

Abstract
Activation of androgen receptor (AR) may play a role in the development of castration resistant prostate cancer. Two intracellular tyrosine kinases, Ack1 (activated cdc42-associated kinase) and Src, phosphorylate and enhance AR activity and promote prostate xenograft tumor growth in castrated animals. However, the upstream signals that activate these kinases and lead to AR activation are incompletely characterized. In this study, we investigated AR phosphorylation in response to non-androgen ligand stimulation using phospho-specific antibodies. Treatment of LNCaP and LAPC-4 cells with epidermal growth factor (EGF), heregulin, Gas6 (ligand binding to Mer receptor tyrosine kinase and activating Ack1 downstream), interleukin (IL)-6 or bombesin stimulated cell proliferation in the absence of androgen. Treatment of LNCaP and LAPC-4 cells with EGF, heregulin, or Gas6 induced AR phosphorylation at Tyr-267; IL-6 or bombesin treatment did not. AR phosphorylation at Tyr-534 was induced by treatment with EGF, IL-6 or bombesin, but not by heregulin or Gas6. siRNA-mediated knockdown of Ack1 or Src showed that Ack1 mediates heregulin- and Gas6-induced AR Tyr-267 phosphorylation whereas Src mediates Tyr-534 phosphorylation induced by EGF, IL-6, and bombesin. Dasatinib, a Src inhibitor, blocked EGF-induced Tyr-534 phosphorylation. In addition, we show dasatinib also inhibited Ack1 kinase. Dasatinib inhibited heregulin-induced Ack1 kinase activity and AR Tyr-267 phosphorylation. Dasatinib inhibited heregulin-induced AR-dependent reporter activity. Dasatinib also inhibited heregulin-induced expression of endogenous AR target genes. Dasatinib inhibited Ack1-dependent colony formation and prostate xenograft tumor growth in castrated mice. Interestingly, Ack1 or Src knockdown or dasatinib did not inhibit EGF-induced AR Tyr-267 phosphorylation or EGF-stimulated AR activity, suggesting the existence of an additional tyrosine kinase that phosphorylates AR at Tyr-267. These data suggest that specific tyrosine kinases phosphorylate AR at distinct sites and that dasatinib may exert anti-tumor activity in prostate cancer through inhibition of Ack1.
Keywords: androgen receptor, phosphorylation, Ack1 kinase, dasatinib, prostate cancer
Introduction
Androgen deprivation therapy through surgical or medical castration is commonly used as a systemic therapy of advanced prostate cancer because it is initially effective in causing tumor regression and palliating cancer-related symptoms. However, virtually all patients eventually progress and develop hormone refractory prostate cancer or castration resistant prostate cancer (CRPC), a terminal disease with poor prognosis. Substantial evidence indicates that androgen receptor (AR) activation in the environment of low circulating testosterone plays a critical role in the development of CRPC. Studies using xenograft tumor models demonstrate that prostate xenograft tumors that recur following castration express AR-dependent genes (Gregory et al 1998). In these cells, the AR protein is stabilized and constitutively localized to the nucleus and is hypersensitive to low levels of androgen (Gregory et al 2001). Overexpression of AR enhanced the ability of androgen-dependent prostate xenografts to form tumor in castrated animals while knockdown of AR inhibited their tumorigenicity (Chen et al 2004). Multiple mechanisms have been shown to activate AR in CRPC. These include AR gene amplification, increased AR expression, AR point mutations frequently occurring in the ligand binding domain that broaden ligand specificity, overexpression of AR coactivators, and intratumoral production of androgen (Scher and Sawyers 2005).
In addition to these mechanisms, AR may be activated by crosstalk with signaling pathways initiated by cell surface receptors binding to growth factors, cytokines, and neuropeptides. Intracellular pathways involving mitogen activated protein kinase (MAPK) or phosphatidylinositol 3-kinase/Akt have been proposed to mediate activation of AR downstream of receptor tyrosine kinases (Gregory et al 2004, Wang et al 2007). For example, epidermal growth factor (EGF) receptor enhances AR transcriptional activity through increasing the interaction between AR and the steroid receptor coactivator TIF2 secondary to MAPK-dependent phosphorylation of TIF2 (Gregory et al 2004). HER-2 (ErbB2) activates the AR transcriptional function by increasing protein stability, recruitment and DNA binding (Liu et al 2005, Mellinghoff et al 2004). AR protein is phosphorylated at multiple serine/threonine residues and phosphorylation at some of these sites has been proposed to regulate nuclear localization and export (Gioeli et al 2002, Gioeli et al 2006, Ponguta et al 2008). Recently, several groups reported that tyrosine phosphorylation of AR protein by nonreceptor tyrosine kinases Src and Ack1 (activated cdc42-associated kinase) may play a role in AR activation in the low androgen environment, thereby promoting the development of CRPC (Guo et al 2006, Kraus et al 2006, Mahajan et al 2007). Src-mediated phosphorylation of AR at Tyr-534 resulted in activation of AR and nuclear translocation and DNA binding in the absence of androgen (Guo et al 2006, Kraus et al 2006). Our previous work identified Ack1 as a protein activated downstream of Mer receptor tyrosine kinase in prostate cancer cells (Mahajan et al 2005). Furthermore, we demonstrated that expression of activated Ack1 increased prostate xenograft tumor growth in castrated animals as well as AR target gene expression and AR recruitment to the enhancers of target genes at suboptimal androgen concentrations (Mahajan et al 2007). Ack1 interacted with and phosphorylated AR protein at Tyr-267 and Ack1 was shown to be required for optimal AR target gene expression and AR recruitment. Tyrosine phosphorylated AR protein was expressed in 8 out of 18 primary CRPC tumor samples and its expression was correlated with Ack1 activation in the same tumors. In this report, we investigate the upstream signals activating AR phosphorylation and we show that Src and Ack1 kinases each contribute to site-specific phosphorylation of AR.
Results
Site-specific phosphorylation of androgen receptor by Ack1 and Src kinases
Previous reports demonstrated that AR protein may be phosphorylated at Tyr-534 or Tyr-267 residues (Guo et al 2006, Kraus et al 2006, Mahajan et al 2007). To elucidate signaling pathways leading to AR tyrosine phosphorylation, phospho-specific antibodies against AR protein phosphorylated at these residues were generated. Their specificity was tested against wildtype and phosphorylation-site mutant AR. Wildtype AR, Y267F AR, or Y534F AR expression vectors were co-transfected along with the activated Ack1 or activated Src kinase expression vector. The phospho-Y267 specific antibody detected AR only when Ack1 was co-expressed and did not detect the AR Y267F mutant protein in cell lysates co-expressing Ack1 (Fig. 1A). The phospho-Y534 specific antibody detected AR only when Src was co-expressed and did not recognize the AR Y534F mutant protein in the presence of Src (Fig. 1B). Co-expression of Ack1 led to phosphorylation of Tyr-267 but not Tyr-534 of AR. Conversely, co-expression of Src resulted in phosphorylation of Tyr-534 but not Tyr-267 of AR. These data suggest that Ack1- and Src-mediated phosphorylation of AR is site-specific (i.e. Ack1 targets Tyr-267 but not Tyr-534 and vice versa for Src).
Figure 1. Phospho-specific antibodies demonstrate that AR is phosphorylated at Tyr-267 and Tyr-534 after ligand stimulation.
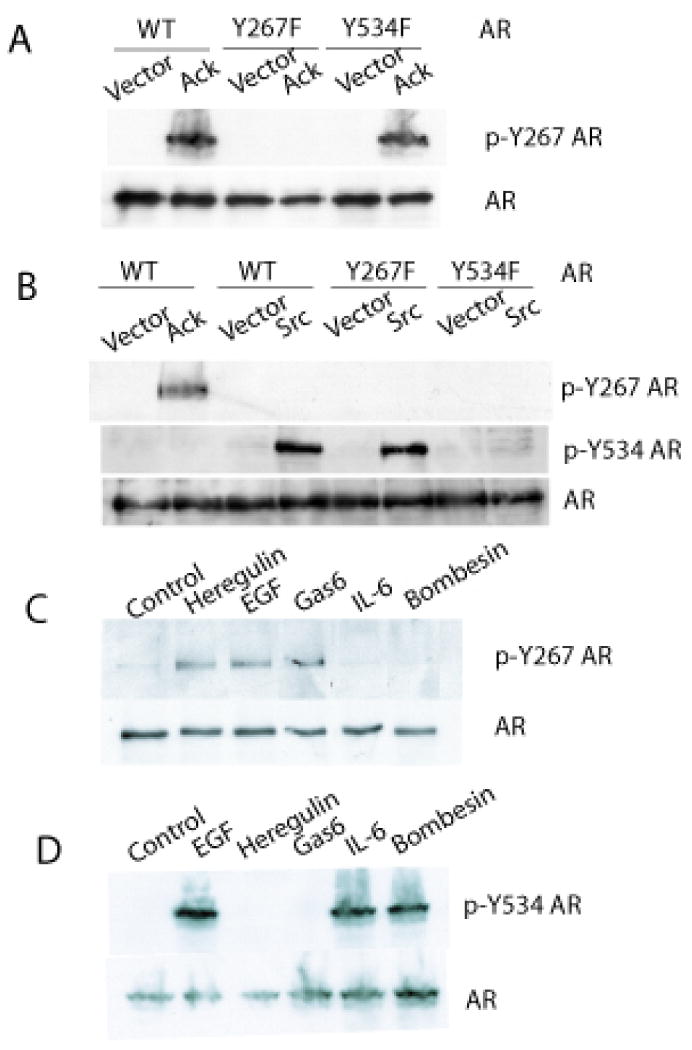
(A) 293T cells were transfected with the expression vector encoding wildtype AR or Y267F AR or Y534F AR along with constitutively active Ack1 or empty vector. After 24 hours, protein extracts were immunoblotted with the antibody specific for phospho-Y267 AR or total AR. (B) COS7 cells were transfected with the vector encoding wildtype AR or Y267F AR or Y534F AR along with constitutively active Ack1 or activated Src or empty vector, as indicated. After 24 hours, protein extracts were immunoprecipitated with the AR antibody, then immunoblotted with the antibody specific for phospho-Y267 AR or phospho-Y534 AR or total AR, as indicated. (C) LNCaP cells were treated with EGF (100 ng/ml), heregulin (10 ng/ml), Gas6 (100 ng/ml), IL-6 (10 ng/ml), or bombesin (1 nM) for 60 min. Protein extracts were immunoblotted with the antibody specific for phospho-Y267 AR or total AR. (D) LNCaP cells were treated as above. Protein extracts were immunoprecipitated with the AR antibody, then immunoblotted with the antibody specific for phospho-Y534 AR. Blots shown are representative of three independent experiments.
Phosphorylation of androgen receptor by ligand stimulation
Tyrosine phosphorylation of endogenous AR in prostate cancer cells induced by physiologic ligand stimulation was characterized using the phospho-specific antibodies. Extracellular ligands that bind to the cell surface receptors and have previously been reported to enhance AR activity or may potentially enhance AR activity were tested for their ability to induce AR tyrosine phosphorylation. Gas6 is the ligand for Mer receptor tyrosine kinase, which we have previously shown to activate Ack1 in prostate cells (Mahajan et al 2005). Heregulin binds to HER-3 and activates HER-2 kinase through the formation of the HER-2/HER-3 heterodimer. Bombesin (also known as gastrin-releasing peptide) is a neuropeptide produced by neuroendocrine cells, which has been reported to support growth of androgen-dependent LNCaP cells in an androgen-depleted condition through Src-mediated activation of AR (Desai et al 2006, Gong et al 2006, Yang et al 2009). Interleukin-6 leads to ligand-independent activation of the AR N-terminal domain that involves MAPK and steroid receptor coactivator-1 (Ueda et al 2002b). Treatment of LNCaP cells with EGF, heregulin, or Gas6 induced phosphorylation of the AR protein at Tyr-267 (Fig. 1C). AR phosphorylation was transient and peaked at 60-90 min. and returned to the basal level by 4 hrs (data not shown). Treatment with IL-6 or bombesin did not induce phosphorylation of the AR at Tyr-267. Phosphorylation of the AR at Tyr-534 was induced by treatment with EGF, IL-6 or bombesin, but not with heregulin or Gas6 (Fig. 1D). EGF was the only ligand tested that induced AR phosphorylation at both Tyr-267 and Tyr-534 sites, in contrast to other ligands that produced site-specific AR phosphorylation (i.e. heregulin and Gas6 at Tyr-267 and IL-6 and bombesin at Tyr-534). LAPC-4 prostate cancer cells that express wildtype AR endogenously (Klein et al 1997) were treated with these ligands and AR phosphorylation at Tyr-267 and Tyr-534 sites was analyzed similarly. LAPC-4 cells exhibited the same pattern of AR phosphorylation by these ligands as LNCaP cells (Supplementary Fig. 1).
Stimulation of cell proliferation by non-androgen ligands
Since cell proliferation requires AR activation in androgen-dependent prostate cancer cells, the effect of these ligands inducing AR phosphorylation on cell proliferation was determined. In LNCaP cells, treatment with EGF, heregulin, Gas6, bombesin, or IL-6 all stimulated cell proliferation to similar levels as the androgen dihydrotestosterone (DHT) (Fig. 2). In LAPC-4 cells, all ligands stimulated cell proliferation although Gas6 and IL-6 were less effective than DHT or other ligands. These results are in agreement with previous reports of stimulation of proliferation in the absence of androgen by heregulin (Gregory et al 2005), bombesin (Desai et al 2006, Lee et al 2001), or IL-6 (Ueda et al 2002a). These data demonstrate that these non-androgen ligands inducing AR phosphorylation promote prostate cancer cell proliferation in the absence of androgen.
Figure 2. EGF, heregulin, Gas6, bombesin, and IL-6 stimulate proliferation of androgen-dependent prostate cancer cell lines in the absence of androgen.
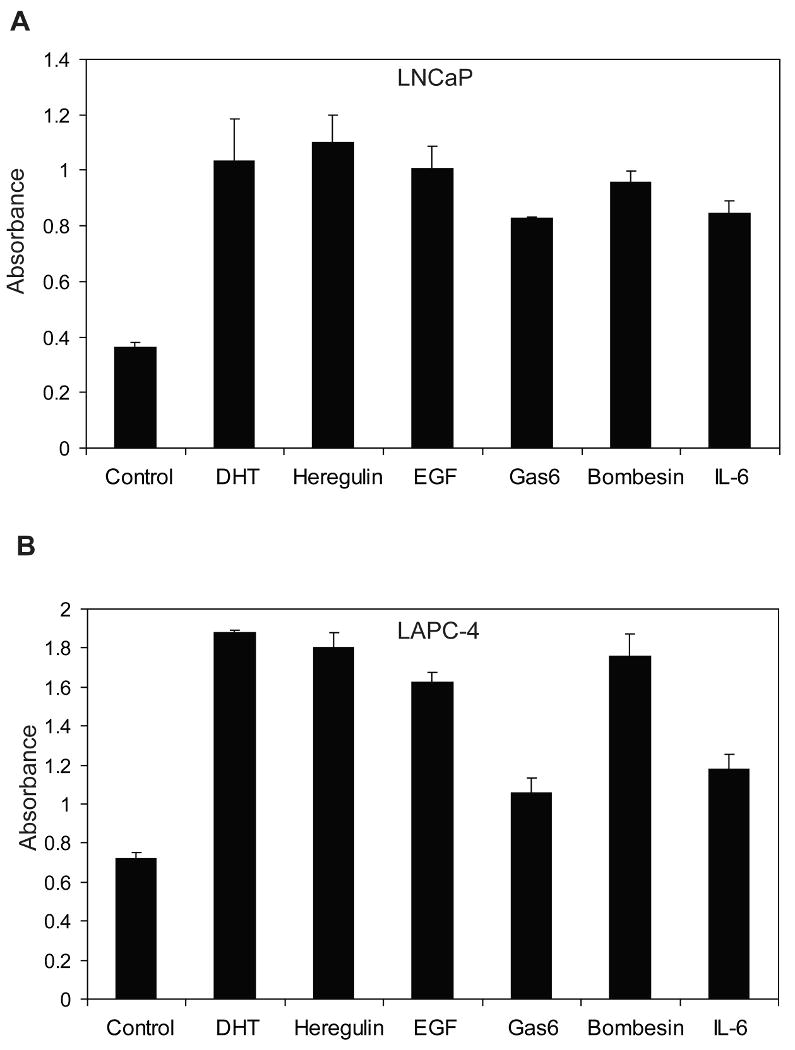
Cells were seeded in serum-free medium in triplicate wells of a 96-well plate and treated with EGF (100 ng/ml), heregulin (10 ng/ml), Gas6 (100 ng/ml), IL-6 (10 ng/ml), or bombesin (1 nM) for 72 hours. Relative cell proliferation was determined by addition of colorimetric dye WST-8 and measuring absorbance at 450 nM. (A) LNCaP cells. (B) LAPC-4 cells. Data shown are representative of three similar independent experiments. All treatment conditions are significantly different from untreated control with p <0.05 by t-test.
Dasatinib inhibits Ack1 kinase activity but does not inhibit EGF-induced AR phosphorylation at Tyr-267
For characterization of signaling pathways and therapeutic applications, a small molecule kinase inhibitor of Ack1 would be desirable. A previous report indicated that dasatinib, a potent inhibitor of Src and Abl kinases, interacted with several additional tyrosine kinases in a competition binding assay, including Ack1 (Carter et al 2005). The ability of dasatinib to inhibit Ack1 autophosphorylation as a marker of kinase activity was tested in 293T cells transfected to express activated Ack1 (Fig. 3A). Dasatinib inhibited Ack1 autophosphorylation in a dose dependent manner, with dasatinib doses of 10 nM or greater demonstrating nearly complete inhibition of Ack1 phosphorylation, consistent with the previously reported Kd of 6 nM (Carter et al 2005). Treatment of LNCaP cells with dasatinib inhibited heregulin-induced Ack1 phosphorylation almost completely at doses of 10 nM or greater (Fig. 3B). Dasatinib treatment inhibited heregulin-induced AR phosphorylation at Tyr-267. The concentration of dasatinib required for inhibition of AR phosphorylation at Tyr-267 was similar to that for inhibition of heregulin-induced Ack1. This result is consistent with the notion that heregulin-induced activation of Ack1 mediates AR phosphorylation at Tyr-267. The effect of dasatinib on EGF-induced phosphorylation of AR was tested. EGF-induced AR phosphorylation at Tyr-534 was inhibited by dasatinib (Fig. 4A). Since dasatinib inhibits Src, this finding is consistent with the idea that Src activation downstream of EGF receptor leads to AR phosphorylation at Tyr-534. Dasatinib had no effect on EGF-induced AR phosphorylation at Tyr-267, even at doses as high as 1000 nM (Fig. 4B). However, Src activation after EGF treatment was inhibited by dasatinib at a dose as low as 1 nM (Fig. 4D). In LNCaP cells, Ack1 was not activated after EGF treatment while heregulin-induced Ack1 activation was detected (Fig. 4C). These results suggest that at least one other tyrosine kinase (other than Src and Ack1) mediates AR phosphorylation at Tyr-267 after EGF treatment in LNCaP cells and that this kinase (or kinases) is insensitive to dasatinib.
Figure 3. Dasatinib inhibits Ack1 kinase activity and inhibits heregulin-induced AR phosphorylation at Tyr-267.
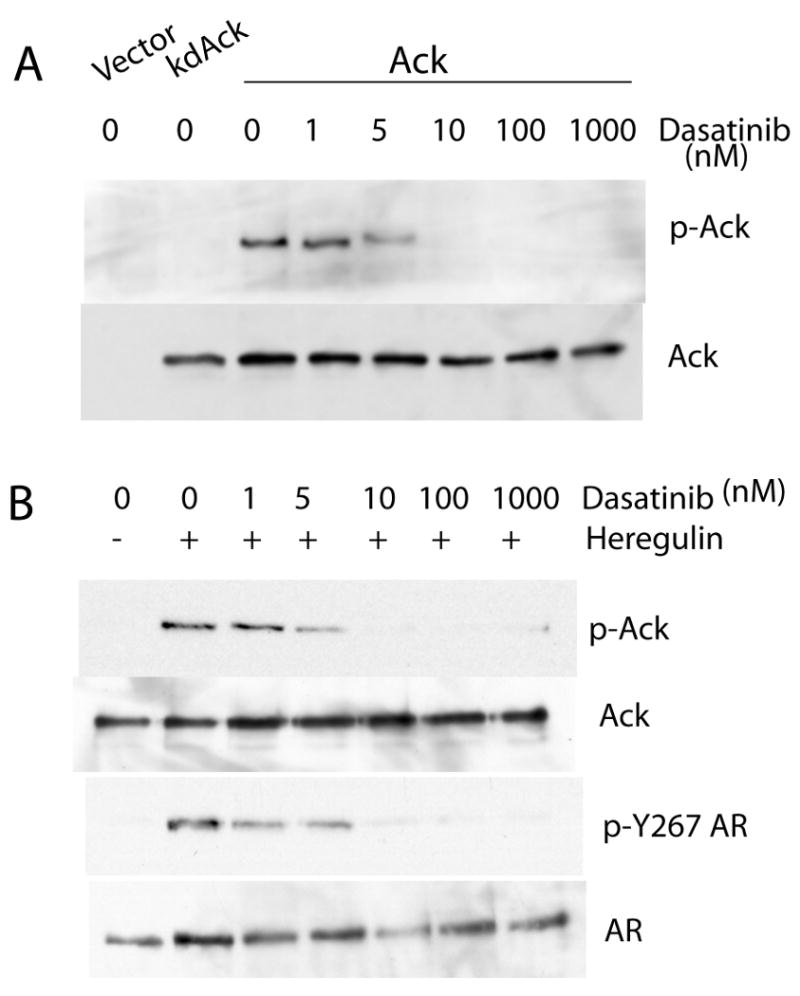
(A) 293T cells were transfected with empty vector or kinase-dead Ack1 or constitutively active Ack1. After 24 hours, cells were treated with increasing concentrations of dasatinib for 2 hours. Protein extracts were immunoblotted with the antibody against phospho-Ack1 or total Ack1. (B) LNCaP cells were pre-treated with dasatinib at increasing concentrations for 2 hours, then treated with heregulin (10 ng/ml) for 60 min. Protein extracts were immunoblotted with the antibody against phospho-Y267 AR or total AR or phospho-Ack1 or total Ack1. Blots shown are representative of three independent experiments.
Figure 4. Dasatinib inhibits EGF-induced AR phosphorylation at Tyr-534 but not Tyr-267.
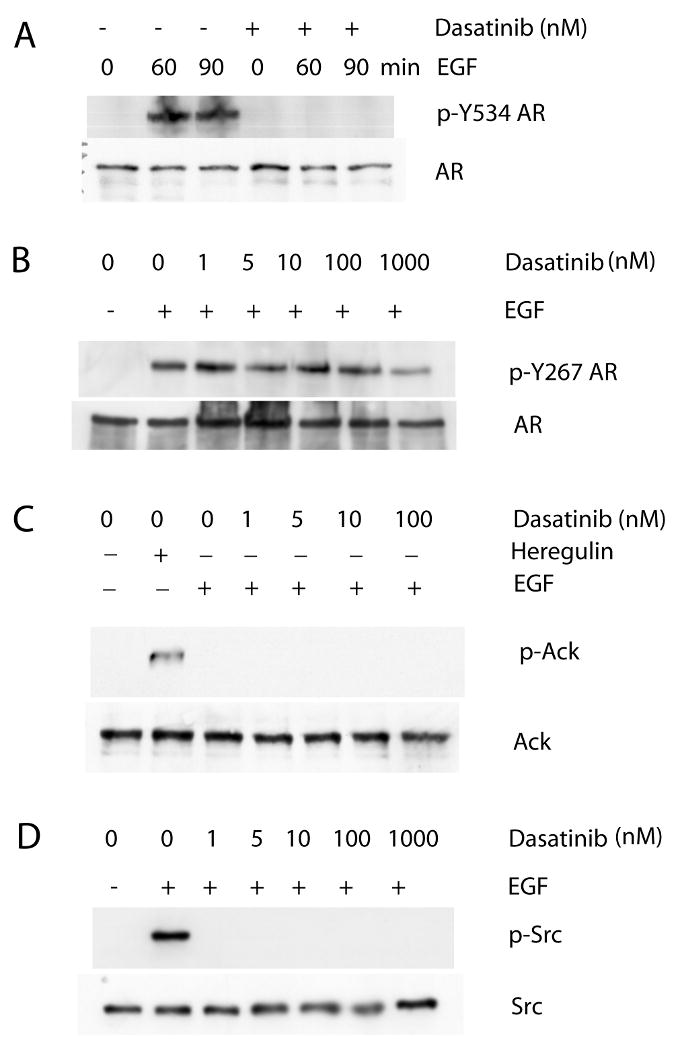
(A) LNCaP cells were pre-treated with dasatinib (10 nM) for 2 hours, then treated with EGF (100 ng/ml) for 60 or 90 min. Protein extracts were immunoprecipitated with the AR antibody, then immunoblotted with the antibody specific for phospho-Y534 AR or total AR. (B) LNCaP cells were pre-treated with dasatinib at increasing concentrations for 2 hours, then treated with EGF (100 ng/ml) for 60 min. Protein extracts were immunoblotted with the antibody against phospho-Y267 AR or total AR. (C, D) LNCaP cells were treated as above. Cells were also treated with heregulin (10 ng/ml) for 60 min as a positive control of Ack1 activation. Protein extracts were immunoblotted with the antibody against phospho-Ack1 or total Ack1 or phospho-Src or total Src. Blots shown are representative of three independent experiments.
Role of Ack1 and Src in ligand-stimulated AR phosphorylation
To investigate more directly the role of Ack1 or Src in AR phosphorylation after ligand stimulation, the effect of Ack1 or Src knockdown by siRNA was tested (Fig. 5). Ack1 knockdown inhibited AR phosphorylation at Tyr-267 induced by heregulin or Gas6 treatment. Src knockdown inhibited AR phosphorylation at Tyr-534 induced by EGF or IL-6 or bombesin treatment. However, EGF-induced AR phosphorylation at Tyr-267 was not inhibited by Ack1 knockdown or Src knockdown, despite markedly reduced expression levels of Ack1 or Src by siRNA confirmed by immunoblotting. These data suggest that downstream of cell surface receptors, Ack1 mediates AR tyrosine phosphorylation at Tyr-267 and Src mediates AR tyrosine phosphorylation at Tyr-534. However, EGF-induced AR phosphorylation at Tyr-267 likely involves an additional kinase(s) that is not inhibited by dasatinib.
Figure 5. Knockdown of Src or Ack1 demonstrates the existence of an additional kinase that induces AR phosphorylation at Tyr-267 after EGF stimulation.
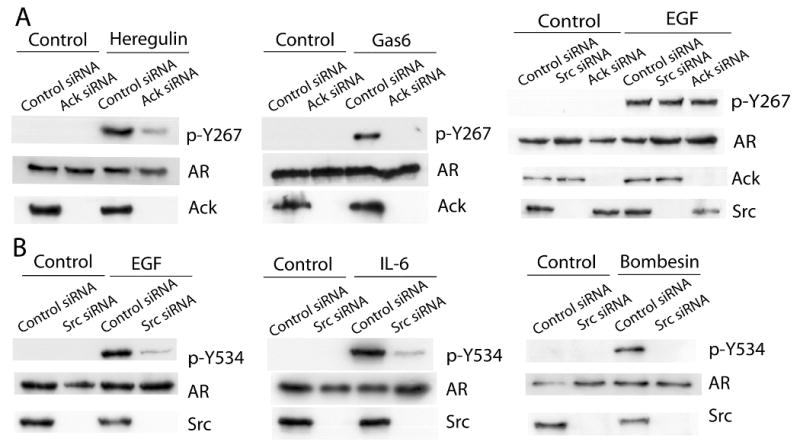
LNCaP cells were transfected with control or Src-specific or Ack1-specific siRNA. After 24 hours, cells were treated with heregulin, Gas6, EGF, IL-6, or bombesin, as indicated, for 60 min. (A) Protein extracts were immunoblotted with the antibody specific for phospho-Y267 AR or total AR. (B) Protein extracts were immunoprecipitated with the AR antibody, then immunoblotted with the antibody specific for phospho-Y534 AR. Protein extracts were also immunoblotted with the antibody specific for Src or Ack1. Blots shown are representative of at least three independent experiments.
Effect of dasatinib on Ack1-induced AR activation
The biological effect of dasatinib on prostate cancer cells was investigated. First, LNCaP cells transfected with the AR-dependent reporter ARR2-PB-luciferase were investigated. Treatment of these cells with heregulin or EGF (without androgen) increased luciferase activity. AR antagonist bicalutamide inhibited androgen-induced AR reporter activity but did not inhibit heregulin- or EGF-induced AR reporter activity (Supplementary Fig. 2). Therefore, heregulin- or EGF-induced AR activation likely reflects ligand-independent activation of AR via receptor tyrosine kinase-mediated pathways. Dasatinib treatment inhibited heregulin-induced AR reporter activity with the IC50 of 4.9 nM, consistent with the hypothesis that the stimulatory effect of heregulin is mediated by downstream Ack1 activation (Fig. 6A). However, dasatinib treatment had no effect on EGF-induced AR reporter activity, even at doses as high as 1000 nM, in contrast to its inhibitory effect on heregulin-mediated AR reporter activity (Fig. 6B). The effect of dasatinib on expression of endogenous AR target genes prostate specific antigen (PSA) and human kallikrein 2 (hK2) (also known as KLK2) was tested. Treatment of LNCaP cells with EGF or heregulin increased PSA and hK2 mRNA levels by 4-6 fold. Dasatinib treatment partially blocked heregulin-induced expression of PSA and hK2 mRNA but not EGF-induced expression of PSA and hK2 mRNA (Fig. 6C and D). Similar results were seen in LAPC-4 cells (Supplementary Fig. 3). Since heregulin-induced Tyr-267 phosphorylation is blocked by dasatinib, but EGF-induced Tyr-267 phosphorylation is not blocked by dasatinib, these results suggest that the stimulatory effect of growth factors on AR is correlated with induction of Tyr-267 phosphorylation, either by Ack1 or an unidentified tyrosine kinase downstream of EGF.
Figure 6. Dasatinib inhibits heregulin-induced AR activity, but not EGF-induced AR activity.
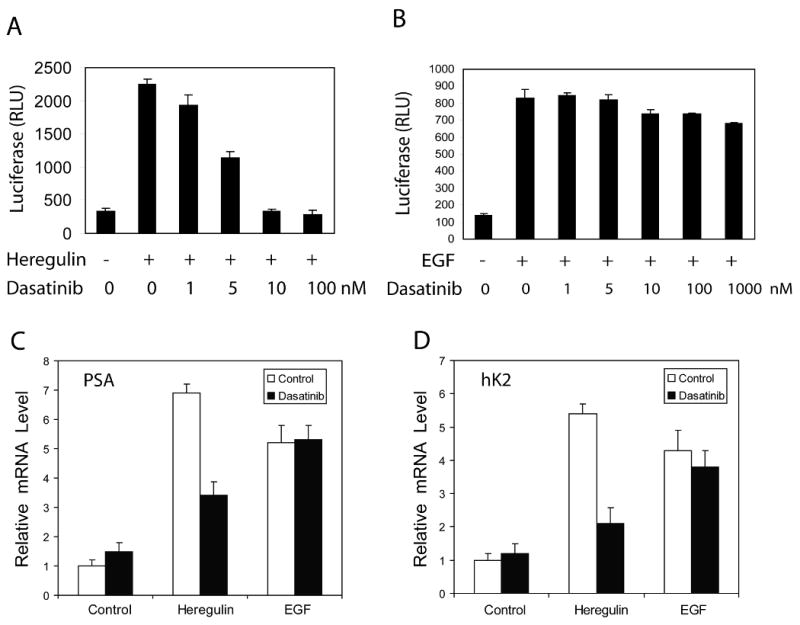
(A, B) LNCaP cells were transfected with the AR reporter ARR2-PB-luciferase. After overnight incubation, cells were pre-treated with dasatinib for 2 hrs, then with heregulin (10 ng/ml) (A) or EGF (100 ng/ml) (B) for 20 hrs. Cell extracts were assayed for luciferase activity. The mean and standard deviation of triplicate samples of a representative experiment are shown from three independent experiments with similar results. (C, D) LNCaP cells were pre-treated with dasatinib (10 nM) for 2 hrs, then with heregulin (10 ng/ml) or EGF (100 ng/ml) for 20 hrs. RNA was isolated and quantitative RT-PCR was performed to determine the levels of PSA (C) or hK2 (D) mRNA. Data shown are representative of two independent experiments with similar results.
Effect of dasatinib on Ack1-induced colony formation and xenograft tumor growth
Activated Ack1 enhances the ability of LNCaP cells to form colonies in soft agar and grow as subcutaneous xenograft tumor in castrated mice (Mahajan et al 2005, Mahajan et al 2007). The effect of dasatinib on Ack1-induced colony formation was determined. Dasatinib treatment decreased the number of soft agar colonies in LNCaP cells expressing activated Ack1 in a dose-dependent manner (Fig. 7A). Dasatinib also inhibited soft agar colony growth of LNCaP vector control cells, likely through inhibition of Src kinase expressed in LNCaP (Park et al 2008). We investigated the effect of dasatinib treatment in vivo on the xenograft tumor growth of LNCaP-Ack1 cells in castrated mice. Castrated mice with xenograft tumors were treated with dasatinib orally or vehicle control and the tumor volume was monitored. Administration of dasatinib resulted in significant inhibition of xenograft tumor growth. At day 25 and 29, the tumor volume of the dasatinib treatment group was decreased by approximately 50% compared to the tumor volume of the control group (Fig. 7B). The phosphorylation status of Ack1 and AR proteins was determined by immunoblotting of protein extracts of harvested tumor tissues at the conclusion of the experiment. In tumors harvested from control animals, Ack1 and AR at Tyr-267 were constitutively phosphorylated, whereas in tumors from dasatinib treated animals, Ack1 and AR proteins were not phosphorylated (Fig. 7C). These data suggest that inhibition of Ack1 kinase by dasatinib leads to loss of AR phosphorylation and decreased xenograft tumor growth.
Figure 7. Dasatinib inhibits anchorage-independent colony formation and xenograft tumor growth of LNCaP-Ack1 cells in castrated mice.
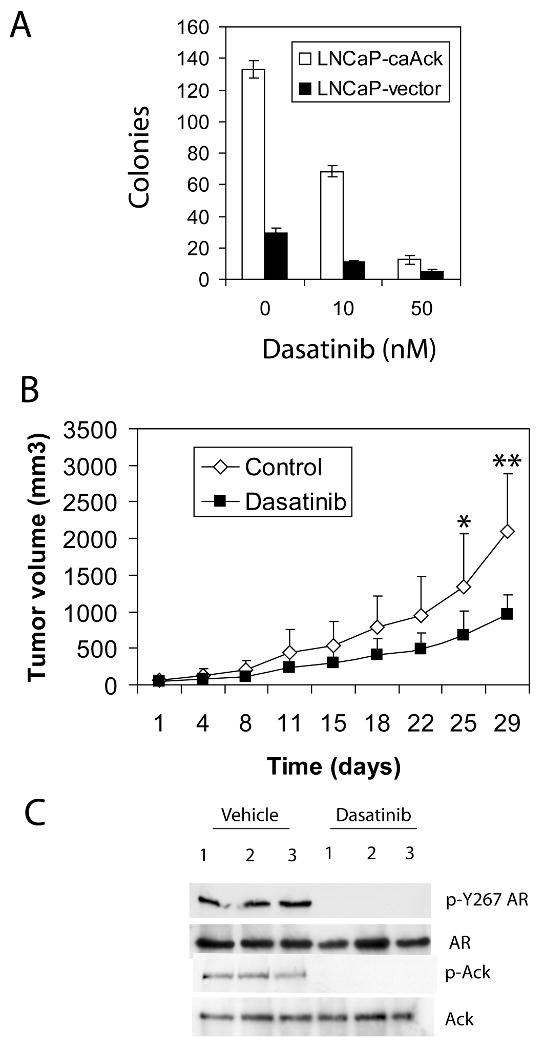
(A) LNCaP-Ack1 cells and vector control cells were plated in soft agar with indicated concentrations of dasatinib in the top layer and incubated for 21 days. Colonies were stained and counted. The mean and standard deviation of triplicate wells of a representative experiment are shown from three independent experiments with similar results. (B) Castrated mice were implanted subcutaneously with LNCaP-Ack1 cells. When tumors became palpable, mice were treated with vehicle control (n=7) or dasatinib (n=8) via oral gavage at a dose of 30 mg/kg bid and the tumor volume was measured twice per week. (* indicates p<0.04 and ** indicates p<0.009.) (C) Three independent tumors were harvested from mice given vehicle only or dasatinib at the conclusion of the experiment. Protein extracts were immunoblotted for expression of total Ack1, phospho-Ack1, total AR and phospho-Y267 AR.
Discussion
In this report, we demonstrate that Ack1 and Src tyrosine kinases target distinct AR phosphorylation sites (i.e. Tyr-267 by Ack1 and Tyr-534 by Src) after activation by cell surface receptors. EGF, IL-6, and bombesin-induced AR phosphorylation at Tyr-534 was inhibited by Src knockdown and dasatinib, a potent Src inhibitor, suggesting Src involvement downstream of these ligands. Activation of AR by EGF and bombesin through Src had been reported previously (Desai et al 2006, Guo et al 2006, Kraus et al 2006). A recent report indicated that the neuroendocrine-derived peptide parathyroid hormone-related protein stabilizes AR by reducing interaction with the ubiquitin ligase CHIP through Src-mediated phosphorylation of AR at Tyr-534 (DaSilva et al 2009). Involvement of Ack1 downstream of heregulin and Gas6 in AR phosphorylation at Tyr-267 was confirmed by Ack1 knockdown as well as dasatinib, which has been shown to be an inhibitor of Ack1 in this work. However, inhibition of Src and Ack1 did not prevent EGF-induced AR tyrosine phosphorylation at Tyr-267, suggesting the existence of an additional unidentified tyrosine kinase (or kinases) capable of phosphorylating AR at Tyr-267 (Fig. 8). Although identification and understanding the functional role of this kinase on AR signaling will require further work, it is clear that Tyr-267 phosphorylation is involved in some modes of AR transactivation no matter which tyrosine kinase is involved. Inhibition of growth factor-induced AR activity by dasatinib correlates with loss of AR phosphorylation at Tyr-267, as dasatinib inhibits heregulin-induced AR Tyr-267 phosphorylation and AR reporter activity and target gene expression. In contrast, EGF-induced AR Tyr-267 phosphorylation and AR reporter activity and target gene expression are resistant to dasatinib. This result raises a possibility that phosphorylation of Tyr-267 is required for AR activation downstream of receptor tyrosine kinases.
Figure 8. Model of AR phosphorylation by non-androgen ligands.
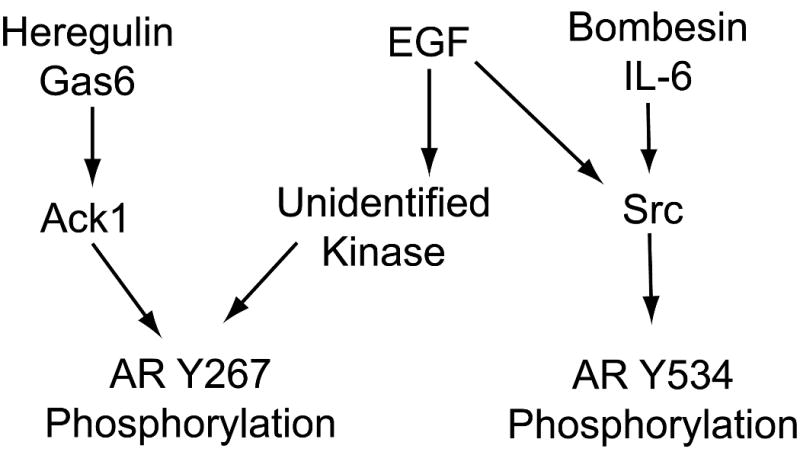
Heregulin and Gas6 activate Ack1 kinase, which leads to AR Tyr-267 phosphorylation. EGF, bombesin, and IL-6 activate Src kinase, leading to AR Tyr-534 phosphorylation. Additionally, EGF activates an unidentified dasatinib-resistant tyrosine kinase that phosphorylates AR at Tyr-267.
The functional consequences of AR phosphorylation at Tyr-267 and Tyr-534 appear similar in that phosphorylation at both sites has been linked to enhanced AR recruitment and expression of AR target genes at suboptimal androgen concentrations (Guo et al 2006, Mahajan et al 2007). However, chromatin immunoprecipitation analysis demonstrated that Src-induced AR activation involves AR recruitment preferentially to the proximal promoter elements of the canonical target gene prostate specific antigen and that there is no increase in the binding of AR to the distal enhancer where androgen-induced and Ack1-driven AR recruitment takes place (Desai et al 2006, Mahajan et al 2007, Yang et al 2009). Whether this difference in the AR recruitment sites stems from the distinct AR phosphorylation sites or possibly due to other components of the transcriptional complex regulated by these kinases is unclear. The differential effect of AR phosphorylation sites on target gene expression and binding of coactivators and corepressors and other proteins involved in the assembly of the active transcriptional complex requires further elucidation.
Dasatinib inhibits a wide spectrum of kinases such as Abl and Src and its ability to inhibit Bcr-Abl kinase (unmutated and mutated forms) led its approval for treatment of imatinib-resistant chronic myelogenous leukemia (Brave et al 2008). In addition to the ability of Src to promotes castration resistant progression and AR activation, Src is involved in regulating prostate cancer cell migration, invasion, and metastasis and affects bone remodeling (Araujo and Logothetis 2009, Park et al 2008). Therefore, dasatinib is currently being studied in treatment of CRPC as a single agent and in combination with docetaxel chemotherapy (Araujo et al 2009, Yu et al 2009). Our data show that Ack1 kinase is inhibited by dasatinib at clinically relevant concentrations, and preclinical xenograft studies demonstrate the feasibility of inhibiting Ack1 in vivo as tumors exhibit loss of constitutive Ack1 and AR phosphorylation after oral treatment with dasatinib. This raises a possibility that dasatinib may have clinical activity against Ack1-driven malignancies. Ack1 binds and is activated by several receptor tyrosine kinases such as EGFR, HER-2, Mer, Axl, platelet derived growth factor receptor, LTK (leucocyte receptor tyrosine kinase belonging to the insulin receptor family) and ALK (anaplastic lymphoma kinase) (Galisteo et al 2006, Mahajan et al 2005, Pao-Chun et al 2009). A recent study demonstrated that the Ack1 gene is amplified and overexpressed in several tumor types, including castration resistant prostate cancer, and this was correlated with cancer progression and poor prognosis (van der Horst et al 2005). Additionally, Ack1 may also be activated by oncogenic mutations. The current release (version 42) of the Catalogue of Somatic Mutation in Cancer database reported 5 out of 229 tumor samples containing point mutations in Ack1, some of which are likely to lead to constitutive activation of kinase (Forbes et al 2006). In a subset of primary CRPC tumor specimens (8 out of 18), expression of tyrosine-phosphorylated AR and Ack1 was detected by immunoprecipitation and immunoblotting of tumor lysates (Mahajan et al 2007). Our findings provide additional mechanisms by which dasatinib may exert anti-tumor activity in CRPC. Although both Src and Ack1 phosphorylate AR protein, they target distinct sites. Therefore, phospho-Tyr-267 and phospho-Tyr-534 AR expression in CRPC tumors may serve as a predictive biomarker of tyrosine kinase inhibitor therapy.
Materials and Methods
Cells and reagents
LNCaP cells were obtained from the American Type Culture Collection (Manassas, VA). LAPC-4 cells were provided by Dr. Charles Sawyers (Klein et al 1997). EGF (R&D Systems, Minneapolis, MN), IL-6 (R&D), Gas6 (R&D), and bombesin (Sigma-Aldrich, St. Louis, MO) were purchased. Heregulin was a gift from Genentech (South San Francisco, CA). Dasatinib was obtained from Bristol-Myers-Squibb (Princeton, NJ). Phospho-specific polyclonal antibody against Tyr-267 of AR was generated by a commercial vendor (21st Century Biochemicals, Marlboro, MA). Rabbits were immunized with carrier-conjugated phospho-peptides spanning Tyr-267. Immunodepletion using a nonphospho-peptide column and affinity purification using the phospho-peptide column were performed by the vendor. Phospho-specific antibody against Tyr-534 of AR was raised in rabbits using standard methods and affinity purified in a similar fashion; its characterization has been reported (DaSilva et al 2009). A mouse monoclonal antibody against total AR (F39.4.1, Biogenex, San Ramon, CA) was used for immunoblotting. A polyclonal antibody against AR (C-19, Santa Cruz) was used for immunoprecipitation. The antibody against total Ack1 was described previously (Mahajan et al 2005). A phospho-specific antibody against Ack1 p-Tyr-284 (# 09-142) was obtained from Millipore (Billerica, MA). Antibodies against total Src (#2108) and phospho-specific Src p-Tyr-416 (#2101) were obtained from Cell Signaling Technology (Beverly, MA).
Transfections and knockdown
293T cells and COS7 cells were transfected with AR or Ack1 or Src expression vectors using Effectene (Qiagen, Valencia, CA) according to the manufacturer's direction. siRNA sequences against Ack1 were previously described (Mahajan et al 2007). For knocking down Src, Validated Stealth RNAi™ siRNA against Src (Invitrogen, Carlsbad, CA) was used according to the manufacturer. LNCaP cells were transfected using siPort Lipid (Ambion, Austin, TX) with 100 nM of siRNA or negative control scrambled siRNA. After 24 hrs, cells were treated with ligands as indicated. All experiments were repeated at least three times.
Proliferation Assays
LNCaP cells or LAPC-4 cells were seeded at a density of 104 cells per well in triplicate in a 96-well plate in serum-free medium (RPMI 1640 for LNCaP and IMDM for LAPC-4). On day 0, cells were treated with DHT (10 nM), EGF (100 ng/ml), heregulin (10 ng/ml), Gas6 (100 ng/ml), bombesin (1 nM), or IL-6 (10 ng/ml). After 3 days of incubation, relative cell proliferation was measured using colorimetric dye WST-8 (Cell Counting Kit-8, Dojindo, Rockville, MD) according to the manufacturer's direction.
Reporter assays and quantitative RT-PCR
LNCaP cells (8 × 105 cells per 6-cm plate) were transfected with the ARR2-PB-luciferase reporter (500 ng) (Zhang et al 2000) along with the AR expression vector (50 ng), using Effectene, as described (Mahajan et al 2007). After overnight incubation, cells were pre-treated with dasatinib as indicated for 2 hrs, then EGF (100 ng/ml) or heregulin (10 ng/ml) for 20 hrs. Luciferase activity was determined, as described (Mahajan et al 2007). The IC50 concentration of dasatinib required for inhibition of heregulin-driven luciferase activity was calculated using the dose-response variable slope (four parameters) function of the GraphPad Prism 5 software (GraphPad, La Jolla, CA). For measurement of PSA and hK2 mRNA levels, LNCaP or LAPC-4 cells were incubated in serum-free medium and were pre-treated with dasatinib (10 nM) for 2 hrs, then treated with heregulin (10 ng/ml) or EGF (100 ng/ml) for 20 hrs. Total RNA was isolated and the mRNA levels of PSA and hK2 were determined by quantitative RT-PCR, as described (Mahajan et al 2007).
Colony formation in soft agar
LNCaP cells expressing Ack1 or vector control (104 cells per well) were suspended in 0.45% Noble agar along with the indicated concentration of dasatinib and placed above a layer of solidified 0.9% Noble agar in 6 well plates in triplicates. After 3 weeks, colonies were visualized by staining with MTT.
Xenograft tumor growth
LNCaP cells stably expressing activated Ack1 by retroviral transduction have been described (Mahajan et al 2005). LNCaP-Ack1 cells (2 × 106) were mixed with an equal volume of Matrigel (BD Biosciences, Franklin Lakes, NJ) and implanted subcutaneously in the flank of castrated nude male mice, as described (Mahajan et al 2005, Mahajan et al 2007). When tumors became palpable, mice were randomly divided into two groups. Dasatinib was dissolved in citrate buffer (80 mM, pH 3.1) and administered to mice via oral gavage at a dose of 30 mg/kg twice daily. Tumor size was measured with calipers twice per week. These procedures were approved by the Institutional Animal Use and Care Committee.
Statistical Methods
The nonparametric Wilcoxon rank-sum test (using Van der Waerden normal scores) was used for the two-group comparisons of tumor volumes of the treated group (n=8) to the control group (n=7) at days 25 and 29 of treatment. Exact nominal (unadjusted for multiple comparisons) two-sided p-values were reported. Statistical analyses were performed using SAS statistical software, Version 9.2 (SAS Institute, Inc., Cary, NC).
Supplementary Material
Acknowledgments
We thank Francis Lee of Bristol-Myers-Squibb for providing dasatinib, Charlene Ross and staff of the UNC Lineberger Animal Studies Facility for assistance in xenograft tumor experiments, Dominic Moore for statistical analysis, and Nupam Mahajan for discussions and sharing unpublished manuscripts. This work was supported by grants from NIH R01CA120921 (Y.E.W.), NIH R01CA120304 (H.S.E.) and NIH T32ES007017 (M.K.).
Grant Support: NIH R01CA120921 (Y.E.W.), NIH R01CA120304 (H.S.E.) and NIH T32ES007017 (M.K.)
References
- Araujo J, Armstrong AJ, Braud EL, Posadas E, Lonberg M, Gallick GE, et al. Dasatinib and docetaxel combination treatment for patients with castration-resistant progressive prostate cancer: A phase I/II study (CA180086) J Clin Oncol. 2009;27(suppl) abstr 5061. [Google Scholar]
- Araujo J, Logothetis C. Targeting Src signaling in metastatic bone disease. Int J Cancer. 2009;124:1–6. doi: 10.1002/ijc.23998. [DOI] [PubMed] [Google Scholar]
- Brave M, Goodman V, Kaminskas E, Farrell A, Timmer W, Pope S, et al. Sprycel for chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia resistant to or intolerant of imatinib mesylate. Clin Cancer Res. 2008;14:352–359. doi: 10.1158/1078-0432.CCR-07-4175. [DOI] [PubMed] [Google Scholar]
- Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK, et al. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci U S A. 2005;102:11011–11016. doi: 10.1073/pnas.0504952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- DaSilva J, Gioeli D, Weber MJ, Parsons SJ. The neuroendocrine-derived peptide parathyroid hormone-related protein promotes prostate cancer cell growth by stabilizing the androgen receptor. Cancer Res. 2009;69:7402–7411. doi: 10.1158/0008-5472.CAN-08-4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai SJ, Ma AH, Tepper CG, Chen HW, Kung HJ. Inappropriate activation of the androgen receptor by nonsteroids: involvement of the Src kinase pathway and its therapeutic implications. Cancer Res. 2006;66:10449–10459. doi: 10.1158/0008-5472.CAN-06-2582. [DOI] [PubMed] [Google Scholar]
- Forbes S, Clements J, Dawson E, Bamford S, Webb T, Dogan A, et al. Cosmic 2005. Br J Cancer. 2006;94:318–322. doi: 10.1038/sj.bjc.6602928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galisteo ML, Yang Y, Urena J, Schlessinger J. Activation of the nonreceptor protein tyrosine kinase Ack by multiple extracellular stimuli. Proc Natl Acad Sci U S A. 2006;103:9796–9801. doi: 10.1073/pnas.0603714103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioeli D, Ficarro SB, Kwiek JJ, Aaronson D, Hancock M, Catling AD, et al. Androgen receptor phosphorylation. Regulation and identification of the phosphorylation sites. J Biol Chem. 2002;277:29304–29314. doi: 10.1074/jbc.M204131200. [DOI] [PubMed] [Google Scholar]
- Gioeli D, Black BE, Gordon V, Spencer A, Kesler CT, Eblen ST, et al. Stress kinase signaling regulates androgen receptor phosphorylation, transcription, and localization. Mol Endocrinol. 2006;20:503–515. doi: 10.1210/me.2005-0351. [DOI] [PubMed] [Google Scholar]
- Gong J, Zhu J, Goodman OB, Jr, Pestell RG, Schlegel PN, Nanus DM, et al. Activation of p300 histone acetyltransferase activity and acetylation of the androgen receptor by bombesin in prostate cancer cells. Oncogene. 2006;25:2011–2021. doi: 10.1038/sj.onc.1209231. [DOI] [PubMed] [Google Scholar]
- Gregory CW, Hamil KG, Kim D, Hall SH, Pretlow TG, Mohler JL, et al. Androgen receptor expression in androgen-independent prostate cancer is associated with increased expression of androgen-regulated genes. Cancer Res. 1998;58:5718–5724. [PubMed] [Google Scholar]
- Gregory CW, Johnson RT, Jr, Mohler JL, French FS, Wilson EM. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001;61:2892–2898. [PubMed] [Google Scholar]
- Gregory CW, Fei X, Ponguta LA, He B, Bill HM, French FS, et al. Epidermal growth factor increases coactivation of the androgen receptor in recurrent prostate cancer. J Biol Chem. 2004;279:7119–7130. doi: 10.1074/jbc.M307649200. [DOI] [PubMed] [Google Scholar]
- Gregory CW, Whang YE, McCall W, Fei X, Liu Y, Ponguta LA, et al. Heregulin-induced activation of HER2 and HER3 increases androgen receptor transactivation and CWR-R1 human recurrent prostate cancer cell growth. Clin Cancer Res. 2005;11:1704–1712. doi: 10.1158/1078-0432.CCR-04-1158. [DOI] [PubMed] [Google Scholar]
- Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006;10:309–319. doi: 10.1016/j.ccr.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Klein KA, Reiter RE, Redula J, Moradi H, Zhu XL, Brothman AR, et al. Progression of metastatic human prostate cancer to androgen independence in immunodeficient SCID mice. Nat Med. 1997;3:402–408. doi: 10.1038/nm0497-402. [DOI] [PubMed] [Google Scholar]
- Kraus S, Gioeli D, Vomastek T, Gordon V, Weber MJ. Receptor for activated C kinase 1 (RACK1) and Src regulate the tyrosine phosphorylation and function of the androgen receptor. Cancer Res. 2006;66:11047–11054. doi: 10.1158/0008-5472.CAN-06-0596. [DOI] [PubMed] [Google Scholar]
- Lee LF, Guan J, Qiu Y, Kung HJ. Neuropeptide-induced androgen independence in prostate cancer cells: roles of nonreceptor tyrosine kinases Etk/Bmx, Src, and focal adhesion kinase. Mol Cell Biol. 2001;21:8385–8397. doi: 10.1128/MCB.21.24.8385-8397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Majumder S, McCall W, Sartor CI, Mohler JL, Gregory CW, et al. Inhibition of HER-2/neu kinase impairs androgen receptor recruitment to the androgen responsive enhancer. Cancer Res. 2005;65:3404–3409. doi: 10.1158/0008-5472.CAN-04-4292. [DOI] [PubMed] [Google Scholar]
- Mahajan NP, Whang YE, Mohler JL, Earp HS. Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: role of Ack1 in polyubiquitination of tumor suppressor Wwox. Cancer Res. 2005;65:10514–10523. doi: 10.1158/0008-5472.CAN-05-1127. [DOI] [PubMed] [Google Scholar]
- Mahajan NP, Liu Y, Majumder S, Warren MR, Parker CE, Mohler JL, et al. Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc Natl Acad Sci U S A. 2007;104:8438–8443. doi: 10.1073/pnas.0700420104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellinghoff IK, Vivanco I, Kwon A, Tran C, Wongvipat J, Sawyers CL. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell. 2004;6:517–527. doi: 10.1016/j.ccr.2004.09.031. [DOI] [PubMed] [Google Scholar]
- Pao-Chun L, Chan PM, Chan W, Manser E. Cytoplasmic ACK1 interaction with multiple receptor tyrosine kinases is mediated by Grb2: an analysis of ACK1 effects on Axl signaling. J Biol Chem. 2009;284:34954–34963. doi: 10.1074/jbc.M109.072660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SI, Zhang J, Phillips KA, Araujo JC, Najjar AM, Volgin AY, et al. Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res. 2008;68:3323–3333. doi: 10.1158/0008-5472.CAN-07-2997. [DOI] [PubMed] [Google Scholar]
- Ponguta LA, Gregory CW, French FS, Wilson EM. Site-specific androgen receptor serine phosphorylation linked to epidermal growth factor-dependent growth of castration-recurrent prostate cancer. J Biol Chem. 2008;283:20989–21001. doi: 10.1074/jbc.M802392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–8261. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- Ueda T, Bruchovsky N, Sadar MD. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. J Biol Chem. 2002a;277:7076–7085. doi: 10.1074/jbc.M108255200. [DOI] [PubMed] [Google Scholar]
- Ueda T, Mawji NR, Bruchovsky N, Sadar MD. Ligand-independent activation of the androgen receptor by interleukin-6 and the role of steroid receptor coactivator-1 in prostate cancer cells. J Biol Chem. 2002b;277:38087–38094. doi: 10.1074/jbc.M203313200. [DOI] [PubMed] [Google Scholar]
- van der Horst EH, Degenhardt YY, Strelow A, Slavin A, Chinn L, Orf J, et al. Metastatic properties and genomic amplification of the tyrosine kinase gene ACK1. Proc Natl Acad Sci U S A. 2005;102:15901–15906. doi: 10.1073/pnas.0508014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kreisberg JI, Ghosh PM. Cross-talk between the androgen receptor and the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer. Curr Cancer Drug Targets. 2007;7:591–604. doi: 10.2174/156800907781662248. [DOI] [PubMed] [Google Scholar]
- Yang JC, Ok JH, Busby JE, Borowsky AD, Kung HJ, Evans CP. Aberrant activation of androgen receptor in a new neuropeptide-autocrine model of androgen-insensitive prostate cancer. Cancer Res. 2009;69:151–160. doi: 10.1158/0008-5472.CAN-08-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu EY, Wilding G, Posadas E, Gross M, Culine S, Massard C, et al. Phase II study of dasatinib in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res. 2009;15:7421–7428. doi: 10.1158/1078-0432.CCR-09-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Thomas TZ, Kasper S, Matusik RJ. A small composite probasin promoter confers high levels of prostate-specific gene expression through regulation by androgens and glucocorticoids in vitro and in vivo. Endocrinology. 2000;141:4698–4710. doi: 10.1210/endo.141.12.7837. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.