

Abstract
Biologically active membrane gangliosides, expressed and released by many human tumors, are hypothesized to significantly impact tumor progression. Lack of a model of complete and specific tumor ganglioside depletion in vivo, however, has hampered elucidation of their role. Here we report the creation of a novel, stable, genetically induced tumor cell system resulting in specific and complete blockade of ganglioside synthesis. Wild type (WT) and GM3 synthase/GM2 synthase double knockout (DKO) murine embryonic fibroblasts were transformed using amphotropic retrovirus-transduced oncogenes (pBABE-c-MycT58A+H-RasG12V). The transformed cells, WTt and DKOt respectively, evidenced comparable integrated copy numbers and oncogene expression. Ganglioside synthesis was completely blocked in the DKOt cells, importantly without triggering an alternate pathway of ganglioside synthesis. Ganglioside depletion (to <0.5 nmol/107 cells from 9-11 nmol/107 WTt or untransfected normal fibroblasts) did not adversely affect cell proliferation kinetics but did reduce cell migration on fibronectin-coated wells, consistent with our previous observations in ganglioside-depleted normal human fibroblasts. Strikingly, despite similar oncogene expression and growth kinetics, DKOt cells evidenced significantly impaired tumor growth in syngeneic immunocompetent mice, underscoring the pivotal role of tumor cell gangliosides and providing an ideal system for probing their mechanisms of action in vivo.
Keywords: ganglioside synthesis, ganglioside depletion, tumor model, c-Myc/H-Ras transformation, tumorigenesis
Introduction
Gangliosides are biologically active glycosphingolipids, ubiquitous in mammalian cells and consisting of a negatively charged sialic acid-containing carbohydrate head group attached to a ceramide that anchors the molecule predominantly in the outer leaflet of the lipid bilayer of the cell membrane. These molecules are recognized to have multiple effects—e.g., acting as cell surface receptors and markers, participating in intercellular communication, and modulating cell signaling, cell cycling, and cell mobility (Allende and Proia, 2002; Regina Todeschini and Hakomori, 2008).
Gangliosides of tumors are particularly important. They are both highly expressed (Hakomori, 1973) and actively shed from the tumor cell surface both in vitro and in vivo (Ladisch et al., 1983). Their shedding can alter the function of normal cells present in the tumor microenvironment, as initially suggested by findings of potent immunosuppressive activity of the shed gangliosides of a murine lymphoma (Ladisch et al., 1983). Subsequently, immunosuppressive activity of tumor gangliosides shed by many different tumor types has been reported (Buggins et al., 2001; Floutsis et al., 1989; Shurin et al., 2001; Uzzo et al., 1999) further supporting the concept that these shed gangliosides may interfere with cellular immune responses critical to effective antitumor immunity. Contrasting these suppressive effects, more recently enhancing effects on the function of other cells found in the tumor microenvironment have been delineated. For example, enrichment of the membranes of normal fibroblasts with complex gangliosides enhanced growth factor-induced receptor activation, signaling, and cell proliferation (Li et al., 2001; Liu et al., 2004). And, membrane ganglioside enrichment of vascular endothelial cells amplified VEGF-induced signaling and the associated cellular responses of proliferation and migration important for angiogenesis (Liu et al., 2006).
In vivo studies also link tumor gangliosides with tumor formation and progression. Two examples are (i) dramatically increased tumorigenicity of poorly malignant ganglioside-poor murine tumor cells by enrichment of their membranes with gangliosides purified from related highly tumorigenic tumor cells (Ladisch et al., 1987) and in humans (ii) high levels of circulating tumor gangliosides at diagnosis being associated with more rapid neuroblastoma tumor progression (Valentino et al., 1990). Altogether, these studies suggest that tumor cell gangliosides could play a critical facilitative role in tumor formation and progression.
What has been lacking to definitively test this hypothesis, however, is a tumor model in which the alteration in tumor cell ganglioside expression is constitutive, complete, and selective. To achieve this, we embarked upon a new, genetic, approach, seeking to stably and maximally deplete tumor cell ganglioside synthesis. By combined knockout (Yamashita et al., 2005) of two key ganglioside synthesis enzymes, GM3 synthase (GM3S) and GM2 synthase (GM2S), shown in supplemental Fig. 1, we found murine embryonic fibroblasts (MEF) to become almost completely depleted of gangliosides. We transformed these cells and wild type MEF with the c-Myc and H-Ras oncogenes (Thompson et al., 1989). The transformed wild type cells maintained the normal ganglioside profile while the transformed double knockout cells were ganglioside-depleted and exhibited strikingly impeded tumor growth. This genetically ganglioside-depleted tumor cell system provides a valuable model for in vivo and in vitro studies directed to clearly defining the role of gangliosides in tumor formation/progression. The findings may ultimately provide the basis for a new targeted therapeutic approach to human cancer.
Results
c-MycT58A/H-RasG12V transformation of MEFs
Fibroblasts (MEF) were cultured from E11.5 embryos of GM3S/GM2S double knockout and littermate wild type mice, which we bred by crossing GM3S knockout with GM2S knockout mice. The oncogenes c-Myc and H-Ras were combined in one plasmid (pBABE-c-MycT58A+H-RasG12V). Amphotropic retroviruses containing the plasmid were generated from the AmphoPack™-293 cell line by transfection (Kendall et al., 2005). These were used to infect MEF from low passage (p4-5) cultures of GM3S/GM2S double knockout and littermate wild type mice. The infected MEF were observed for characteristic changes in morphology indicating successful transduction. Colonies with changed morphology and loss of contact inhibition were cultured for more than 30 passages to eliminate untransfected cells. Colonies randomly selected for further evaluation and referred to as WTt (transformed wild type MEF) and DKOt (transformed GM3S/GM2S double knockout MEF) were expanded for use in the subsequent experiments and aliquots frozen.
Morphology of wild type and GM3S/GM2S double knockout MEF before transformation was similar (Fig. 1A,1C) while the oncogene-transformed cells (WTt and DKOt) differed from the untransformed MEF and also from each other. Both had less cytoplasm and were smaller than the untransformed cells (Fig. 1B,1D), while the DKOt cells had a more flattened morphology and were less refractile than the WTt cells.
Figure 1. Confirmation of transformation of MEF.
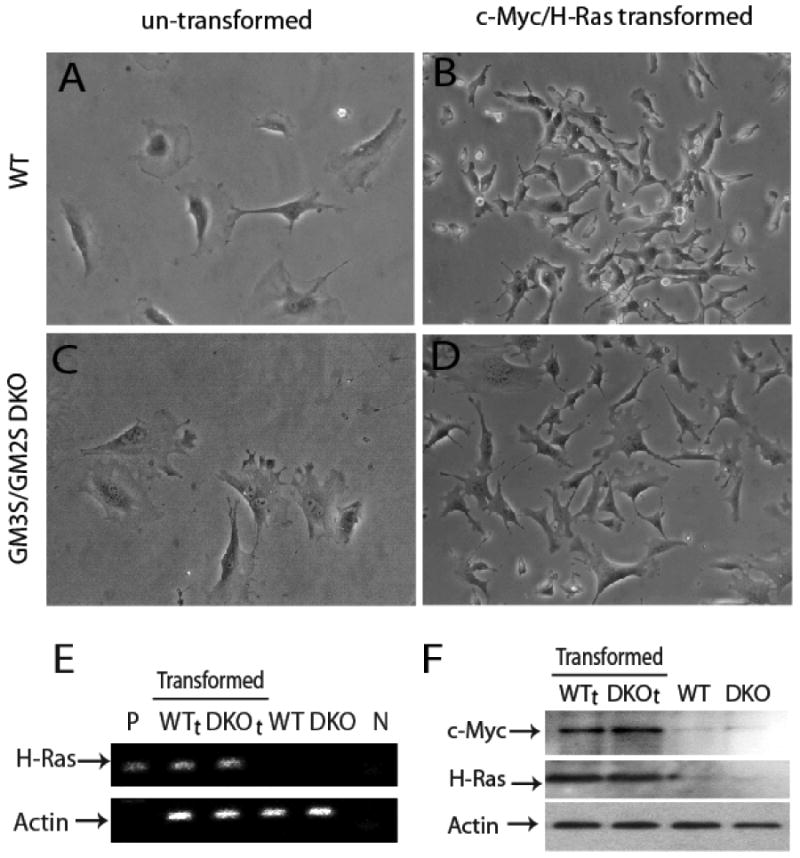
Phase contrast photomicroscopy of cultured MEFs. Key: untransformed WT MEF (panel A) and DKO MEF (panel C) and c-MycT58A/H-RasG12V transformed wild type (WTt, panel B) and GM3S/GM2S double knockout (DKOt, panel D) cells. RT-PCR analysis (Panel E) demonstrates the transcribed H-RasG12V mutation in transformed WTt and DKOt cells but not in untransformed cells (P, N, are positive and negative controls for H-RasG12V). Western blot analysis shows overexpression of the oncogene proteins, c-Myc and H-Ras, in the transformed WTt and DKOt cells but not in untransformed cells (Panel F, right lanes).
RT-PCR amplification showed the H-RasG12V oncogene to be expressed in both WTt and DKOt cells, but not in the untransformed MEF (Fig. 1E), as expected. Western blots documented overexpression of c-Myc/H-Ras in the transformed but not the untransformed MEFs (Fig. 1F), with similar expression levels in the two transformed populations (WTt and DKOt). To independently verify these results, we quantified H-RasG12V DNA and mRNA in the transformed MEF by real-time PCR and quantitative RT-PCR. The relative numbers of integrated copies of H-RasG12V were similar (1.0 and 1.20, WTt and DKOt respectively; Table 1). Moreover, relative expression of the transduced H-RasG12V was also comparable albeit slightly higher in the DKOt cells (1.58 vs. 1.0 in WTt cells, Table 1). From these quantitative studies we conclude that integration and expression of the transduced oncogenes are similar in the WTt and DKOt cells.
Table 1.
Quantification of H-RasG12V expression in transformed MEFs
| Untransformed Fibroblasts | + H-Ras/c-Myc | |||
|---|---|---|---|---|
| WT | GM3S/GM2S Knockout (DKO) | WTt | DKOt (GM3S/GM2S K/O) | |
| H-RasG12V DNA | <10-3 | <10-2 | 1.0 | 1.20 |
| H-RasG12V RNA | <10-5 | <10-5 | 1.0 | 1.58 |
Expression levels of mutant human Ras (H-RasG12V) in murine fibroblasts as detected by real-time PCR (DNA) or real-time RT-PCR (RNA), relative to β-actin expression and normalized (WTt=1.0).
Cellular gangliosides
Two approaches were used to assess gangliosides—highly sensitive metabolic (14C-galactose/glucosamine) radiolabeling/HPTLC autoradiography to detect ganglioside neosynthesis and thereby the activity of the target enzymes, and direct chemical detection/HPTLC densitometry to determine changes in cellular ganglioside content induced by the knockout. GM3S/GM2S double knockout MEF, prior to oncogenic transformation, showed virtually complete depletion of cellular gangliosides (0.4 nmol/107 DKO MEF versus 8.5 nmol/107 WT MEF, Fig 2A), confirming that these cells would be useful for the planned oncogenic transformations. Following c-Myc/H-Ras oncogenic transformation, ganglioside synthesis and expression were conserved in the WTt cells while knockout of GM3S and GM2S enzyme activity was maintained in the DKOt cells (Fig. 2B), in which radiolabeled newly synthesized gangliosides were absent and, as detected chemically, cellular gangliosides remained essentially completely depleted (0.5 nmol/107 cells vs. 11 nmol/107 WTt cells, Fig. 2B). Preservation of the ganglioside-depleted phenotype upon in vivo passage of the DKOt cells, an essential characteristic of a useful model cell system for in vivo study, was also tested. Growing DKOt tumors (vide infra) were explanted and the recultured tumor cells analyzed. Once again the absence of ganglioside synthesis and expression was demonstrated (Fig. 2C), as was maintenance of oncogenic transformation (by PCR amplification of H-RasG12V, not shown).
Figure 2. Glycosphingolipid content of c-Myc/H-Ras transformed GM3S/GM2S double knockout DKOt and transformed wild type WTt MEFs.
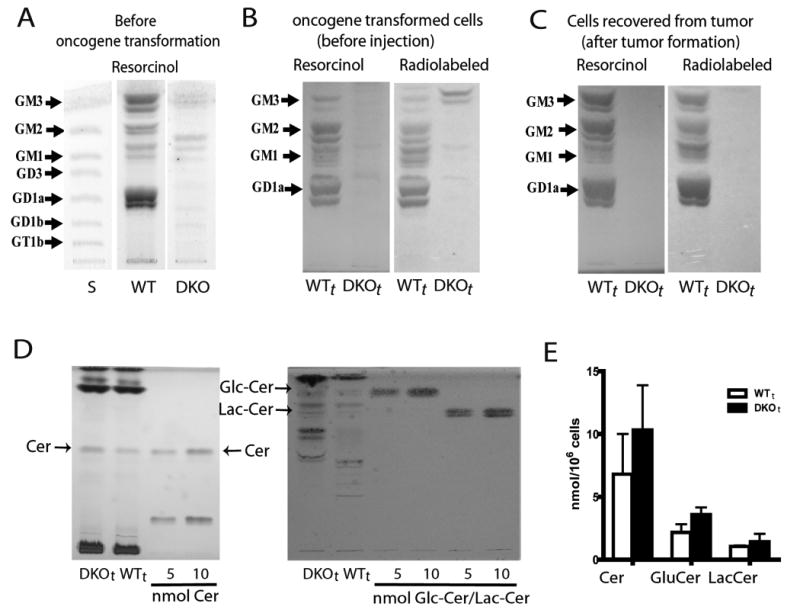
Panel A: HPTLC of gangliosides of untransformed DKO and WT control MEFs visualized by resorcinol; faint bands seen in DKO cells by resorcinol stain may be gangliosides adsorbed from the FCS in the medium. Panel B: Gangliosides of the transformed cells, WTt and DKOt, metabolically radiolabeled with 14C-galactose/ glucosamine, purified, and visualized by resorcinol stain and autoradiography of the same HPTLC plate. DKOt samples show almost complete ganglioside depletion (resorcinol stain) and blockade of ganglioside synthesis (metabolic radiolabeling); the doublet migrating above GM3 in the DKOt autoradiogram is resorcinol-negative, thus non-ganglioside, contaminant. Panel C: Gangliosides of WTt and DKOt tumor cells after in vivo passage. Cells were recultured from tumor explants and processed as in Panel B. Panel D: HPTLC detection of the ganglioside precursors ceramide, glucosylceramide, and lactosylceramide in DKOt and WTt cells. Ceramide (left) and GlcCer/LacCer (right), compared to standards, were visualized by charring and orcinol stain respectively. Panel E: The bar graph depicts mean±SD concentrations (n=3) of these ganglioside precursors. Comparing DKOt with WTt cells, glucosylceramide was mildly increased (65%, p=0.05); increases in ceramide (30%) and lactosylceramide (36%) were not significant.
Critical to validation of a specifically ganglioside-depleted model is confirmation of only minimal changes in concentrations of other related molecules as a result of changes in enzyme activity in the metabolic pathway being blocked. These could be caused either by activation of an alternate pathway not unlike that seen in GM3S knockout mouse fibroblasts (Shevchuk et al., 2007) or by accumulation of upstream ganglioside precursors, ceramide and the neutral glycolipids glucosylceramide and lactosylceramide (suppl. Fig. 1). Gangliosides synthesis by an alternate pathway was not detected, either by metabolic radiolabeling or by direct HPTLC staining (Figs. 2A-C), and there were only minor increases of the neutral glycolipids and ceramide (Figs 2D and 2E).
Attachment, morphology, proliferation, and migration of oncogene c-Myc/H-Ras transformed MEF
The ability of WTt and DKOt cells to adhere was equal and efficient. Of 8.5×105 cells plated/well in six-well plates, 93% of WTt and 96% of DKOt cells had attached within 4 hours after plating, indicating that the ganglioside depletion of DKOt cells did not interfere with their ability to adhere (suppl. Fig. 2). Culture on various extracellular matrices (ECM) highlighted some morphologic differences between the WTt and DKOt cells before and after reaching confluence (Fig. 3). On all matrices tested the DKOt cells appeared more flat, maintaining a nearly perfect single cell monolayer, whereas the WTt cells were arranged in a criss-crossing pattern evident particularly at low cell density and were highly refractile with multi-layered oriented patterns and piling up of cells at high cell density.
Figure 3. Morphology of the c-Myc/H-Ras transformed WTt and DKOt cells.
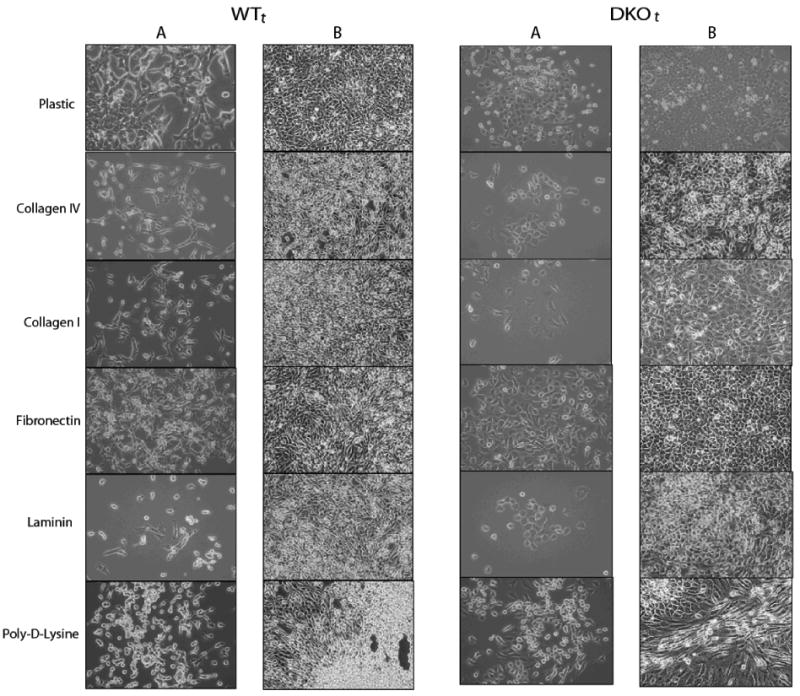
Photomicrographs (100×) of transformed WTt and DKOt cells taken in areas of low cell density (columns A, 24 hours after plating) and high cell density (columns B, after confluence). Clear differences are seen in morphology and growth patterns of oncogene-transformed cells, both on uncoated plastic and on several extracellular matrices (ECMs)—collagens IV and I, fibronectin, laminin, and poly-D-Lysine.
Overall proliferation kinetics, analyzed by cell counting and by 3H-thymidine uptake, were essentially identical for the WTt and DKOt cells, both on plastic and on fibronectin-coated plates (Fig. 4). Population doubling times (∼11.3 hours) and log phase growth rates (k=2.1/day) were equal. This, and similar saturation densities for the two populations, indicated that there was no adverse effect of ganglioside depletion on the intrinsic ability of DKOt cells to autonomously proliferate. Also, there was no sign of increased apoptosis by the ganglioside-depleted cells; in a four-day culture analyzed by FACS with 7-AAD and annexin-V, there were 10.3±6.4% vs.8.7±1.5% apoptotic cells, WTt vs. DKOt, respectively (p=0.56).
Figure 4. Proliferation of WTt and DKOt cells.
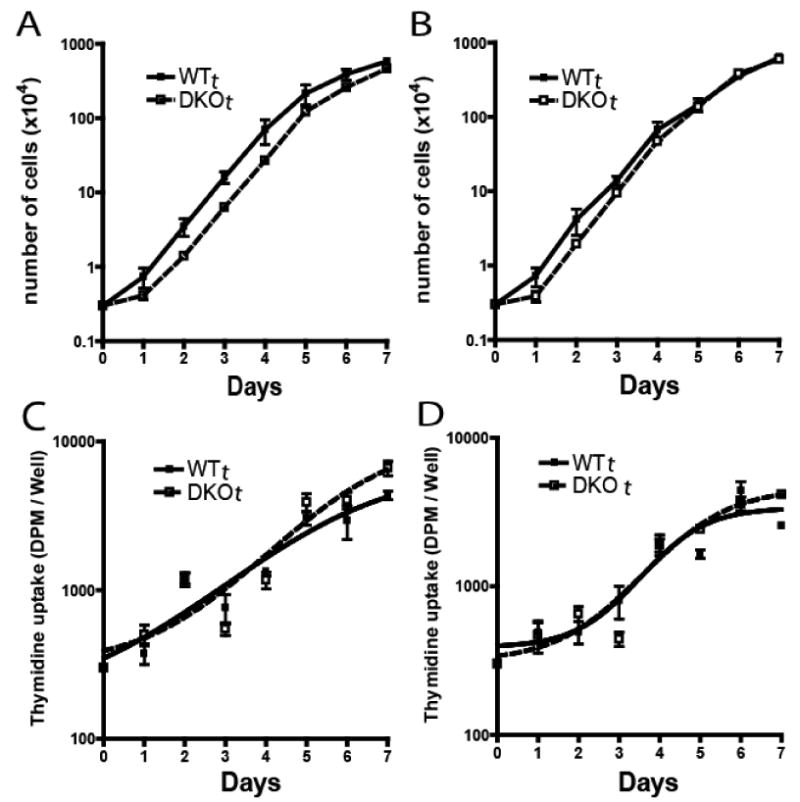
Panels A & B: 3×103 cells were seeded/well in 12-well plates and counted daily for 7 days. Panels C and D: cells were seeded at 300/well in 96 well plates and 3H-thymidine uptake quantified daily. Panels A and C: fibronectin coated plates. Panels B and D: uncoated plastic plates. Each point represents the mean±SD of 6 experiments.
Cell migration was assessed on various matrices. The most substantial migration of WTt cells occurred on fibronectin and was several fold that of baseline migration on an uncoated surface (Fig. 5). The fibronectin-related enhanced migration was essentially absent in the ganglioside-depleted (DKOt) cells. Fibronectin-associated migration was also completely inhibited by an anti-integrin beta-1 antibody, antiCD29, implicating an integrin-related ganglioside requirement for this migration (Fig. 5). Migration of WTt vs. DKOt cells on several other matrices (BSA, collagen-1, laminin) was similar and only slightly greater than migration on uncoated surfaces (suppl. Fig. 3). These results further support a particularly strong effect of ganglioside absence on integrin-mediated migration.
Figure 5. Cell migration of WTt and DKOt cells.
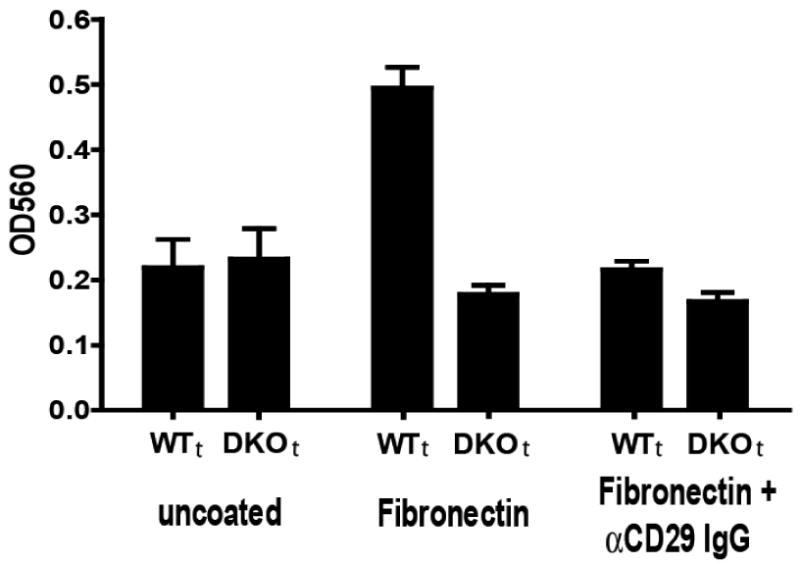
3×103 WTt or DKOt cells were seeded/well and 4-h migration on uncoated (left) or fibronectin-coated wells (middle), or fibronectin-coated wells with anti-CD29 antibody (right) was assessed. Migration in the Transwell cultures was quantified by spectrophotometry as described in Methods. Bars represent the mean±SD of multiple assays in 2-5 separate experiments. WTt cell migration on fibronectin was abrogated by anti-integrin beta-1 antibody. DKOt cell migration was significantly reduced on fibronectin-coated wells (p<0.0001).
Tumorigenicity of oncogene-transformed MEF
An initial in vitro test of the potential ability of the transformed cells to form tumors was to assess the effect of tumor cell ganglioside depletion on tumorigenic potential by quantifying the formation of colonies by WTt and DKOt cells in soft agar (Villar et al., 2007). These experiments gave a mixed picture—while WTt cells formed slightly more colonies than did DKOt cells (431±27 vs. 366±12/well, p=0.02), the DKOt cells appeared to form larger colonies (Fig. 6A).
Figure 6. Tumorigenicity of oncogene-transformed WTt and DKOt cells.
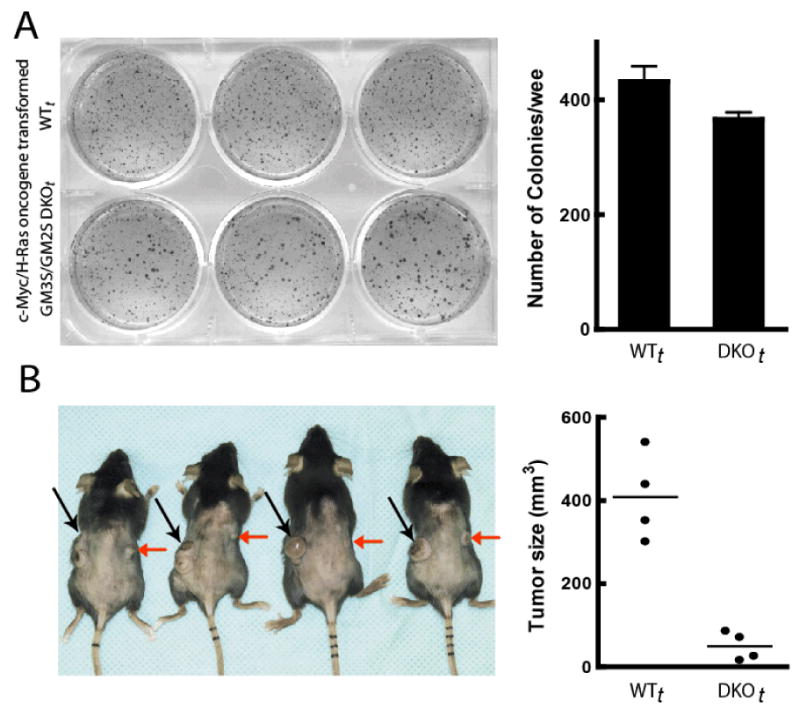
(A) In vitro, 5×104 cells/well were seeded in soft agar in 6-well plates, incubated for 2 weeks, and then photographed. Colony numbers were. The bar graph depicts the mean±SD number of colonies/well quantified using Quantity One 4.6.7 software (BIO RAD Hercules, CA) at settings of sensitivity=1.0 and average=1. (B) Tumor formation was documented for 100% of both DKOt and WTt cell injections. Tumors were induced in mice by i.d. injection of 105 WTt cells (left flank, black arrows) and 105 DKOt cells (right flank, red arrows). Tumors and tumor volumes 18 days post-injection are shown. The mean DKOt tumor volume was significantly smaller than that of WTt tumors (p=0.008).
The findings with respect to tumor formation in this new model of genetic tumor cell ganglioside depletion were highly interesting. All mice injected with 105 WTt cells i.d. (in the left flank) and 105 genetically ganglioside-depleted DKOt cells (in the right flank) formed tumors (Fig. 6A) of both the WTt and the DKOt cells (100% incidence), demonstrating that oncogenic transformation was effective and not counteracted by the genetic ganglioside depletion. Importantly, however, the difference in tumor growth was striking: 16 days after inoculation, the mean WTt tumor volume (408±104mm3) was eight times that of the DKOt tumors (50±34 mm3, p=0.002, Fig. 6A). This was seen also in a separate independent experiment (suppl. Fig. 4). Substantially impaired tumor growth of DKOt cells in syngeneic immunocompetent mice shows that tumor gangliosides impact tumor formation in vivo. Thus this model of stable and specific ganglioside depletion in oncogene-transformed cells will constitute an ideal system for probing molecular mechanisms of tumor ganglioside action, including the effects of tumor gangliosides on the tumor-host interaction in the tumor microenvironment.
Discussion
Gangliosides, a widely distributed class of molecules, have been extensively studied to define their cellular biological functions. This has been prompted by their prominence in brain tissue (Ledeen, 1978), the severe genetic diseases resulting from mutations in the biosynthetic and catabolic enzymes which alter ganglioside expression (Jeyakumar et al., 2002), and the frequently altered ganglioside expression in tumor cells (Hakomori, 1996). The latter in turn is accompanied by striking ganglioside shedding into the tumor microenvironment (Ladisch et al., 1983), which raised the intriguing question of whether tumor gangliosides have a role in tumor formation. In fact, in vitro evidence suggests a major action of gangliosides across the tumor microenvironment—that these molecules shed by tumor cells affect host cell function. But, lack of an adequate in vivo model had been a formidable gap in testing this possibility. Here, by transforming fibroblasts that are genetically depleted of gangliosides, we sought to create a syngeneic murine tumor cell model in which ganglioside biosynthesis is constitutively, completely, and specifically inhibited.
We initially explored the GM3S knockout mouse model (Yamashita et al., 2003) because of its almost complete depletion of brain gangliosides and because a naturally occurring point mutation in the human GM3S gene (Simpson et al., 2004) completely blocks ganglioside synthesis of human fibroblasts (Liu et al., 2008). Fibroblasts of the GM3S knockout mouse, however, had unexpected activation of the normally silent alternate “O” synthetic pathway resulting in substantial synthesis of complex gangliosides (Shevchuk et al., 2007), negating potential usefulness of the model to study effects of ganglioside depletion on tumor formation. Combined knockout of GM3S and GM2S (see suppl. fig 1), on the other hand, blocks that “O” pathway as well, and a mouse created by crossbreeding the two single knockouts had completely blocked brain ganglioside synthesis (Yamashita et al., 2005).
Since these mice die early in life, with white matter pathology and CNS axon degeneration, we isolated embryonic fibroblasts. These MEF were almost completely ganglioside depleted, allowing us to create ganglioside-depleted tumor cells by an amphotropic retrovirus derived plasmid (pBABE-c-MycT58A+H-RasG12V). Both WTt and DKOt cells had stable transformed genotypes, and compared to untransformed cells, stable ganglioside phenotypes, confirming that oncogenic c-Myc/H-Ras transformation itself did not alter ganglioside expression or activate an alternate ganglioside synthesis pathway in either the WTt or the DKOt cells. Stable, selective, and constitutive inhibition of ganglioside synthesis and depletion of cellular gangliosides—without either alternate pathway activation or a significant increase in other precursor molecules (especially ceramide) in DKOt cells—overcomes several problems of previous approaches, including incomplete inhibition of glycosphingolipid synthesis, e.g., by antisense transfection of glucosylceramide synthase (Deng et al., 2002), and only transient and less than specific ganglioside inhibition by available pharmacological inhibitors, such as PDMP (Inokuchi et al., 1989). Stable in vivo, the model provides an ideal system for studies of tumor growth and of mechanisms of action.
We next examined cell biological characteristics of these novel constitutively ganglioside-depleted DKOt cells. Cell proliferation kinetics were not different between WTt and DKOt cells and there was no increase in apoptosis of DKOt cells. This was important because it excludes direct cytotoxic effects of ganglioside depletion on the transformed cells as a potential cause of decreased tumorigenicity, i.e., that reduced ganglioside-depleted tumor growth is not simply related to increased toxicity of ganglioside depletion on cell viability or proliferation potential. The absence of direct cytotoxic effects fits with the absence of any substantial increase in DKOt cells of ganglioside precursors (especially ceramide), increases that have been linked to cell apoptosis (Schenck et al., 2007). The only slight increase in ceramide in the DKOt cells is consistent with findings (Yamashita et al., 2005) of no increase in brain ceramide in DKO mice.
One in vitro cell biological finding that was significantly altered in the DKOt cells was their ability to migrate. Because tumor cell migration is important for the metastatic process, numerous previous studies have evaluated effects of qualitative and quantitative ganglioside alterations on the in vitro migration of tumor cells of different cell types and on different matrices. At this point it is difficult to draw a single uniform conclusion since ganglioside enhancement of migration was found in some studies (Cazet et al., 2009; Saha et al., 2005) and inhibition in others (Hyuga et al., 1999; Mitsuzuka et al., 2005). The different findings may well be cell type-related or a consequence of specific different alterations in ganglioside composition in these previous studies. This will require further examination. Here, the greatest enhancement of migration of WTt cells was observed on a fibronectin surface, was abrogated when the cell gangliosides were absent, and was inhibited by anti-integrin beta-1 antibody. Together the findings point to an integrin-related ganglioside requirement for this migration (a concept consistent with previous thought) and point to the altered migration as possibly relating to the subsequent observations regarding tumor growth of these cells, below.
Our ultimate purpose in developing this model is to be able to fully explore the effects of specific tumor cell ganglioside depletion in vivo. While DKOt cell behavior in soft agar (only mildly affected) did not give a very strong hint, the striking impairment of DKOt tumor growth clearly shows the inhibitory consequence of tumor cell ganglioside depletion on tumorigenic potential in vivo. Several novel and important inferences can be drawn: First, minimal in vitro consequences despite complete absence of all gangliosides suggest that gangliosides are not absolutely necessary for tumor cell survival. Nevertheless, tumor growth was impeded, suggesting that host-derived gangliosides cannot compensate for this absence in the tumor cells. Second, the apparent mildness of the in vitro alterations in the DKOt cell contrasts sharply to the inhibitory effect on tumor growth in vivo, supporting the concept that a critical factor in tumor promotion may be effects of shed tumor gangliosides on normal host cell function in the host tumor microenvironment. Mechanisms could include, for example, direct modifying effects of shed gangliosides on normal cell function (by binding to these normal cells in the microenvironment) or a “protective” role of gangliosides on the tumor cell surface, against host defenses such as antitumor immune responses. In vitro findings support both of these mechanisms – amplified responses to VEGF of normal vascular endothelial cells enriched with gangliosides, as may occur by shedding (Liu et al., 2006) and inhibition of immune responses by tumor gangliosides (Ladisch, 2003). Thus, in addition to looking at the tumor cell itself to understand the role of gangliosides in tumor formation, in our future studies it will be both important and now possible, with our new model, for us to probe in vivo tumor-host interactions in this context.
In conclusion, these studies establish and create a novel model for the study of the mechanisms of effects of gangliosides on both cell biology in vitro and tumor formation in vivo. The advantages of the system are that ganglioside depletion is complete, there is little alteration of upstream precursors of gangliosides of potential biological activity, the system is genetically stable, DKOt cells are able to form tumors, and the ganglioside phenotypes of both WTt and DKOt, are preserved upon in vivo passage. Altogether these characteristics will provide an ideal model syngeneic system to better understand mechanisms by which ganglioside depletion impedes tumor formation, a longstanding question of great interest in cancer biology with implications for the development of new targeted therapies.
Material and Methods
Generation of GM3S/GM2S double knockout embryonic fibroblasts
GM3S/GM2S double knockout mice were bred by crossing GM3S knockout mice (kind gift of Dr. Rick Proia) and GM2S knockout mice (from the Consortium for Functional Glycomics) to yield mice carrying homozygous null mutations in two ganglioside specific glycosyltransferase genes, Siat9 (encoding GM3 synthase) and Galgt1 (encoding GM2 synthase). MEF from E11.5 embryos of these mice and from littermate wild type (WT) mice were cultured in complete medium (used throughout this work) consisting of DMEM with 4.5 g/L glucose (Lonza, Walkersville, MD, USA), 10% FCS, 2 mM L-glutamine, and 1% NEAA. The genotypes of the cultured MEFs were confirmed using 100 ng of genomic DNA, prepared using the DNeasy Tissue Kit (Qiagen, Valencia, CA, USA) and amplified by PCR using Choice™ Taq DNA Polymerase (Denville, Metuchen, NJ, USA). Primers to identify the GM3S knockout mutation (Yamashita et al., 2005) were: 5′ AGC TCA GAG CTA TGC TCA GGA - 3′ (GM3S-WT forward) and 5′ TAC CAC ATC GAA CTG GTT GAG - 3′ (GM3S-WT reverse), 5′ CAA TAG ATC TGA CCC CTA TGC - 3′ (GM3S-Mut forward) and 5′ TCG CCT TCT TGA CGA GTT CTT CTG AG - 3′ (GM3S-Mut reverse). Primers to identify the GM2S knockout mutation were 5′ ACA CGT GGA GCA CTA CTT CAT G - 3′ (GM2S-WT forward), 5′ AGG TCC AGG GGC GTC TTC T - 3′ (GM2S-WT and Mut reverse) and 5′ TGC AAT CCA TCT TGT TCA ATG -3′ (GM2S-Mut forward). The GM3S wild-type allele is ∼400 bp fragment and the mutant allele is ∼300 bp fragment. The GM2S wild-type allele is ∼600 bp fragment and the mutant allele is ∼350 bp fragment. The reaction was amplified by incubation at 95°C (3 min), 35 cycles at 95°C (15 sec), 60° C (1 min), 72°C (1 min) and extension at 72° C (7 min).
Transformation of embryonic fibroblasts
Two oncogenes (c-Myc and H-Ras) carried in one plasmid (pBABE-c-MycT58A+H-RasG12V; Addgene, Cambridge, MA, USA) were used for the cell transformations. The identities of all plasmids were confirmed to be correct by PCR and sequencing. Amphotropic retroviruses were generated from the AmphoPack™-293 cell line by transfection using the CalPhos™ Mammalian Transfection Kit (Clontech, Mountain View, CA, USA). The amphotropic retroviruses produced from this plasmid were used to infect MEF cells as described (Kendall et al., 2005). The cells, cultured in complete medium, were observed for colonies with changed morphology and loss of contact inhibition. These were selected for further study.
Confirmation of transformation by Western blot, PCR, RT-PCR and real-time PCR
The first step in selection of transformed embryonic fibroblasts was Western blot analysis for c-Myc and H-Ras. Cell lysates from oncogene transformed and untransformed MEF were resolved by SDS-PAGE and immunoblotted using anti-Myc (9E10) and anti-Ras(c-20) antibodies (Santa Cruz, Santa Cruz, CA, USA). The c-Myc and H-Ras over-expressing cell cultures were expanded and two cell cultures, WTt and DKOt, which highly expressed the translation products from plasmid pBABE-c-MycT58A+H-RasG12V, were studied further.
The untransformed and WTt and DKOt cells were analyzed for the presence of the human H-RasG12V by PCR, RT-PCR and real time PCR, using primers specific to the transgene H-RasG12V. Genomic DNA (200ng) was hybridized with primers complementary to the human sequences that span the activating mutation site or to β-actin as follows: 5′-GTCGGTGTGGGCAAGAGT-3′ and 5′-ATCAATGACCACCTGCTTCC-3′ (H-RasG12V), 5′-AGAGGGAAATCGTGCGTGAC-3′ and 5′-CAATAGTGATGACCT GGCCGT-3′ (mouse β-actin). Total RNA (1 μg) prepared using the TRIzol® Reagent was reverse transcribed using Oligo dT primer and SuperScript™ III Reverse Transcriptase (RT) (Invitrogen, Carlsbad, CA, USA). Control reactions containing no RT were assayed in parallel. The products were detected by ethidium bromide staining following electrophoresis in 1.6% agarose gels. The sizes of the amplified PCR products were compared to the predicted fragments based upon the plasmid sequence using DNA (100 bp ladder) markers (New England Biolabs, Ipswich, MA, USA). The relative amounts of H-RasG12V gene or mRNA were analyzed by real time PCR or real time RT-PCR, respectively, using the SYBR GreenER Q-PCR SuperMix for the iCycler® kit and protocol (Invitrogen, Carlsbad, CA, USA). Amplification was monitored using the BIO-RAD iCycler detection system. Conservation of knockout of GM3S and GM2S in the transformed DKOt fibroblasts was confirmed by the same procedure.
Assessment of transformed fibroblast ganglioside metabolism
Gangliosides in the transformed cells were assessed by direct chemical HPTLC detection and quantification by densitometry (to determine total cellular ganglioside content and changes induced by the knockout) and also by metabolic radiolabeling to detect newly synthesized gangliosides (assessing the activity of the enzymes knocked out). Ganglioside purification and analysis was performed as described (Ladisch and Gillard, 1985). Briefly, after total lipid extraction and DIPE/1-butanol/saline partition, total gangliosides, in the lower aqueous phase, were further purified by Sephadex G-50 gel filtration. HPTLC analysis was performed using 10×20 cm precoated silica gel 60 HPTLC plates (Merck, Darmstadt, Germany) and developed in chloroform/methanol/0.25% aqueous CaCl2.2H20 (60:40:9, v/v/v). Gangliosides were stained as purple bands with resorcinol reagent. To assess ganglioside synthesis by metabolic radiolabeling, transformed cells were incubated with 1.0 μCi/ml each of D-[1-14C]-glucosamine hydrochloride and D-[1-14C]-galactose for 24 h and the gangliosides purified and analyzed by HPTLC autoradiography (Li and Ladisch, 1991).
To assess ceramide content, the cell pellets were extracted and total lipids partitioned in chloroform:methanol: 0.9%NaCl following the method of (Rani et al., 1995). Ceramides, in the lower phase, were dried, resuspended in chloroform:methanol (1:1), and separated by HPTLC in chloroform:acetic acid (9:1). The neutral glycosphingolipids glucosylceramide and lactosylceramide were recovered in the lower phase after partitioning the total lipids in acidic chloroform:methanol:water (Lavie et al., 1996) and separated by HPTLC in chloroform:methanol:water (60:40:8). The lipids were visualized by charring or orcinol stain and quantified by densitometry using ImageJ software.
Cell adherence, proliferation, and migration
To quantify the effect of ganglioside expression on cell adherence, we counted cells not adhering within 4 h after seeding 8.5×105cells/well in a 6-well plate, and subtracted them from the total number seeded. Differential cell adherence on various matrices was tested by seeding 5×104 cells/well in 6-well plates either uncoated (control) or coated with different ECM components—collagen IV, collagen I, fibronectin, laminin and poly-D-lysine (BD Labware, Bedford, MA, USA). Cell morphological and growth characteristics were observed by phase contrast microscopy.
Cell proliferation kinetics was assessed by seeding 300 WTt or DKOt cells/100μl/well in 96 well plates (Corning, Lowell, MA, USA) and culturing for 7 days. The medium was changed every two days, the cells were harvested daily, and thymidine uptake (0.5 μCi 3H-thymidine per well for 4 h) quantified by β–scintillation counting. Direct daily cell counting was also performed (12-well plates, 3×103 cells seeded/well). The cell doubling time was calculated as Ttotal/n, where n=3.32[log10Nt-log10N0], Ttotal was the total hours observed, and N0 and Nt were number of cells at the beginning and end of this time. The cell growth rate constant, k, was obtained by dividing n by the observation time (Nt - Ni) in days. Apoptosis was assessed by FACS of four day cultures stained with 7-AAD and annexin-V (van Engeland et al., 1996).
Cell migration was assessed using the chamber method (Ronnov-Jessen et al., 2002). Briefly, 1 ml complete medium was added to the bottom well of the 8μm pore size QCM™ 24-well colorimetric cell migration assay kit (Chemicon Int. Temecula, CA) or the CytoSelect™ Assay kit (Cell BioLabs, Inc., San Diego, CA). 3×103 WTt or DKOt cells/0.5ml/well were seeded in 6.5 mm Transwell plates, placed on the CM plates, and cultured at 37°C in 5%CO2 for 4 h. For the blocking assay, the anti-integrin beta1 antibody CD29 (Biolegend, San Diego, CA was mixed with cells before seeding. To quantify cell migration Cell Stain was added for 20 min at room temperature, the chamber washed 3 times with fresh water, and the cells in the upper chamber gently removed by scraping with cotton-tipped swab. The membrane was air dried and transferred to a clean well containing 200μl of extraction buffer for 15 min at room temperature, and 100μl of the resulting dye mixture was quantified by spectrophotometry (OD=560nm).
Anchorage-independent growth and tumorigenicity assays
Anchorage-independent proliferation was quantified by standard soft-agar assays. 5×104 WTt or DKOt cells were seeded/well in soft agar (Villar et al., 2007), cultured for 2 weeks, stained with nitrotetrazolium blue chloride (1 mg/ml), and photographed. Colonies were counted using Quantity One 4.6.7 software (BioRad) at sensitivity=1.0 and average=1 settings. The ability of the transformed cells to form tumors in vivo was assessed by i.d. injection of 105 cells each of WTt and DKOt cells on opposite flanks of each of six syngeneic normal 6-8 week old C57Bl/6 mice. In another experiment, 105 of each cell type were injected into separate mice and tumor incidence and growth monitored. Tumor volume was calculated according to the formula V=1/2(LxWxH). Mouse care was performed in accordance with IACUC protocols. Statistical significance was evaluated by ANOVA or two-tailed Student's t test.
Supplementary Material
Suppl. Figure 1. Major ganglioside synthesis pathways. “0” pathway gangliosides are normally not expressed, but appear upon knockout of GM3 synthase activity and are absent after knockout of GM2 synthase. Combined knockout of both enzymes blocks all ganglioside synthesis.
Suppl. Figure-2: Initial adherence of WTt and DKOt cells. 4×105 cells were seeded/well, the cells not adhering were collected hourly, counted, and subtracted from the total cell count. Results are the mean±SD of four separate experiments.
Suppl. Figure-3: Migration of WTt and DKOt cells. Cell migration on a panel of matrices was assessed. 3×103 WTt or DKOt cells were seeded/well and 4 hour migration was quantified as described in Methods. Bars represent the mean±SD of 4 samples in two separate experiments. Fibronectin-associated migration was robust in WTt cells and abrogated in ganglioside-depleted, i.e. DKOt, cells (p<0.0001. There was lesser migration and no significant differences between WTt and DKOt cells on the other matrices.
Suppl. Figure 4: Tumor growth curve of H-Ras/c-Myc transformed WTt and DKOt. 105 WTt or DKOt cells were injected into syngeneic 6-8 week old C57Bl/6 mice (6 mice per group). Data are the mean±SD tumor volumes; p=0.0001.
Acknowledgments
We thank Rick Proia for the GM3S mice. and Daniel Bobb for excellent assistance in animal care. This work was supported by NIH grants CA61010 and CA42361 to SL, CA64472 to VN, and GM62116 to the Consortium for Functional Glycomics.
Footnotes
Conflict of Interest Statement: The authors declare that they have no conflicts of interest
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- Allende ML, Proia RL. Lubricating cell signaling pathways with gangliosides. Curr Opin Struct Biol. 2002;12:587–92. doi: 10.1016/s0959-440x(02)00376-7. [DOI] [PubMed] [Google Scholar]
- Buggins AG, Milojkovic D, Arno MJ, Lea NC, Mufti GJ, Thomas NS, et al. Microenvironment produced by acute myeloid leukemia cells prevents T cell activation and proliferation by inhibition of NF-kappaB, c-Myc, and pRb pathways. J Immunol. 2001;167:6021–30. doi: 10.4049/jimmunol.167.10.6021. [DOI] [PubMed] [Google Scholar]
- Cazet A, Groux-Degroote S, Teylaert B, Kwon KM, Lehoux S, Slomianny C, et al. G(D3) synthase overexpression enhances proliferation and migration of MDA-MB-231 breast cancer cells. Biol Chem. 2009;390:601–9. doi: 10.1515/BC.2009.054. [DOI] [PubMed] [Google Scholar]
- Deng W, Li R, Guerrera M, Liu Y, Ladisch S. Transfection of glucosylceramide synthase antisense inhibits mouse melanoma formation. Glycobiology. 2002;12:145–52. doi: 10.1093/glycob/12.3.145. [DOI] [PubMed] [Google Scholar]
- Floutsis G, Ulsh L, Ladisch S. Immunosuppressive activity of human neuroblastoma tumor gangliosides. Int J Cancer. 1989;43:6–9. doi: 10.1002/ijc.2910430103. [DOI] [PubMed] [Google Scholar]
- Hakomori S. Glycolipids of tumor cell membrane. Adv Cancer Res. 1973;18:265–315. doi: 10.1016/s0065-230x(08)60755-1. [DOI] [PubMed] [Google Scholar]
- Hakomori S. Tumor malignancy defined by aberrant glycosylation and sphingo(glyco)lipid metabolism. Cancer Res. 1996;56:5309–18. [PubMed] [Google Scholar]
- Hyuga S, Yamagata S, Takatsu Y, Hyuga M, Nakanishi H, Furukawa K, et al. Suppression by ganglioside GD1A of migration capability, adhesion to vitronectin and metastatic potential of highly metastatic FBJ-LL cells. Int J Cancer. 1999;83:685–91. doi: 10.1002/(sici)1097-0215(19991126)83:5<685::aid-ijc20>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Inokuchi J, Momosaki K, Shimeno H, Nagamatsu A, Radin NS. Effects of D-threo-PDMP, an inhibitor of glucosylceramide synthetase, on expression of cell surface glycolipid antigen and binding to adhesive proteins by B16 melanoma cells. J Cell Physiol. 1989;141:573–83. doi: 10.1002/jcp.1041410316. [DOI] [PubMed] [Google Scholar]
- Jeyakumar M, Butters TD, Dwek RA, Platt FM. Glycosphingolipid lysosomal storage diseases: therapy and pathogenesis. Neuropathol Appl Neurobiol. 2002;28:343–57. doi: 10.1046/j.1365-2990.2002.00422.x. [DOI] [PubMed] [Google Scholar]
- Kendall SD, Linardic CM, Adam SJ, Counter CM. A network of genetic events sufficient to convert normal human cells to a tumorigenic state. Cancer Res. 2005;65:9824–8. doi: 10.1158/0008-5472.CAN-05-1543. [DOI] [PubMed] [Google Scholar]
- Ladisch S, Gillard B. A solvent partition method for microscale ganglioside purification. Anal Biochem. 1985;146:220–31. doi: 10.1016/0003-2697(85)90419-1. [DOI] [PubMed] [Google Scholar]
- Ladisch S, Gillard B, Wong C, Ulsh L. Shedding and immunoregulatory activity of YAC-1 lymphoma cell gangliosides. Cancer Res. 1983;43:3808–13. [PubMed] [Google Scholar]
- Ladisch S, Kitada S, Hays EF. Gangliosides shed by tumor cells enhance tumor formation in mice. J Clin Invest. 1987;79:1879–82. doi: 10.1172/JCI113031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladisch S. The role of tumor gangliosides in the immune dysfunction of cancer. In: Finke JH, Bukowski RM, editors. Current Clinical Oncology: Cancer Immunotherapy at the Crossroads: How Tumors Evade Immunity and What Can Be Done. Humana Press; Totowa, NJ: 2003. pp. 145–156. [Google Scholar]
- Lavie Y, Cao H, Bursten SL, Giuliano AE, Cabot MC. Accumulation of glucosylceramides in multidrug-resistant cancer cells. J Biol Chem. 1996;271:19530–6. doi: 10.1074/jbc.271.32.19530. [DOI] [PubMed] [Google Scholar]
- Ledeen RW. Ganglioside structures and distribution: are they localized at the nerve ending? J Supramol Struct. 1978;8:1–17. doi: 10.1002/jss.400080102. [DOI] [PubMed] [Google Scholar]
- Li R, Liu Y, Ladisch S. Enhancement of epidermal growth factor signaling and activation of SRC kinase by gangliosides. J Biol Chem. 2001;276:42782–92. doi: 10.1074/jbc.M101481200. [DOI] [PubMed] [Google Scholar]
- Li RX, Ladisch S. Shedding of human neuroblastoma gangliosides. Biochim Biophys Acta. 1991;1083:57–64. doi: 10.1016/0005-2760(91)90124-z. [DOI] [PubMed] [Google Scholar]
- Liu Y, Li R, Ladisch S. Exogenous ganglioside GD1a enhances epidermal growth factor receptor binding and dimerization. J Biol Chem. 2004;279:36481–9. doi: 10.1074/jbc.M402880200. [DOI] [PubMed] [Google Scholar]
- Liu Y, McCarthy J, Ladisch S. Membrane ganglioside enrichment lowers the threshold for vascular endothelial cell angiogenic signaling. Cancer Res. 2006;66:10408–14. doi: 10.1158/0008-5472.CAN-06-1572. [DOI] [PubMed] [Google Scholar]
- Liu Y, Su Y, Wiznitzer M, Epifano O, Ladisch S. Ganglioside depletion and EGF responses of human GM3 synthase-deficient fibroblasts. Glycobiology. 2008;18:593–601. doi: 10.1093/glycob/cwn039. [DOI] [PubMed] [Google Scholar]
- Mitsuzuka K, Handa K, Satoh M, Arai Y, Hakomori S. A specific microdomain (“glycosynapse 3”) controls phenotypic conversion and reversion of bladder cancer cells through GM3-mediated interaction of alpha3beta1 integrin with CD9. J Biol Chem. 2005;280:35545–53. doi: 10.1074/jbc.M505630200. [DOI] [PubMed] [Google Scholar]
- Rani CS, Abe A, Chang Y, Rosenzweig N, Saltiel AR, Radin NS, et al. Cell cycle arrest induced by an inhibitor of glucosylceramide synthase. Correlation with cyclin-dependent kinases. J Biol Chem. 1995;270:2859–67. doi: 10.1074/jbc.270.6.2859. [DOI] [PubMed] [Google Scholar]
- Regina Todeschini A, Hakomori SI. Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth, through glycosynaptic microdomains. Biochim Biophys Acta. 2008;1780:421–33. doi: 10.1016/j.bbagen.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnov-Jessen L, Villadsen R, Edwards JC, Petersen OW. Differential expression of a chloride intracellular channel gene, CLIC4, in transforming growth factor-beta1-mediated conversion of fibroblasts to myofibroblasts. Am J Pathol. 2002;161:471–80. doi: 10.1016/s0002-9440(10)64203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Mohanty KC, Mallick P. Gangliosides enhance migration of mouse B16-melanoma cells through artificial basement membrane alone or in presence of laminin or fibronectin. Indian J Exp Biol. 2005;43:1130–8. [PubMed] [Google Scholar]
- Schenck M, Carpinteiro A, Grassme H, Lang F, Gulbins E. Ceramide: physiological and pathophysiological aspects. Arch Biochem Biophys. 2007;462:171–5. doi: 10.1016/j.abb.2007.03.031. [DOI] [PubMed] [Google Scholar]
- Shevchuk NA, Hathout Y, Epifano O, Su Y, Liu Y, Sutherland M, et al. Alteration of ganglioside synthesis by GM3 synthase knockout in murine embryonic fibroblasts. Biochim Biophys Acta. 2007;1771:1226–34. doi: 10.1016/j.bbalip.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Shurin GV, Shurin MR, Bykovskaia S, Shogan J, Lotze MT, Barksdale EM., Jr Neuroblastoma-derived gangliosides inhibit dendritic cell generation and function. Cancer Res. 2001;61:363–9. [PubMed] [Google Scholar]
- Simpson MA, Cross H, Proukakis C, Priestman DA, Neville DC, Reinkensmeier G, et al. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat Genet. 2004;36:1225–9. doi: 10.1038/ng1460. [DOI] [PubMed] [Google Scholar]
- Thompson TC, Southgate J, Kitchener G, Land H. Multistage carcinogenesis induced by ras and myc oncogenes in a reconstituted organ. Cell. 1989;56:917–30. doi: 10.1016/0092-8674(89)90625-9. [DOI] [PubMed] [Google Scholar]
- Uzzo RG, Rayman P, Kolenko V, Clark PE, Cathcart MK, Bloom T, et al. Renal cell carcinoma-derived gangliosides suppress nuclear factor-kappaB activation in T cells. J Clin Invest. 1999;104:769–76. doi: 10.1172/JCI6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentino L, Moss T, Olson E, Wang HJ, Elashoff R, Ladisch S. Shed tumor gangliosides and progression of human neuroblastoma. Blood. 1990;75:1564–7. [PubMed] [Google Scholar]
- van Engeland M, Ramaekers FC, Schutte B, Reutelingsperger CP. A novel assay to measure loss of plasma membrane asymmetry during apoptosis of adherent cells in culture. Cytometry. 1996;24:131–9. doi: 10.1002/(SICI)1097-0320(19960601)24:2<131::AID-CYTO5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Villar J, Arenas MI, MacCarthy CM, Blanquez MJ, Tirado OM, Notario V. PCPH/ENTPD5 expression enhances the invasiveness of human prostate cancer cells by a protein kinase C delta-dependent mechanism. Cancer Res. 2007;67:10859–68. doi: 10.1158/0008-5472.CAN-07-2041. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Hashiramoto A, Haluzik M, Mizukami H, Beck S, Norton A, et al. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc Natl Acad Sci U S A. 2003;100:3445–9. doi: 10.1073/pnas.0635898100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Wu YP, Sandhoff R, Werth N, Mizukami H, Ellis JM, et al. Interruption of ganglioside synthesis produces central nervous system degeneration and altered axon-glial interactions. Proc Natl Acad Sci U S A. 2005;102:2725–30. doi: 10.1073/pnas.0407785102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. Figure 1. Major ganglioside synthesis pathways. “0” pathway gangliosides are normally not expressed, but appear upon knockout of GM3 synthase activity and are absent after knockout of GM2 synthase. Combined knockout of both enzymes blocks all ganglioside synthesis.
Suppl. Figure-2: Initial adherence of WTt and DKOt cells. 4×105 cells were seeded/well, the cells not adhering were collected hourly, counted, and subtracted from the total cell count. Results are the mean±SD of four separate experiments.
Suppl. Figure-3: Migration of WTt and DKOt cells. Cell migration on a panel of matrices was assessed. 3×103 WTt or DKOt cells were seeded/well and 4 hour migration was quantified as described in Methods. Bars represent the mean±SD of 4 samples in two separate experiments. Fibronectin-associated migration was robust in WTt cells and abrogated in ganglioside-depleted, i.e. DKOt, cells (p<0.0001. There was lesser migration and no significant differences between WTt and DKOt cells on the other matrices.
Suppl. Figure 4: Tumor growth curve of H-Ras/c-Myc transformed WTt and DKOt. 105 WTt or DKOt cells were injected into syngeneic 6-8 week old C57Bl/6 mice (6 mice per group). Data are the mean±SD tumor volumes; p=0.0001.