

Abstract
Human African trypanosomiasis (HAT), also known as African sleeping sickness, is a neglected tropical disease with inadequate therapeutic options. We have launched a collaborative new lead discovery venture using our repository of extracts and natural product compounds as input into our growth inhibition primary screen against Trypanosoma brucei. Careful evaluation of the spectral data of the natural products and derivatives allowed for the elucidation of the absolute configuration (using the modified Mosher’s method) of two new peroxiterpenes: (+)-muqubilone B (1a) and (−)-ent-muqubilone (3a). Five known compounds were also isolated: (+)-sigmosceptrellin A (4a), (+)-sigmosceptrellin A methyl ester (4b), (−)-sigmosceptrellin B (5), (+)-epi-muqubillin A (6) and (−)-epi-nuapapuin B methyl ester (7). The isolated peroxiterpenes demonstrated activities in the range from IC50 = 0.2 – 2 μg/mL.
The search for chemotherapeutics to treat human African trypanosomiasis (HAT), also known as African sleeping sickness, has not been a major target of industry drug discovery campaigns, even though the currently available therapeutics are inadequate.1 This is surprising as there are 50,000 annual cases of infection, HAT is the world’s third most devastating parasitic disease, and it remains a major threat to more than 60 million people.2–4 Because it is a serious health problem for resource poor regions of Africa, HAT is designated as a neglected tropical disease (NTD). The causative agent is a protozoan parasite, Trypanosoma brucei,5 subdivided in two sub-genera, T. brucei gambiense in West Africa and T. brucei rhodesiense in East Africa.1 Most of the four current chemotherapeutics to combat these parasites at their different development stages are very antiquated. The drugs (by registration date for HAT treatment) consist of suramin4 (1922), pentamidine1 (1941), melarsoprol3 (1949), and eflorithine1 (1990). Another relevant point is that new strains of T. brucei are showing cross-resistance to some of these agents.6 In recent years, only one compound, DB2897 (a synthetic analog of pentamidine), received serious clinical evaluation against HAT, but further development was discontinued in 2008.3
The quest to discover antiparasitic lead structures has rarely focused on the chemical diversity inherent in marine natural products. Alternatively, recent studies have used combinatorial chemistry libraries of purine2 and thiosemicarbazone5 derived structures to seek inhibitors of different T. brucei targets.2,5 In the early 1990’s the University of California Santa Cruz group participated in a collaborative effort with Syntex (with Dr. T. Mathews) to explore marine natural products for their potential to provide anthelminthic lead structures,8 but unfortunately these efforts were prematurely suspended. In this context there is a rather attention-grabbing report showing that psammaplin A,9 first isolated from the marine sponge Psamaplysilla in 1987 and found to possess anthelminthic activity (against Nippostrongylus brasiliensis),8 was recently found to be active against T. brucei (EC50 = 2.6 μM).10 Inspired by this recent development, we have launched a new collaborative lead discovery venture using our repository of extracts and natural product compounds as input into our growth inhibition primary screen against T. brucei. We were encouraged to continue this effort because of an assay hit from the sponge, Diacarnus bismarckensis, which was available in large amounts. The current literature shows over two dozen peroxiterpenes11 reported from Diacarnus taxon and these have been primarily evaluated in anticancer screens.12 Reported in this account are our efforts to isolate and define the total structures of the active principles, presumed to be peroxiterpenes, followed by further biological activity study.
Results and Discussion
Once the decision was made to select the T. brucei assay active crude extracts from D. bismarckensis (coll. no. 03512), the next step was to employ bioassay-guided fractionation. Although the methanol crude extract partition (XFM, 2.06 g, IC50 = 5.0 μg/mL) showed good activity, the hexanes (XFH, 8.80 g, IC50 = 0.02 μg/mL) and dichloromethane (XFD, 0.66 g, IC50 = 0.3 μg/mL) partitions were selected for further study due to their higher potencies. Analysis of their respective 13C NMR spectra confirmed the presence of peroxiterpenes because of prominent resonances in the region δC 80 – 82. Orthogonal chromatography facilitated the isolation work, beginning with silica gel flash chromatography followed by reversed phase HPLC, which eventually afforded seven peroxiterpene compounds. The XFH, provided a mixture of 1a, related to (+)-muqubilone/aikupikoxide A (2),13,14 and (−)-ent-muqubilone (3a), an unreported enantiomer of 2. Preparation of their respective methyl esters was required in the final purification of these compounds. Five additional compounds came from the XFH including (+)-sigmosceptrellin A (4a),15,16 (+)-sigmosceptrellin A methyl ester (4b),15,16 (−)-sigmosceptrellin B (5),16 (+)-epi-muqubilin A (6),12a and (−)-epi-nuapapuin B methyl ester (7).12a The XFD was also a source of four pure compounds: 1a, 4a, 5, and 6. We obtained all compounds as free carboxylic acids, with the exception that minor quantities of methyl ester 4b and 7 that are most likely artifacts of isolation. The known compounds (4 – 7) have also been previously obtained from sponges classified as Diacarnus sp.17
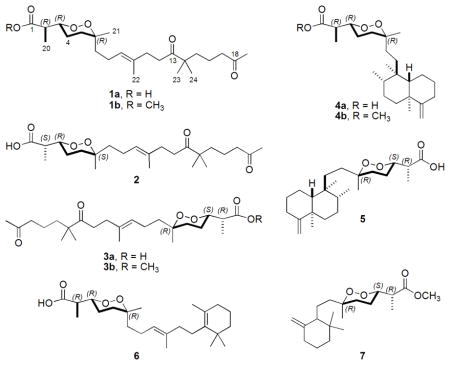
The structure elucidation of the new compound, (+)-muqubilone B (1a), C24H40O6, was initiated by using the characteristic 13C NMR shifts at δC = 81.2 (CH) and 80.0 (C) to indicate the presence of a peroxide functionality with trisubstitution (Table 1).18 Distinct NMR signals for a carboxylic acid (δC = 178.6), an acetyl group (δC = 209.1, 29.8), an isolated ketone (δC = 215.4), a trisubstituted double bond (δC = 124.6, 134.4), the molecular formula obtained from the HR-ESI-MS, and dereplication insights, all supported that 1 was a norsesterterpene containing an endoperoxide ring. Another diagnostic NMR signal was that of an isochronous gem-dimethyl functionality (δC = 24.3; δH = 1.13). At this point it was evident that the C3 containing carboxylate and the C16 acyclic side chains were similar to that of 2.13,14 The side-by-side comparison of the similarities and differences in the NMR data between 1a and 2 plus the 2D NMR data of the former (Figure 1) unequivocally confirmed that this pair had identical planar structures and must be diastereomers.
Table 1.
NMR Data for (+)-Muqubilone B (1a) in CDCl3a
| Position | δC, type | δH, mult. (J in Hz) | HMBC (H to C) | COSY |
|---|---|---|---|---|
| 1 | 178.6, C | - | ||
| 2 | 42.4, CH | 2.55, quint. (7.2) | 3, 20 | 3, 20 |
| 3 | 81.2, CH | 4.25, ddd (8.4, 7.2, 4.2) | 2, 4 | |
| 4ax | 23.8, CH2 | 1.77, dtd (12.0, 8.4, 4.2) | 21 | 5 |
| 4eq | 1.70, dtd (12.0, 4.2, 3) | 3 | 3 | |
| 5 | 32.5, CH2 | 1.66, m | 6, 21 | 4 |
| 6 | 80.0, C | - | ||
| 7 | 34.7, CH2 | 1.49, m | 21 | 8 |
| 8a | 22.0, CH2 | 2.04, m | 7, 9 | |
| 8b | 1.94, m | |||
| 9 | 124.6, CH | 5.16, td (7.2, 1.2) | 8 | |
| 10 | 134.4, C | - | ||
| 11 | 33.4, CH2 | 2.21, t (7.8) | 9, 10, 12, 22 | 12 |
| 12 | 35.6, CH2 | 2.56, t (7.8) | 10, 11, 13 | 11 |
| 13 | 215.4, C | - | ||
| 14 | 47.5, C | - | ||
| 15a | 39.1, CH2 | 1.51, m | 16 | 16 |
| 15b | 1.46, m | |||
| 16 | 18.9, CH2 | 1.42, m | 15 | 15, 17 |
| 17 | 43.9, CH2 | 2.42, t (7.2) | 16, 18 | 16 |
| 18 | 209.1, C | - | ||
| 19 | 29.8, CH3 | 2.14, s | 17, 18 | |
| 20 | 12.7, CH3 | 1.20, d (7.2) | 1, 2, 3 | 2 |
| 21 | 22.6, CH3 | 1.12, s | 4, 5, 6, 8 | |
| 22 | 16.0, CH3 | 1.62, d (1.2) | 9, 10, 11 | |
| 23,24 | 24.3, CH3 | 1.13, s | 13, 14, 15 | |
| OH | - | 3.71, s | 1 | |
Measured at 600 MHz (1H) and 150 MHz (13C).
Figure 1.
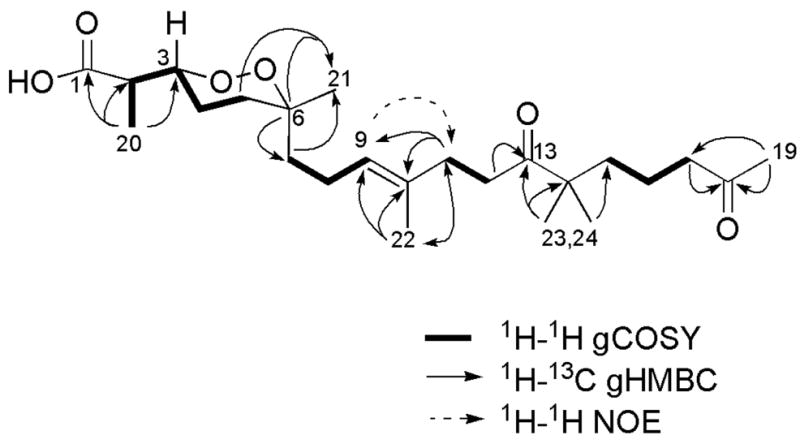
Key 2D NMR correlations for (+)-muqubilone B (1a).
Establishing the complete absolute configuration of 1a, while reasonably straightforward, required a multi-step analysis. The E double bond geometry was confirmed by the NOE data shown in Figure 1 (correlation from H-9 to H2-11) and the characteristic C-22 shift.19 We have previously used the empirical observations laid out by Capon and MacLeod18 to set the relative configuration at each of the three chiral centers of the molecular fragment present in Diacarnus derived peroxiterpenes and repeated this process here,12a but it first required the preparation of the methyl ester 1b to obtain a data set consistent with the models.20 The carbon substituent at C-3 was set as equatorial because H-3 exhibited vicinal axial coupling to H-4, J3,4 = 8.0 Hz (J = 3 – 4 Hz is expected for equatorial). The methyl at C-6 (δC = 24.2) was next assigned as equatorial because of its diagnostic 13C NMR shift (δC,eq = 23.5 – 24.0; δC,ax = 20.5 – 20.9). Finally the erythro (R*/R*) configuration at C-2/C-3 was designated based on the proton chemical shift position at H3-20, δH 1.14 (standard values: δH,erythro = 1.13 – 1.14; δH,threo = 1.22 – 1.24).18 The resulting all R* configuration at each of the chiral centers confirmed that 1a ( ) was an unreported diastereomer of 2, whose relative configuration were previously independently reported as 2R*, 3S*, 6R* for both (+) muqubilone ( )13 and aikupikoxide A ( ).14
We next concluded it was essential to push forward and ascertain the absolute configuration of 1a with the expectation such findings could provide similar insights for 2. Hydrogenation of 1b 21 afforded diol 8 that was further transformed into Mosher’s esters 9a and 9b as shown in Scheme 1.21,22 The circumstance that the Δ9 double bond was also reduced during the hydrogenation did not affect the outcome in evaluating the Δδ values (δS - δR) to verify C-3 as R. Thus, the 2R, 3R, 6R final absolute configuration is now assigned for (+)-muqubilone B (1a). It is notable that (+)-2R, 3R, 6R-epi-muqubilin A12a (6) ( ) also isolated from this sponge represents an obvious biosynthetic precursor of (+)-1a, especially in view of their identical chirality. Our further discussion on the absolute configuration of 2 shown here as 2S, 3R, 6S will accompany the characterization of 3 which follows next.
Scheme 1.

Modified Mosher’s method analysis of (+)-muqubilone B (1a). i) TMS-CHN2 (2M in hexanes), CH3OH, r.t., 30 min.; ii) H2 (1atm), Pd/C, EtOH, r.t., 1 hr; iii) (+)- or (−)-MTPA-Cl, pyr., r.t., 18 hr.
Noted above was that the XFH provided a mixture of 1a and an additional new compound (−)-ent-muqubilone (3a). The separation of these compounds was accomplished by first subjecting the mixture to methylation using TMS-diazomethane followed by isocratic HPLC purification to cleanly afford 1b ( ) and 3b ( ) in a 2:1 ratio. The absolute stereostructure assigned for 3a was based on data obtained for 3b using the process parallel to that described above for 1. Both 1b and 3b possessed identical molecular formulas and afforded nearly identical NMR shifts with 2.13 The following observations of H-3, Jax = 8.0 Hz; CH3-21, δC,ax = 20.9; CH3-20, δH,threo = 1.25 indicated that the relative configurations reported for 3b were equivalent with that of 2, but these compounds possessed opposite rotation data (see above) and must be enantiomers. A review of the current literature (see Table S1, Supporting Information)16,18,23 affirms that the variations in the absolute configurations at C-2 and C-6 within the peroxide containing substructure comprised of the chain from C-1 to C-6 do not influence the rotation sign. Additionally, the sign of optical rotation does not change upon substitution of the free carboxylate. Thus, the absolute configuration at C-3 can be used is an empirical anchor point. For example, when H-3 is in the axial orientation, a (−) [α]D is observed for 3S configuration and the (+) rotation is observed for 3R (in CHCl3). Consequently, the compound (−)-3b can be concluded to have the stereostructure of 2R, 3S, 6R. Furthermore (+)-muqubilone/aikupikoxide A13,14 (2), whose rotation data were discussed above, can be assigned as 2S, 3R, 6S.
The peroxiterpenes isolated in this work and evaluated against T. brucei can be grouped into four structural types based simply on the side-chain attached at C-6 of the 1,2-dioxane ring. The first three types each have the C16 carbon side chain that is either acyclic (1 – 3), monocyclic (6), or bicyclic (4 – 5). The final type (7) has a C11 monocyclic carbon side chain. The muqubilones (1a, 1b, 3b) showed similar bioactivities independent of the nature of the carboxylate: free acid (1a, IC50 = 2 μg/mL) or methyl ester 1b (IC50 = 5 μg/mL) and 3b (IC50 = 3 μg/mL). Consistent with the supposition that bioactivity hinges on the presence of the 1,2-dioxane ring was that diol 8 (IC50 > 25 μg/mL) prepared from 1b was inactive. A change in the side chain to that containing either a monocyclic or bicyclic ring imparts a barely observable change as shown by the data for sigmosceptrellin A (4a, IC50 = 1 μg/mL) and its methyl ester (4b, IC50 = 2 μg/mL), sigmosceptrellin B (5, IC50 = 0.2 μg/mL), and epi-muqubilin A (6, IC50 = 0.9 μg/mL). Finally, the length of the side chain does not greatly influence the activity as shown by epi-nuapapuin B methyl ester (7, IC50 = 2 μg/mL).
It is relevant to overlay our observations with those from recent studies involving 1,2-dioxane ring containing terpenoids or polyketides and somewhat analogous peroxide ring compounds studied by others. An investigation of Diacarnus megaspinorhabdosa afforded a series of peroxiterpenes and one diol product with structures parallel to those reported here.12 There were varying EC50 cytotoxicity effects observed from the evaluation of these compounds against three cancer cell lines and those possessing a C16 side chain at C-6 were generally more cytotoxic than those with a C11 appendage. Curiously, the one natural product evaluated where the peroxide ring was not present, but replaced by a diol, displayed mild activity against one cell line and inactivity against the other two cell types. In another study, the cytotoxic and antiparasitic properties were compared for 1,2-dioxane and furan ketides isolated from the sponge Plakortis angulospiculatus.24 These results showed that both heterocyclic ring types displayed modest cytotoxicity effects while one of the 1,2-dioxane structures potently inhibited Leishmania chagasi and Trypanosoma cruzi. These preceding literature findings, and those not discussed here showing that other polyketide 1,2-dioxanes have antileishmanial activity,25 alongside our results together suggest that the polyalkylated 1,2-dioxane frameworks are of promise for further development as lead structures. In addition, the therapeutic index of antiparasitic to cytotoxicity action for these structures is in the correct direction.
The responses of Trypanosoma brucei rhodesiense have been evaluated for two other sets of peroxide containing structures as follows. The wormwood tree peroxisesquiterpene, artemisinin, exhibits nM action against Plasmodium falciparum.26 This compound (T. b. rhodesiense IC50 = 627 or 2526 μg/mL) and its close analog deoxyartemisinin (T. b. rhodesiense IC50 = 3426) possess similar mild activity against T. b. rhodesiense and the latter, devoid of the cyclic peroxide functionality, is inactive vs. P. falciparum. A similar SAR activity pattern is shown for another matched set. This includes the 1,2,4-trioxolane containing synthetic polycyclic OZ277 (T. b. rhodesiense IC50 = 0.6 μg/mL)26 vs. the 1,3-dioxolane containing synthetic polycyclic carbaOZ277 (T. b. rhodesiense IC50 = 0.9 μg/mL)26 which are equipotent against T. b. rhodesiense (and have not been tested vs. P. falciparum). Overall, it would appear that structural features of 5 (T. brucei IC50 = 0.2 μg/mL) would be an excellent starting point for further SAR modifications as it showed similar potencies on par with a current HAT therapeutic, pentamidine (T. b. rhodesiense IC50 = 0.4 μg/mL).27
Conclusions
This study has revealed new dimensions to the structural and biological activity properties of peroxiterpenes isolated from Diacarnus sponges. First, careful evaluation of the spectral data of the natural products and derivatives allowed for the elucidation of the absolute configuration of compounds we isolated including 1a and 3a plus a proposal for the absolute configuration of 2. Secondly, the observation of enantiomeric structures (2 vs. 3) from the same genus, Diacarnus, but different species from different oceans (2 from a Red Sea specimen, D. erythaeanus and 3 from the Indo-Pacific specimen D. bismarckensis) is a stunning and rare observation in marine natural products chemistry. Thirdly, we have further demonstrated the value in vigorously considering peroxiterpenes, especially the known compound, (−)-sigmosceptrellin B (5) as template for the development of therapeutic leads against T. brucei.
Experimental Section
General Experimental Procedures
All NMR experiments were run on Varian UNITY 500 (500 and 125 MHz for 1H and 13C, respectively) and Varian INOVA 600 spectrometers (600 and 150 MHz for 1H and 13C, respectively) using standard pulse sequences with residual solvent protons and carbons as references (CDCl3: 7.27 and 77.23 ppm for 1H and 13C, respectively). Mass measurements were obtained on a bench top Mariner ESI-TOF-MS. Crude extractions were obtained using an Accelerated Solvent Extractor (1500 psi, 100 °C, 25 min). Flash chromatography was done on a CombiFlash, using prepacked 40 g silica columns. HPLC was performed with Phenomenex Synergi RP-Max preparative (10 μm, C18, 21.2 mm × 250 mm) and Luna semipreparative (5 μm, C18, 10 mm × 250 mm) columns.
Collection and Identification
The sponge materials were collected in December 2003 by SCUBA at a depth range of 31 – 40 ft near Sanaroa, Papua New Guinea (GPS = 9° 37.214 S: 150° 57.332 E). The sponge samples were ramose in shape with very small oscules throughout and had a purple colored endosome with a grey colored ectosome. The collection was identified as Diacarnus bismarckensis (Kelly-Borges & Vacelet, 1995) by Dr. R. W. M. van Soest. Voucher samples have been deposited at UCSC and the Zoological Museum of Amsterdam (UCSC coll. no. 03512 = ZMAPOR 18576). Photographs are also available from the Crews laboratory.
Extraction and Isolation of 03512
Following standard Crews laboratory field protocol, the sponge was soaked for over 24 hours in an ethanol:seawater (1:1) preservative solution which was discarded in the field. The samples were then packed in bottles and shipped to UCSC where the sponge was cut into 1.5 inch segments and dried under air for 2 days. 73.4 g of dried sponge was extracted using an accelerated solvent extractor (ASE) (100° C, 1500 psi, 25 min) to yield hexanes (XFH, 8.80 g), CH2Cl2 (XFD, 0.6615 g), and CH3OH (XFM, 2.06 g) crude extracts. A bioassay guided fraction then followed on the XFH and XFD fractions. The large quantity of XFH crude warranted a further Kupchan-like solvent partition to yield hexanes (XFHFH, 4.08 g), CH2Cl2 (XFHFD, 4.08 g) and CH3OH (XFHFM, 0.518 g) fractions. The XFHFH crude was subjected to normal phase automated flash chromatography using a linear gradient on silica gel (CH2Cl2 to CH3OH, 40 min) yielding 6 fractions (XFHFHC1 – XFHFHC6). An orthogonal chromatographic approach was used on the C3 (1.10 g) and C4 (942 mg) fractions with reversed-phase preparative HPLC (80% aq to 100% CH3CN linear gradient, 0.1% formic acid, 20 min, ELSD) followed by semipreparative HPLC (70% aq CH3CN isocratic, 0.1% formic acid, UV = 200 nm). From these fractions, (−)-epi-nuapapuin B methyl ester12a (7, 2.9 mg), (+)-sigmosceptrellin A15,16 (4a, 68.6 mg), (−)-sigmosceptrellin B16 (5, 33.8 mg), (+)-epi-muqubillin A12a (6, 23.9 mg) and (+)-sigmosceptrellin A methyl ester15,16 (4b, 8.2 mg) were isolated. The XFHFD was treated in a similar manner to yield a 2:1 mixture (21.4 mg) of (+)-muqubilone B (1a) and (−)-ent-muqubilone (3a) which were separable after methyl esterification and HPLC (isocratic 85% aq CH3CN, 0.1% formic acid, UV = 200 nm). The XFD crude fraction was subjected to orthogonal chromatography as previously described. These fractions yielded (+)-muqubilone B (1a, 14.3 mg), (+)-sigmosceptrellin A15,16 (4a, 34.3 mg), (−)-sigmosceptrellin B12a (5, 6.1 mg), and (+)-epi-muqubillin A12a (6, 6.9 mg).
Trypanosoma brucei brucei Assay
Trypanosoma brucei brucei strain 221 was grown in complete HMI-9 medium containing 10% FBS, 10% Serum Plus medium (Sigma Inc. St. Louis Mo. USA) and 1X penicillin/streptomycin. The trypanosomes were diluted to 1×105 per mL in complete HMI-9 medium. 95 μL per well of the diluted trypanosomes was added to sterile Greiner 96-well flat white opaque culture plates that contained 5 μL of test samples (in 10% DMSO). Control wells contained 95 μL of the diluted trypanosomes and 5 μL of 10% DMSO while control wells for 100% inhibition contained 95 WL of the diluted trypanosomes and 5 μL of 1 mM thimerosal (in 10% DMSO). Trypanosomes were incubated with test samples for 48 h at 37 °C with 5% CO2 before monitoring viability. Trypanosomes were then lysed in the wells by adding 50 μL of CellTiter-Glo™ (Promega Inc., Madison, WI, USA). Lysed trypanosomes were placed on an orbital shaker at room temperature for 2 min. The resulting ATP-bioluminescence of the trypanosomes in the 96-well plates was measured at room temperature using an Analyst HT plate reader (Molecular Devices, Sunnyvale, CA, USA). All IC50 curve fittings were performed with Prism 4 software (GraphPad, San Diego, CA).
(+)-Muqubilone B (1a)
A clear oil; (c = 0.046, CHCl3); NMR on Table 1; HR-ESI-MS 425.2934 [M + H]+ (calcd for C24H41O6, 425.2898).
(+)-Sigmosceptrellin A (4a)
(+)-Sigmosceptrillin A methyl ester (4b)
(−)-Sigmosceptrellin B (5)
(+)-Epi-muqubilin A (6)
Physical data in accordance with published data.12a
(−)-Epi-nuapapuin B methyl ester (7)
Physical data in accordance with published data.12a
Methylation of (+)-muqubilone B (1a)
6.5 mg of (+)-muqubilone B (1a) was dissolved into 1 mL of CH3OH to which 2 mL of trimethylsilyldiazomethane (2M in hexanes) was added and stirred at room temperature for 30 min. The solvent was evaporated under nitrogen and the reaction mixture was purified by HPLC (C18 column, 85%aq CH3CN isocratic, UV = 200 nm) to yield 6.2 mg of (+)-muqubilone B methyl ester (1b). 1b: (c = 0.011, CHCl3); 1H NMR (CDCl3, 500 MHz) δH 1.11 (3H, s, H3-21), 1.12 (6H, s, H3-23, H3-24), 1.14 (3H, d, J = 7.5 Hz, H3-20), 1.41 (2H, m, H2-7), 1.47 (2H, m, H2-16), 1.55 (2H, m, H2-15), 1.62 (3H, d, J = 1 Hz, H3-22), 1.63 (2H, m, H2-5), 1.76 (1H, dtd, J = 13, 4.5, 2.5 Hz, Heq-4), 1.85 (1H, dtd, J = 13, 8, 4.5 Hz, Hax-4), 1.94 (1H, m, Hb-8), 2.04 (1H, m, Ha-8), 2.13 (3H, s, H3-19), 2.21 (2H, t, J = 8 Hz, H2-11), 2.41 (2H, t, J = 7 Hz, H2-17), 2.55 (2H, t, J = 8 Hz, H2-12), 2.58 (1H, quint., J = 7.5 Hz, H-2), 3.70 (3H, s, OCH3), 4.24 (1H, ddd, J = 8.0, 7.5, 4.5 Hz, H-3), 5.16 (1H, tq, J = 6.5 Hz, 1, H-9); 13C NMR (CDCl3, 125 MHz) δc 13.1 (CH3-20), 16.3 (CH3-22), 19.2 (CH2-16), 22.3 (CH2-8), 22.9 (CH2-4), 24.2 (CH3-21), 24.6 (CH3-23), 24.6 (CH3-24), 30.2 (CH2-7), 32.7 (CH3-11), 33.7 (CH2-19), 35.0 (CH2-12), 35.9 (CH2-5), 39.4 (CH2-15), 42.9 (CH-2), 44.2 (CH2-17), 44.2 (C-14), 52.2 (O-CH3), 80.1 (C-6), 81.5 (CH-3), 124.8 (CH-9), 134.6 (C-10), 174.6 (C-1), 208.8 (C-18), 215.4 (C-13); HR-ESI-MS m/z 461.2906 [M + Na]+ (calc. for C25H42O6Na, 461.2874).
Hydrogenation of (+)-muqubilone B methyl ester (1b)
4 mg of (+)-muqubilone B methyl ester (1b) was dissolved into 2 mL of ethanol. 10 mg of palladium on carbon was then added and the slurry was stirred under 1 atm of H2 for 1 hr at room temperature. The reaction mixture was quenched with water and the palladium catalyst was filtered through Celite to afford 4 mg of (+)-muqubilone B diol (8). 8: (c = 0.017, CHCl3); 1H NMR (CDCl3, 500 MHz) δH 0.87 (3H, d, J = 6.5 Hz, H3-22), 1.11 (6H, s, H3-23, H3-24), 1.16 (3H, s, H3-21), 1.21 (3H, d, J = 7 Hz), 1.30 (2H, m, H2-8), 1.37 (1H, m, Hb-5), 1.40 (2H, m, H2-11), 1.43 (1H, m, Hb-15), 1.44 (2H, m, H2-7), 1.45 (2H, m, H2-16), 1.46 (1H, m, Ha-15), 1.50 (1H, m, Ha-5), 1.59 (1H, m, Hb-9), 1.60 (1H, m, Ha-10), 1.66 (1H, m, Ha-9), 1.67 (1H, m, Ha-4), 2.13 (3H, s, H3-19), 2.41 (2H, t, J = 6.5 Hz, H2-17), 2.46 (2H, t, J = 8.5 Hz, H2-12), 2.56 (H, quint., J = 7 Hz, H-2), 3.70 (H, ddd, J = 8, 6, 2 Hz, H-3), 3.72 (3H, s, O-CH3); 13C NMR (CDCl3, 125 MHz) δc 14.5 (CH3-20), 19.2 (CH3-22), 19.8 (CH2-16), 21.5 (CH2-8), 24.6 (CH3-23), 24.6 (CH3-24), 27.1 (CH3-21), 29.0 (CH2-4), 30.1 (CH3-19), 30.9 (CH-10), 32.6 (CH2-11), 34.7 (CH2-12), 37.6 (CH2-9), 39.5 (CH2-15), 42.3 (CH2-7), 42.6 (CH2-5), 44.1 (CH2-17), 45.5 (CH-2), 47.7 (C-14), 52.0 (O-CH3), 72.6 (C-6), 74.0 (CH-3), 176.7 (C-1), 208.9 (C-18), 216.1 (C-13); HR-ESI-MS m/z 465.3224 [M + Na]+ (calc. for C25H46O6Na, 465.3192).
(R)- and (S)-MTPA esterification of (+)-muqubilone B diol (8)
2 mg of (+)-muqubilone B diol (8) was dissolved in 200 μL of dry pyridine to which 15 μL of (−)-(R)-MTPA-Cl was added. This was stirred at room temperature, under nitrogen, overnight. After the solvent was evaporated under nitrogen, the reaction mixture was purified by HPLC (C18 column, 85%aq CH3CN isocratic, UV = 254 nm) to yield 1.8 mg of (S)-MTPA ester (9a). Similarly, 1.8 mg of the diol (8) was derivatized with (+)-(S)-MTPA-Cl to afford 1.7 mg of the (R)-MTPA ester (9b). 9a: 1H NMR (CDCl3, 500 MHz) δH 0.863 (3H, d, J = 6 Hz, H3-22), 1.110 (3H, s, H3-21), 1.117 (6H, s, H3-23, H3-24), 1.126 (3H, d, J = 6 Hz, H3-20), 2.123 (3H, s, H3-19), 2.410 (2H, t, J = 6.5 Hz, H2-17), 2.855 (2H, t, J = 6.5 Hz, H2-12), 2.855 (H, quint., J = 7 Hz, H-2), 3.522 (3H, s, MTPA-OCH3), 3.586 (3H, s, O-CH3), 5.394 (H, td, J = 7.5, 4 Hz, H-3), 7.425 (4H, m, MTPA-Ar), 7.561 (1H, m, MTPA-Ar); ESI-MS m/z 681 [M + Na].+ 9b: 1H NMR (CDCl3, 500 MHz) δH 0.873 (3H, d, J = 6.5 Hz, H3-22), 1.036 (3H, s, H3-21), 1.121 (6H, s, H3-23, H3-24), 1.186 (3H, d, J = 7.5 Hz, H3-20), 2.125 (3H, s, H3-19), 2.413 (2H, t, J = 6.5 Hz, H2-17), 2.467 (2H, t, J = 6.5 Hz, H2-12), 2.865 (1H, quint., J = 7.5 Hz, H-2), 3.540 (3H, s, MTPA-O-CH3), 3.644 (3H, s, O-CH3), 5.382 (1H, td, J = 7.5, 4 Hz, H-3), 7.428 (4H, m, MTPA-Ar), 7.560 (1H, m, MTPA-Ar); ESI-MS m/z 681 [M + Na].+
Methylation of (−)-ent-muqubilone (3a)
5 mg of an inseparable mixture of (+)-muqubilone B (1a) and (−)-ent-muqubilone (3a) (2:1) was dissolved into 1 mL of CH3OH to which 2 mL of trimethylsilyldiazomethane (2 M in hexanes) was added and stirred at room temperature for 30 min. The solvent was evaporated under nitrogen and the reaction mixture was purified by HPLC (C18 column, 85%aq CH3CN isocratic, UV = 200 nm) to yield 3.2 mg of (+)-muqubilone B methylester (1b) and 1.6 mg of (−)-ent-muqubilone methyl ester (3b). 3b: (c = 0.004, CHCl3); 1H NMR (CDCl3, 500 MHz) δH 1.11 (6H, s, H3-23, H3-24), 1.25 (3H, d, J = 7.0 Hz, H3-20), 1.29 (3H, s, H3-21), 1.41 (2H, m, H2-16), 1.44 (2H, m, H2-7), 1.47 (2H, m, H2-15), 1.60 (3H, s, H3-22), 1.63 (2H, m, H2-5), 1.71 (2H, m, H2-4), 2.00 (2H, m, H2-8), 2.13 (3H, s, H3-19), 2.20 (2H, t, J = 8.0 Hz, H2-11), 2.41 (2H, t, J = 7.0 Hz, H2-17), 2.54 (2H, t, J = 8.0 Hz, H2-12), 2.66 (1H, q, J = 7.5 Hz, H-2), 3.70 (3H, s, O-CH3), 4.12 (1H, ddd, J = 8.0, 7.5, 4.5 Hz, H-3), 5.10 (1H, t, J = 7.0 Hz, H-9); 13C NMR (CDCl3, 125 MHz) δc 13.8 (CH3-20), 16.3 (CH3-22), 19.2 (CH2-16), 20.9 (CH3-21), 21.9 (CH2-8), 23.7 (CH2-4), 24.5 (CH3-23), 24.5 (CH3-24), 30.1 (CH3-19), 32.2 (CH2-5), 33.7 (CH2-11), 35.8 (CH2-12), 39.4 (CH2-15), 39.8 (CH2-7), 43.2 (CH-2), 44.1 (CH2-17), 47.7 (C-14), 52.7 (O-CH3), 80.3 (C-6), 81.6 (CH-3), 124.6 (CH-9), 134.8 (C-10), 174.5 (C-1), 208.8 (C-18), 215.3 (C-13); HR-ESI-MS m/z 461.2898 [M + Na]+ (calc. for C25H42O6Na, 461.2874).
Supplementary Material
Acknowledgments
Large thanks to our long standing collaborator, Dr. Rob W. M. van Soest, for his expertise with sponge taxonomy. Funding for these projects is provided by the Sandler Family Foundation, the National Institutes of Health (RO1 CA052855 and U01 AI075641), the NIGMS (MBRS GM058903), and the California Institute for Quantitative Biosciences. We also thank Dr. W. Inman for his insightful conversations and support of this study, the Captain and Crew of the M/V Golden Dawn for their assistance in sponge collection, and Dr. T. Matainaho of the University of Papua New Guinea for his assistance with collection permits.
Footnotes
Supporting Information Available: NMR and MS spectra, sponge photographs, isolation scheme and table of optical rotations and absolute configurations of known norterpene peroxides. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.World Health Organization: Human African Trypanosomiasis. 2008 Sept 12; http://www.who.int/trypanosomiasis_african/en/index.html.
- 2.Mallari JP, Shelat AA, Obrien T, Caffrey CR, Kosinski A, Connelly M, Harbut M, Greenbaum D, McKerrow JH, Guy RK. J Med Chem. 2008;51:545–552. doi: 10.1021/jm070760l. [DOI] [PubMed] [Google Scholar]
- 3.Kennedy PG. Annal Neurol. 2008;64:116–126. doi: 10.1002/ana.21429. [DOI] [PubMed] [Google Scholar]
- 4.Barrett MP, Boykin DW, Brun R, Tidwell RR. Br J Pharmacol. 2007;152:1155–1171. doi: 10.1038/sj.bjp.0707354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mallari JP, Shelat A, Kosinski A, Caffrey CR, Connelly M, Zhu F, McKerrow JH, Guy RK. Bioorg Med Chem Lett. 2008;18:2883–2885. doi: 10.1016/j.bmcl.2008.03.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Koning HP. Trends Parasitol. 2008;24:345–349. doi: 10.1016/j.pt.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 7.Das BP, Boykin DW. J Med Chem. 1977;20:531–536. doi: 10.1021/jm00214a014. [DOI] [PubMed] [Google Scholar]
- 8.Crews P, Hunger TM. Marine Biotechnology. In: Attaway DH, Zaborsky OR, editors. Pharmaceutical and Bioactive Natural Products. Vol. 1. Plenum Press; New York: 1993. pp. 343–389. [Google Scholar]
- 9.a) Quinoa E, Crews P. Tetrahedron Lett. 1987;28:3229–3232. [Google Scholar]; b) Arabshahi L, Schmitz FJ. J Org Chem. 1987;57:3584–3586. [Google Scholar]
- 10.Urbaniak MD, Tabudravu JN, Msaki A, Matera KM, Brenk R, Jaspars M, Ferguson MA. Bioorg Med Chem Lett. 2006;16:5744–1547. doi: 10.1016/j.bmcl.2006.08.091. [DOI] [PubMed] [Google Scholar]
- 11.The term “peroxiterpene” derived from “peroxide” and “terpene” is defined here for the first time. It unifies a growing class of stable, often bioactive, terepenes possessing such functionality.
- 12.a) Sperry S, Valeriote FA, Corbett TH, Crews P. J Nat Prod. 1998;61:241–247. doi: 10.1021/np970467w. [DOI] [PubMed] [Google Scholar]; b) Ibrahim SRM, Ebel R, Wray V, Mueller WEG, Edrada-Ebel R, Proksch P. J Nat Prod. 2008;71:1358–1364. doi: 10.1021/np800102u. [DOI] [PubMed] [Google Scholar]
- 13.El Sayed KA, Hamann MT, Hashish NE, Shier WT, Kelly M, Khan AA. J Nat Prod. 2001;64:522–524. doi: 10.1021/np000529+. [DOI] [PubMed] [Google Scholar]
- 14.Youssef DTA, Yoshida WY, Kelly M, Scheuer PJ. J Nat Prod. 2001;64:1332–1335. doi: 10.1021/np010184a. [DOI] [PubMed] [Google Scholar]
- 15.Albericci M, Collartlempereur M, Braekman JC, Daloze D, Tursch B, Declercq JP, Germain G, Vanmeerssche M. Tetrahedron Lett. 1979;20:2687–2690. [Google Scholar]
- 16.Albericci M, Braekman JC, Daloze D, Tursch B. Tetrahedron. 1982;38:1881–1890. [Google Scholar]
- 17.vanSoest RWM, Boury-Esnault N, Hooper JNA, Rützler K, de Voogd NJ, Alvarez B, Hadju E, Pisera AB, Vacelet J, Manconi R, Schoenberg C, Janussen D, Tabanick KR, Klautau M. World Porifera Database. 2008 Available online at http://www.marinespecies.org/porifera. Consulted on 2008-10-19.b) According to the World Porifera Database, sponges classified as Sigmosceptrella laevis are now accepted as Diacarnus laevis.
- 18.Capon RJ, Macleod JK. Tetrahedron. 1985;41:3391–3404. [Google Scholar]
- 19.Crews P, Rodriguez J, Jaspars M. Organic Structure Analysis. Oxford University Press, Inc; New York, NY: 1998. [Google Scholar]
- 20.Kuehnel E, Laffan DDR, Lloyd-Jones GC, del Campo TM, Shepperson IR, Slaughter JL. Angew Chem, Int Ed. 2007;46:7075–7078. doi: 10.1002/anie.200702131. [DOI] [PubMed] [Google Scholar]
- 21.Yanai M, Ohta S, Ohta E, Hirata T, Ikegami S. Bioorg Med Chem. 2003;11:1715–1721. doi: 10.1016/s0968-0896(03)00030-0. [DOI] [PubMed] [Google Scholar]
- 22.Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]
- 23.a) D’Ambrosio M, Guerriero A, Deharo E, Debitus C, Munoz V, Pietra F. Helv Chim Acta. 1998;81:1285–1292. [Google Scholar]; b) Ovenden SPB, Capon RJ. J Nat Prod. 1999;62:214–218. doi: 10.1021/np980223r. [DOI] [PubMed] [Google Scholar]; c) Phuwapraisirisan P, Matsunaga S, Fusetani N, Chaitanawisuti N, Kritsanapuntu S, Menasveta P. J Nat Prod. 2003;66:289–291. doi: 10.1021/np020417d. [DOI] [PubMed] [Google Scholar]; d) Tanaka J, Higa T, Suwanborirux K, Kokpol U, Bernardinelli G, Jefford CW. J Org Chem. 1993;58:2999–3002. [Google Scholar]; e) Capon RJ, Macleod JK, Coote SJ, Davies SG, Gravatt GL, Dordorhedgecock IM, Whittaker M. Tetrahedron. 1988;44:1637–1650. [Google Scholar]; f) Capon RJ, Macleod JK, Willis AC. J Org Chem. 1987;52:339–342. [Google Scholar]
- 24.Kossuga MH, Nascimento AM, Reimao JQ, Tempone AG, Taniwaki NN, Veloso K, Ferreira AG, Cavalcanti BC, Pessoa C, Moraes MO, Mayer AMS, Hajdu E, Berlinck RGS. J Nat Prod. 2008;71:334–339. doi: 10.1021/np0705256. [DOI] [PubMed] [Google Scholar]
- 25.Lim CW, Kim YK, Youn HD, Park HY. Agric Chem Biotechnol (Engl Ed) 2006;49:21–23. [Google Scholar]
- 26.Kaiser M, Wittlin S, Nehrbass-Stuedli A, Dong Y, Wang X, Hemphill A, Matile H, Brun R, Vennerstrom JL. Antimicrob Agents Chemother. 2007;51:2991–2993. doi: 10.1128/AAC.00225-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mishina YV, Krishna S, Haynes RK, Meade JC. Antimicrob Agents Chemother. 2007;51:1852–1854. doi: 10.1128/AAC.01544-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.