

Abstract
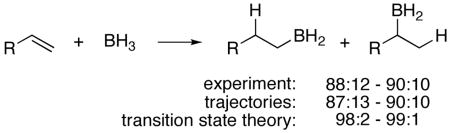
Transition state theory fails to accurately predict the selectivity in an example where it is ubiquitously invoked, hydroboration. The hydroboration of terminal alkenes with BH3 is moderately regioselective, affording an 88:12 - 90:10 ratio of anti-Markovnikov:Markovnikov adducts. High-level ab initio calculations predict too large of an energy difference between anti-Markovnikov and Markovnikov transition structures to account for the observed product ratio, and the consideration of calculational error, solvent, tunneling, and entropy effects does not resolve the discrepancy. Trajectory studies, however, predict well the experimental selectivity. The decreased selectivity versus transition state theory arises from the excess energy generated as the BH3 interacts with the alkene, and the observed selectivity is proposed to result from a combination of low selectivity in direct trajectories, moderate RRKM selectivity, and high selectivity after thermal equilibration.
The hydroboration of simple alkenes with BH3 preferentially occurs in an “anti-Markovnikov”1 fashion. The standard explanation for this preference in the literature,1,2 reproduced in some form in all general textbooks of organic chemistry, is that the selectivity arises from a greater stability for the anti-Markovnikov transition state over the alternative “Markovnikov” transition state. We find here that transition state theory fails to accurately account for the regioselectivity of hydroboration. Instead, a consideration of dynamic trajectories allows understanding of the selectivity.
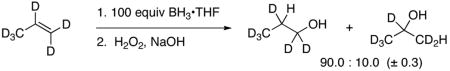
The addition of BH3 to terminal alkenes is only moderately regioselective. With simple terminal alkenes such as 1-hexene, the ratio of primary to secondary alcohol products after hydroboration with BH3 at 0 – 25 °C followed by oxidation is approximately 94:6.1,3 This ratio as it has been observed is a composite of the regioselectivity in three separate steps – hydroboration by BH3, hydroboration by RBH2, and hydroboration by R2BH. When unhindered, the intermediate alkylboranes are more reactive,4 but they are more regioselective as well, so the initial reaction of BH3 is less selective than the composite ratio. Using the observed 1-hexanol/2-hexanol ratio of 97:3 for the reaction of n-butylborane with 1-hexene5 as a measure of the selectivity of the second and third steps of hydroboration, the regioselectivity for reaction of 1-hexene with BH3 itself would be ≈88:12. In our hands, the hydroboration of propene-d6 at 21 °C with 100 equiv of BH3•THF (to minimize the contribution of hydroboration by alkylboranes) affords a 90.0:10.0 ratio of primary and secondary alcohols (based on direct analysis of the oxidized reaction mixture by 2H NMR). Assuming the applicability of transition state theory, the ΔΔG‡ for the transition states leading to the two products would be 1.1 – 1.3 kcal/mol.
A variety of gas-phase computational approaches were explored in an attempt to predict this ΔΔG‡.6 CCSD(T)/aug-cc-pvdz calculations were used to locate transition structures 1‡ and 2‡ for formation of the regioisomeric products from the precursor complex 3. The relative energetics of the anti-Markovnikov transition structure 1‡ and the Markovnikov structure 2‡ in high-level single-point calculations, as well as analogous structures optimized in other ways, are summarized in Table 1. All of the calculations, including particularly a converged series of CCSD(T) single-point energies employing very large basis sets, predict an energetic preference for 1‡ that greatly exceeds that implied by the experimental selectivity. In other words, the experimental reaction is considerably less selective than the 98:2 – 99:1 expected from the calculations.
Table 1.
Calculated ΔΔE‡ or ΔΔG‡ for Transition Structures 1‡ versus 2‡ for the Hydroboration of Propene with BH3.
| Method/Basis Set | ΔΔE‡a or ΔΔG‡b (kcal/mol) |
|---|---|
| B3LYP/6-31G* | 2.4b |
| G3 | 2.4b |
| CBS-QB3 | 2.3b |
| CCSD(T)/cc-pvtzc | 2.4a |
| CCSD(T)/aug-cc-pvtzc | 2.4a |
| CCSD(T)/cc-pvqzc | 2.4a |
| CCSD(T)/aug-cc-pvqzc | 2.4a |
| BD(TQ)/aug-cc-pvdzc | 2.6a |
| CCSD(T)/extrapolated to infinitebasis + enthalpy correction – TΔS | 2.5b,d |
| Experiment (assuming transition state theory) | 1.1–1.3 |
ΔΔE‡ (as potential energy).
ΔΔG‡ at 25 °C including harmonic enthalpy and entropy estimates based on the unscaled frequencies.
Single point calculations on the CCSD(T)/aug-cc-pvdz structures.
MP4/cc-pvdz frequencies were used in the enthalpy and entropy estimates.
We considered many possible reasons for this discrepancy. The simplest possibility, unadorned error in the calculated relative energies, seems doubtful based on the similarity of the structures being compared, the convergence of the results from various calculational methods, and the quality of the methods employed. A second possibility is that entropy strongly favors 2‡ in a way that is missed by the often-erring harmonic entropy estimate. Treatment of the most-vulnerable methyl-group rotation as a hindered rotor based on CCSD(T)/cc-pvtz single points on the rotational second-order saddle points favors 2‡, but by only 0.1 kcal/mol at 25 °C. On the other hand, inclusion of anharmonic corrections (B3LYP/6-31G*) increased the preference for 1‡ by 0.1 kcal/mol. A third possibility is that tunneling favors the Markovnikov process since its barrier is higher. Experimentally, however, there was no apparent difference in the regioselectivity of the reaction of BH3 versus BD3 with 1-octene. A one-dimensional infinite-parabola tunneling estimate7 based on the curvature of the transition vectors (301i and 530i cm−1 for 1‡ and 2‡, respectively) does favor 2‡, but by only a rate-factor of 1.22.
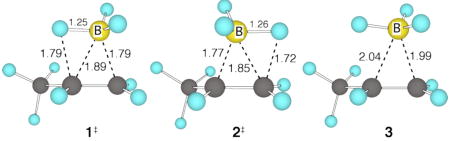
A more complex possibility to consider is the role of solvent. We first consider the effect of solvent polarity. Experimentally, solvent polarity has no discernable effect on the regioselectivity.8 Theoretically, the SCF dipole moments (aug-cc-pvqz) for 1‡ and 2‡ are only 3.34 and 2.95 D, respectively, and because the dipole moment of 1‡ is greater, solvent polarity should favor 1‡, not 2‡. In accord with this, solvent-model calculations (PCM, CPCM, SCIPCM) increase the preference for 1‡ by 0.3–0.4 kcal/mol, further from experiment.
A second possible role for solvent is the mechanistic complication of a direct transfer of the BH3 from the solvent as ligand. This is a debated issue (see the Supporting Information for a discussion). In calculations at various levels,6 loose transition structures for BH3 transfer from THF to propene are readily located, e.g. 4‡ (B3LYP/6-31G*,6 variationally optimized), but the product of these structures is complex 3, uncoordinated by THF. In such circumstances the 1‡/2‡ energy difference would still control the regioselectivity. Notably, the free-energy barrier associated with 4‡ is predicted to be 2.3 kcal/mol above a dissociative pathway followed by variational transition structure 5‡ (CCSD(T)/6-31+G**//B3LYP/6-31G*, after allowing for neat THF as its standard state).
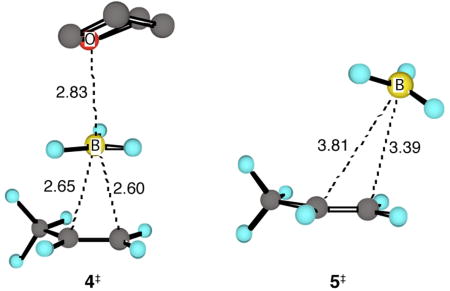
Why does transition state theory fail to account for the product ratio? The formation of 3 from BH3/propene is enthalpically barrierless (experimentally, 2 ± 3 kcal/mol with ethylene in the gas phase9) and downhill by 11.0 kcal/mol (CCSD(T)/aug-cc-pvqz + harmonic enthalpy estimate). Considerable excess energy is thus available from the formation of 3, and the barriers for formation of products from 3 are quite small – 1‡ is only 0.8 kcal/mol above 3 in free energy. Under these circumstances, we considered that trajectories may pass directly to the products or afford the products faster than thermal equilibration with solvent, making transition state theory inappropriately applicable.10
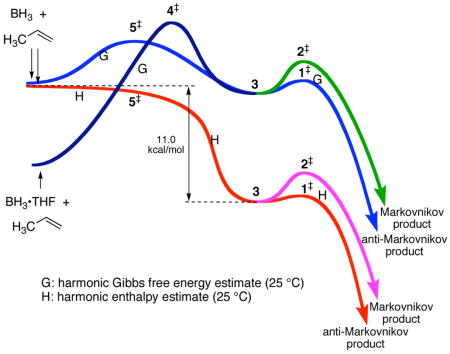
To explore this idea, a series of classical trajectory studies were performed by direct dynamics11 (Table 2), including B3LYP/6-31G* trajectories started directly from 3, 4‡, and 5‡, and ONIOM (B3LYP/6-31G*:AM1) trajectories started from 4‡ or 5‡ in a bath of 18 THF molecules confined to a 14 Å cubic box after a 500 – 8500 fs equilibrations. Trajectories started from 3 statistically at 25 °C, i.e., with no excess energy, underwent addition with a half-life of ≈700 fs and afforded ≈1% of the Markovnikov product. This fits quite well with transition state theory, though not with experiment, and weighs against recrossing or unknown subtle classical entropy effects as the source of the discrepancy. In contrast, trajectories started from points before 3 on the reaction coordinate, thus having excess energy in the area of 3, afforded 10–12% of the Markovnikov product, fitting strikingly well with experiment.
Table 2.
Classical Trajectory Studies.
| Starting point | anti- Markovnikov | Markovnikov | Unreactive in 5000 fs |
|---|---|---|---|
| 3 | 875 | 10 (1%) | 3 |
| 4‡ | 338 | 47 (12 %) | 496a |
| 5‡ | 739 | 110 (13%) | 465 |
| 5‡ + 18 THF, equilibratedb | 76 | 9 (11%) | 17a |
| 4‡ + 17 THF, equilibratedc | 19 | 2 (10%) | 3a |
Includes trajectories affording THF•BH3
500 fs equilibrations before releasing a fixed B–C distance.
1000 –8500 fs equilibrations.
A more detailed understanding of the regioselectivity can be gleaned from considering the time-course of the selectivity for trajectories started from 5‡. For the 25% of trajectories that afford product within 800 fs, the amount of Markovnikov product is particularly high (21%). The selectivity is higher for the 39% of trajectories that afford product in the 800 – 5000 fs range, with 7.5% Markovnikov addition. An RRKM calculation based on the energy difference between 5‡ and 3 predicts 7.4%. From these observations, we envision the selectivity in solution as involving three stages: a ‘direct-trajectory’ stage, with low selectivity, an RRKM stage, with medium selectivity, and a thermally-equilibrated stage, developing over several picoseconds12 as the excess energy is transferred to solvent. This last stage should be quite selective, as evidenced by the results with trajectories starting from 3, but by then the damage is done.
Some unusual experimental observations in hydroboration support the mechanistic picture here. In particular, hydroboration has long been recognized as “remarkably insensitive to major changes in the structure of the olefin”,13 a factor in its broad utility, and it exhibits an equally remarkable low intermolecular H/D isotope effect of 1.18.14 These observations were shoehorned into a conventional mechanistic picture, though in retrospect they clearly contradict it. A new unusual observation is that the regioselectivity is surprisingly temperature-insensitive – hydroboration of propene-d6 at 21, 45 and 70 °C afforded 10.0, 10.6 and 11.2% Markovnikov addition, respectively. This corresponds to a ΔΔH‡ of only 0.5 ± 0.2 kcal/mol.
In this most ‘textbook’ of reactions, transition state theory fails and the selectivity can only be understood by consideration of dynamic trajectories. We are unaware of any more striking example of the role of dynamics in an experimentally preeminent reaction.
Supplementary Material
Acknowledgments
We thank NIH grant # GM-45617 and The Robert A. Welch Foundation for financial support.
Footnotes
Supporting Information Available: Experimental and calculational procedures, energies and geometries of all calculated structures, and technical comments. (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Brown HC, Zweifel G. J Am Chem Soc. 1960;82:4708–4712. [Google Scholar]
- 2.(a) Dewar MJS, McKee ML. Inorg Chem. 1978;17:1075–1082. [Google Scholar]; (b) Graham GD, Freilich SC, Lipscomb WN. J Am Chem Soc. 1981;103:2546–2552. [Google Scholar]; (c) Wang X, Li Y, Wu Y-D, Paddon-Row MN, Rondan NG, Houk KN. J Org Chem. 1990;55:2601–2609. [Google Scholar]
- 3.(a) Pasto DJ, Cumbo CC, Balasubramaniyan P. J Am Chem Soc. 1966;88:2187–2194. doi: 10.1021/ja00962a021. [DOI] [PubMed] [Google Scholar]; (b) Kalbalka GW, Wadgaonkar PP, Shoup TM. Organometallics. 1990;9:1316–1320. [Google Scholar]
- 4.Pasto DJ, Lepeska B, Balasubramaniyan V. J Am Chem Soc. 1972;94:6090–6096. [Google Scholar]
- 5.Srebnik M, Cole TE, Ramachandran PV, Brown HC. J Am Chem Soc. 1989;54:6085–6096. [Google Scholar]
- 6.See the Supporting Information for calculational details and validation studies. Interestingly, of diverse combinations of basis sets and DFT functionals, B3LYP/6-31G* most closely reproduced CCSD(T)/aug-cc-pvtz energetics in this system.
- 7.Bell RP. The Tunnel Effect in Chemistry. Chapman & Hall; London: 1980. pp. 60–63. [Google Scholar]
- 8.Lane CF. J Org Chem. 1974;39:1437–1438. [Google Scholar]
- 9.Fehlner TP. J Am Chem Soc. 1971;93:6366–6373. [Google Scholar]
- 10.Vayner G, Addepalli SV, Song K, Hase WL. J Chem Phys. 2006;125:014317. doi: 10.1063/1.2206785. [DOI] [PubMed] [Google Scholar]
- 11.Hase WL, Song KH, Gordon MS. Comp Sci Eng. 2003;5:36–44. [Google Scholar]
- 12.Elles CG, Cox MJ, Crim FF. J Chem Phys. 2004;120:6973–79. doi: 10.1063/1.1676292. [DOI] [PubMed] [Google Scholar]
- 13.Brown HC, Moerikofer AW. J Am Chem Soc. 1963;85:2063–65. [Google Scholar]
- 14.Pasto DJ, Lepeska B, Cheng T-C. J Am Chem Soc. 1972;94:6083–6090. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.