0. [DOI] [PubMed] [Google Scholar]
- 17.DiNardo S, Kuner J, Theis J, O’Farrell PH. Cell. doi: 10.1016/0092-8674(85)90012-1. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akam ME. EMBO J. 1983;2:2075–2084. doi: 10.1002/j.1460-2075.1983.tb01703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hafen E, Kuroiwa A, Gehring WJ. Cell. 1984;37:833–841. doi: 10.1016/0092-8674(84)90418-5. [DOI] [PubMed] [Google Scholar]
- 20.Struhl G. Proc natn Acad Sci USA. 1982;79:7380–7384. doi: 10.1073/pnas.79.23.7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hafen E, Levine M, Gehring W. Nature. 1984;307:287–289. doi: 10.1038/307287a0. [DOI] [PubMed] [Google Scholar]
- 22.Harding K, Wedeen C, McGinnis W, Levine M. Science. doi: 10.1126/science.3898362. in the press. [DOI] [PubMed] [Google Scholar]
- 23.O’Farrell PH, et al. UCLA Symp molec cell Biol, new Ser. 1985;31 [Google Scholar]
- 24.Wakimoto BT, Turner RF, Kaufman TC. Devl Biol. 1984;102:147–172. doi: 10.1016/0012-1606(84)90182-9. [DOI] [PubMed] [Google Scholar]
- 25.McGinnis W, Garber RL, Wirz J, Kuroiwa A, Gehring WJ. Cell. 1984;37:403–408. doi: 10.1016/0092-8674(84)90370-2. [DOI] [PubMed] [Google Scholar]
- 26.Scott MP, Weiner AJ. Proc natn Acad Sci USA. 1984;81:4115–4119. doi: 10.1073/pnas.81.13.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laughon A, Scott MP. Nature. 1984;310:25–31. doi: 10.1038/310025a0. [DOI] [PubMed] [Google Scholar]
- 28.Levine M, Rubin G, Tjian R. Cell. 1984;38:667–673. doi: 10.1016/0092-8674(84)90261-7. [DOI] [PubMed] [Google Scholar]
- 29.McGinnis W, Hart CP, Gehring WJ, Ruddle F. Cell. 1984;38:675–680. doi: 10.1016/0092-8674(84)90262-9. [DOI] [PubMed] [Google Scholar]
- 30.Carrasco AE, McGinnis W, Gehring WJ, DeRobertis EM. Cell. 1984;37:409–414. doi: 10.1016/0092-8674(84)90371-4. [DOI] [PubMed] [Google Scholar]
- 31.Johnson A, Herskowitz I. Cell. 1985;42:237–247. doi: 10.1016/s0092-8674(85)80119-7. [DOI] [PubMed] [Google Scholar]
- 32.Tatchell K, Nasmyth K, Hall B, Astell C, Smith M. Cell. 1981;27:25–35. doi: 10.1016/0092-8674(81)90357-3. [DOI] [PubMed] [Google Scholar]
- 33.Shepherd JCW, McGinnis W, Carrasco AE, DeRobertis EM, Gehring WJ. Nature. 1984;310:70–71. doi: 10.1038/310070a0. [DOI] [PubMed] [Google Scholar]
- 34.Pabo CO, Sauer RTA. Rev Biochem. 1984;53:293–321. doi: 10.1146/annurev.bi.53.070184.001453. [DOI] [PubMed] [Google Scholar]
- 35.Poole SJ, Kauvar LM, Drees B, Kornberg T. Cell. 1985;40:37–43. doi: 10.1016/0092-8674(85)90306-x. [DOI] [PubMed] [Google Scholar]
- 36.McKay R. J molec Biol. 1981;145:471–488. doi: 10.1016/0022-2836(81)90540-4. [DOI] [PubMed] [Google Scholar]
- 37.Ross W, Landy A. Proc natn Acad Sci USA. 1982;79:7724–7728. doi: 10.1073/pnas.79.24.7724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuner JM, et al. Cell. 1985;42:309–315. doi: 10.1016/s0092-8674(85)80126-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin S-Y, Riggs AD. J molec Biol. 1972;72:671–690. doi: 10.1016/0022-2836(72)90184-2. [DOI] [PubMed] [Google Scholar]
- 40.Better M, Lu C, Williams RC, Echols H. Proc natn Acad Sci USA. 1982;79:5837–5841. doi: 10.1073/pnas.79.19.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banerji J, Rusconi S, Schaffner W. Cell. 1981;27:299–308. doi: 10.1016/0092-8674(81)90413-x. [DOI] [PubMed] [Google Scholar]
- 42.Gillies SD, Morrison SL, Oi VT, Tonegawa S. Cell. 1983;33:717–728. doi: 10.1016/0092-8674(83)90014-4. [DOI] [PubMed] [Google Scholar]
- 43.Stuart GW, Searle PF, Chen HY, Brinster RL, Palmiter RD. Proc natn Acad Sci USA. 1984;81:7318–7322. doi: 10.1073/pnas.81.23.7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller AM, MacKay VL, Nasmyth KA. Nature. 1985;314:598–603. doi: 10.1038/314598a0. [DOI] [PubMed] [Google Scholar]
- 45.Dynan WS, Tijan R. Nature. 1985;316:774–778. doi: 10.1038/316774a0. [DOI] [PubMed] [Google Scholar]
- 46.Kassis J, Wong ML, O’Farrell PH. Molec cell Biol. 1985;5:3600–3609. doi: 10.1128/mcb.5.12.3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ruther U, Muller-Hill B. EMBO J. 1983;2:1791–1794. doi: 10.1002/j.1460-2075.1983.tb01659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Farrell P. Focus. 1981;3:1–3. [Google Scholar]
- 49.Weiner AJ, Scott MP, Kaufman TC. Cell. 1984;37:843–851. doi: 10.1016/0092-8674(84)90419-7. [DOI] [PubMed] [Google Scholar]