

Abstract
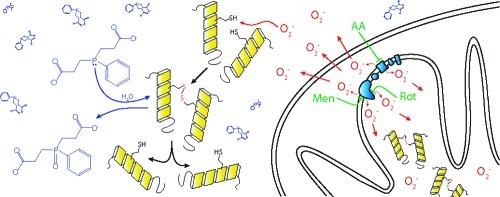
Central neurons undergo cell death after axotomy. One of the signaling pathways for this process is oxidative modification of one or more critical sulfhydryls in association with superoxide generation within mitochondria. Agents that reduce oxidized sulfhydryls are neuroprotective of axotomized retinal ganglion cells, and we hypothesized that this occurs via reversal of the effects of mitochondrial-produced superoxide. To study this, we measured the ability of the novel borane−phosphine complex drugs bis(3-propionic acid methyl ester)phenylphosphine borane complex (PB1) and (3-propionic acid methyl ester)diphenylphosphine borane complex (PB2) to inhibit the death of neuron-like RGC-5 cells induced by perturbation of the mitochondrial electron transport chain. We found that borane−phosphine complexes prevent neuronal cell death from superoxide produced by the redox-cycling agent menadione and the complex III inhibitor antimycin A, which produce superoxide toward the cytoplasm and matrix, but not the complex I inhibitor rotenone, which produces superoxide in the matrix alone. The ability of these disulfide reductants to prevent cell death may be predicted by the topology of superoxide production with respect to the mitochondrial matrix and extramitochondrial space.
Keywords: Superoxide, mitochondria, axonal injury, retinal ganglion cells, sulfhydryl modification
Retinal ganglion cells (RGCs) are archetypal central neurons that undergo apoptosis after axonal injury (1,2). RGCs have an optimal redox state for survival after axotomy, in that a reduced redox state is associated with increased survival compared with neutral or more oxidized conditions (3,4). The disulfide-reducing agent tris(2-carboxyethyl)phosphine (TCEP) dramatically increases survival of cultured retinal ganglion cells after axotomy in vitro(4) and in vivo(5), even in the absence of exogenous neurotrophic factors. Similar but quantitatively lower levels of neuroprotection are seen with dithiothreitol, which also reduces disulfides (4). Together, these findings are consistent with a mechanism whereby oxidation of one or more critical sulfhydryls is a signaling step for the induction of apoptosis in RGCs after axotomy. One of the possible sources of oxidative species is the generation of superoxide anion after axonal injury to the retinal ganglion cell (6), probably of mitochondrial origin (6,7). Supporting this, menadione-induced mitochondrial superoxide causes changes in the redox status of neuronal sulfhydryls (8).
Given that (i) oxidative modification of one or more critical sulfhydryls on signaling proteins is part of the signal pathway for neuronal death, (ii) superoxide is generated in mitochondria after axotomy, and (iii) mitochondria induce apoptosis by release of cytochrome c and other apoptosis inducing factors, we hypothesized that reduction of oxidatively modified sulfhydryls by TCEP and related molecules could be the mechanism by which this and similar drugs are neuroprotective. To study this further, we developed novel forms of disulfide reducing agents, with molecular features that increased penetration of cell membranes, increased retention within the cell, and protected the drugs from degradation by oxidation (9). This class of drugs is neuroprotective of RGCs in vitro, being active at picomolar to nanomolar concentrations (9).
To test the hypothesis that the mechanism of action for this class of drugs is via reversal of the effects of oxidative species generated in mitochondria, we measured their ability to inhibit the death of neuron-like RGC-5 cells induced by perturbation of the mitochondrial electron transport chain (METC). We found that the ability to prevent cell death due to mitochondrial disruption was not due to direct scavenging of superoxide within the cell and was specific to the site of superoxide generation with respect to the mitochondrial matrix.
Results and Discussion
Dose-Dependent Toxicity of Mitochondrial Electron Transport Chain Inhibition and Reactive Oxygen Induction in RGC-5 Cells
RGC-5 cells plated in 96-well plates were treated for 24 h with the redox cycling agent menadione, the complex I inhibitor rotenone, or the complex III inhibitor antimycin A at final concentrations ranging from 100 nM to 31.6 μM (menadione) or 1−316 μM (rotenone, antimycin A). There was a dose−response relationship between these METC-active drugs and RGC-5 viability (Figure 1).
Figure 1.

Cell death from METC inhibition is dose-dependent. Increasing concentrations of (A) menadione, (B) rotenone, and (C) antimycin A increase cell death in neuronal precursor RGC-5 cells.
To assess neuroprotection with PB1 and PB2 compounds, the initial dose−response curves were used to choose a single concentration of each METC-active drug that would achieve 50−70% cell death (menadione and rotenone at 3.16 μM; antimycin A at 1 μM). RGC-5 cells were then coincubated with the METC-active drugs and a range of concentrations of the borane−phosphines PB1 or PB2 for 24 h, followed by assessment of cell viability by calcein/propidium iodide assay. TCEP (100 μM) served as a positive control.
PB1 and PB2 showed significant rescue of RGC-5 cells from death induced by METC-active drugs menadione, rotenone, and antimycin A. PB1 was highly neuroprotective when given to RGC-5 cells at a final concentration of 100 μM against menadione (83% ± 5% vs 34% ± 4%; p < 0.0001) and against antimycin A (74% ± 4% vs 34% ± 2%; p < 0.0001). PB1 rescue from rotenone-induced oxidative stress was significant but substantially less than for the other METC-active drugs (36% ± 3% vs 25% ± 3%; p = 0.01) (Figure 2). PB2 was highly neuroprotective at a final concentration of 1 μM against menadione (95% ± 5% vs 34% ± 4%; p < 0.0001) and antimycin A (68% ± 3% vs 34% ± 2%; p < 0.0001). As with PB1, PB2 showed significant but substantially lower protection from rotenone (34% ± 2% vs 25% ± 3%; p = 0.03) (Figure 3). At higher concentrations, PB1 and PB2 show little or no rescue of RGC-5 cells, most likely due to toxicity.
Figure 2.

PB1 is protective against cell death induced by METC disruption. PB1 is protective of RGC-5 cells against treatment with (A) menadione, (B) rotenone, and (C) antimycin A at concentrations ranging from 10 nM to 100 μM: ∗, p < 0.05; ∗∗, p < 0.01.
Figure 3.

PB2 is protective against cell death induced by METC disruption. PB2 is protective of RGC-5 cells against treatment with (A) menadione, (B) rotenone, and (C) antimycin A at concentrations ranging from 10 nM to 10 μM: ∗, p < 0.05; ∗∗, p < 0.01.
PB1 and PB2 Show Significantly More Rescue than Tris(2-carboxyethyl)phosphine at the Same or Lower Dose
We used PB1 and PB2, members of the borane-protected phosphine class of reducing agents, because they are cell-permeable analogs of TCEP with consequently greater rescue of primary RGCs (9). We compared the ability of TCEP to rescue RGC-5 cells with that of PB1 and PB2. PB1 and PB2 showed significantly more rescue than TCEP in RGC-5 cells cotreated with superoxide-generating compounds (Figure 4). PB1 and PB2 rescued cells to a level indistinguishable from control (menadione, p = 0.45, 0.70; antimycin A, p = 0.79, 0.82), and significantly higher than TCEP for the same treatment for five of the six comparisons (p < 0.05).
Figure 4.

PB1 and PB2 exhibit greater protection than TCEP against METC disruption. PB1 (100 μM) and PB2 (1 μM) significantly rescue RGC-5 cells to levels indistinguishable from control against menadione and antimycin A. TCEP does not result in significant protection against any of the METC-active compounds: ∗∗, p < 0.01.
PB1 and PB2 Do Not Scavenge Superoxide
METC inhibition causes a significant production of mitochondrial superoxide as a result of reduced complexes leaking electrons to free molecular oxygen. The decrease in cell death by PB1 and PB2 following METC inhibition could therefore result either from a direct interaction between the borane−phosphine complex and superoxide, or chemical reduction of oxidized sulfhydryls. To distinguish these two possibilities, we assessed the ability of PB1, PB2, or the unprotected phosphine-based reducing agent TCEP to scavenge superoxide in a cell-free assay. Dihydroethidium (HEt) was used to measure superoxide levels in the presence of xanthine and xanthine oxidase, a superoxide-generating system, in the presence and absence of the phosphines. Superoxide dismutase (SOD) was used as a positive control. While SOD produced the expected dose-dependent reduction of HEt fluorescence (80527 ± 535, 75117 ± 546, 57238 ± 428, and 26321 ± 671 for 0, 1, 10, and 100 U/mL, respectively, in arbitrary fluorescence units; p = 0.002, p < 0.0001, and p < 0.0001 vs control, respectively), PB1 and PB2 produced a minimal reduction in superoxide levels and only at the lower concentrations (77116 ± 501 and 74530 ± 180 for 1 and 10 μM PB1, respectively, and 77342 ± 892 for 1 μM PB2; p = 0.01, 0.004, and 0.05, respectively) (Figure 5). To rule out the possibility that the protective borane group could inhibit superoxide scavenging in cell-free conditions, the compounds were stripped of this group by incubation in the presence of diazabicyclo-2,2,2-octane (DABCO), followed by measurement of superoxide as above. PB1 and PB2 deprotected in this manner still did not significantly scavenge superoxide, with similar lack of activity to TCEP, a phosphine that does not contain borane (Figure 5). These results and the lack of a dose-dependent reduction in superoxide levels imply that the borane−phosphine compounds PB1 and PB2 do not scavenge superoxide in a cell-free system.
Figure 5.

PB1 and PB2 do not substantially scavenge superoxide in a cell-free system. PB1 and PB2 exhibit very low superoxide scavenging activity in a non-dose-dependent manner. Their borane-deprotected analogs (dPB1 and dPB2), as well as TCEP and the borane-deprotecting agent DABCO, do not show significant reduction in superoxide levels at any tested concentrations: ∗, p < 0.05; ∗∗, p < 0.01.
It is possible that PB1 and PB2 undergo reactions within the cell (Figure 6A) that would make them able to scavenge superoxide. In order to rule this out, RGC-5 cells were treated with menadione, following which intracellular superoxide was measured with HEt. Menadione (17.8 μM) caused a significant increase in intracellular superoxide levels at 2 h over background compared with untreated cells (12567 ± 766 vs −360 ± 141; p < 0.0001; Figure 6B), consistent with its action as a redox cycling agent. Incubation of cells with PEG−SOD immediately prior to menadione significantly reduced the menadione-induced increase in HEt fluorescence (3218 ± 603; p < 0.0001). Treatment of the cells with PB1 or PB2 did not result in a substantial decrease in the rate of HEt oxidation (p = not significant for all treatments compared with menadione alone). PB2 actually induced a noticeable but not significant rise in HEt fluorescence at 100 μM. Given that we observe toxicity in the cells at this concentration, this increase is not altogether surprising. Together, these results imply that PB1 and PB2 are not significant scavengers of superoxide.
Figure 6.

(A) Proposed activation of PB1 in the cell. The borane group of the borane−phosphine complexes is removed by a nucleophilic attack by tissue amines. Intracellular esterases cleave the methyl ester groups, resulting in the negatively charged, deprotected species shown. (B) PB1 and PB2 do not substantially scavenge intracellular superoxide. RGC-5 cells were incubated with menadione to generate superoxide via redox cycling, measured by oxidation of HEt. PB1 and PB2 minimally affected intracellular superoxide levels: ∗, p < 0.01.
Discussion
These data demonstrate that disulfide-reducing borane−phosphine complexes prevent neuronal cell death from superoxide produced by the redox-cycling agent menadione and the complex III inhibitor antimycin A but not the complex I inhibitor rotenone. The rescue shown by the borane−phosphine complexes PB1 and PB2 against METC dysfunction in this study was substantial and significant and not due to a direct scavenging of superoxide. These findings suggest that the topology for the chemical reduction of oxidized sulfhydryls is external to the mitochondrial matrix.
The choice of experimental paradigm influences the assessment of topology of mitochondrial superoxide production (Table 1). There is a general consensus that rotenone inhibition of complex I generates superoxide via increased leakage of electrons being transferred to the ubiquinone binding site of NADH dehydrogenase. The latter is physically located inside the matrix, explaining the consistent detection of 90−100% of superoxide at that location. On the other hand, there are conflicting data regarding the directionality of superoxide production via electron leakage from complex III after inhibition with antimycin A, although it is generally believed to originate from binding to the Qi site of the complex. The widely accepted paradigm for many years, drawn from research with submitochondrial particles (SMPs), was of superoxide release into the matrix space (10). This belief shifted toward release into the intermembrane space, based on studies of whole mitochondria, use of alternative detection methods for superoxide and its dismutation byproducts, and improved understanding of the physical location of the subunits of complex III (11). Continued research has failed to definitively answer the question of what proportion of superoxide is produced toward each side of the membrane (Table 1) or how superoxide might translocate from the outer face of the membrane into the matrix after inhibition with antimycin A. Overall, the accumulated data is most consistent with superoxide release toward both sides of the inner membrane after inhibition with antimycin A.
Table 1.
| topology (%) |
|||||
|---|---|---|---|---|---|
| concentration | matrix | IMS | method | model | citation |
| Rotenone | |||||
| 1−10 μM | 100 | 0 | capillary electrophoresis of TPP-HE | 143B cells (mouse) | (12) |
| 5 μM | ∼93 | ∼7 | liver (mouse) | ||
| 5 μM | ∼98 | ∼2 | skeletal muscle (rat) | ||
| 5 μM | >90 | <10 | fluorometric detection of H2O2; aconitase inactivation | Drosophila | (16) |
| 10 μM | 100 | 0 | fluorometric detection of H2O2; aconitase inactivation | skeletal muscle (SOD1 −/− and WT mouse) | (17) |
| 5 μM | 100 | 0 | fluorometric detection of H2O2 | Drosophila | (18) |
| 10 μM | 100 | 0 | fluorometric detection of H2O2 | heart (rat); skeletal muscle (rat) | (11) |
| Menadione | |||||
| 80−90 | 20−10 | capillary electrophoresis of TPP-HE | 143B cells (mouse) | (12) | |
| 5 μM | ∼90 | ∼10 | liver (mouse) | ||
| 5 μM | ∼90 | ∼10 | skeletal muscle (rat) | ||
| 50 μM | 0 | 100 | capillary electrophoresis of HE | skeletal muscle (rat) | (13) |
| Antimycin A | |||||
| 20−80 | 80−20 | capillary electrophoresis of TPP-HE | 143B cells (mouse) | (12) | |
| 5 μM | ∼45 | ∼55 | liver (mouse) | ||
| 5 μM | ∼60 | ∼40 | skeletal muscle (rat) | ||
| 10 μM | ∼7 | ∼93 | capillary electrophoresis of HE | skeletal muscle (rat) | (13) |
| ∼70 | ∼30 | fluorometric detection of H2O2; aconitase inactivation | Drosophila | (16) | |
| 10 μM | ∼50 | ∼50 | fluorometric detection of H2O2; aconitase inactivation | skeletal muscle (SOD1 −/− and WT mouse) | (17) |
| 3 μM | ∼65 | ∼35 | fluorometric detection of H2O2 | Drosophila | (18) |
| 1 μg/mg protein | ∼30 | ∼70 | fluorometric detection of O2−•; Fluorometric detection of H2O2; spin-trapping EPR | heart (rat) | (19) |
| 0.625 nmol/mg protein | ∼20 | ∼80 | fluorometric detection of H2O2 | heart (rat) | (11) |
| ∼40 | ∼60 | skeletal muscle (rat) | |||
| 2 μM | 100 | 0 | adrenochrome formation; ferricytochrome c reduction | submitochondrial particles from heart and lung (bovine) | (10) (review) |
The literature on the topology of superoxide production after menadione treatment is limited but shows a similar diversity of findings. Xu and Arriaga (12) measured superoxide production directed primarily toward the matrix, while Meany et al.13 measured generation exclusively into the intermembrane space. Both groups used highly similar methods and the same model. Differential sensitivity of the fluorescent dyes used (hydroethidium and triphenylphosphonium−hydroethidium) might lead to different sensitivities to inner- and extramitochondrial superoxide production, but most probably the findings suggest superoxide production to both sides of the inner membrane.
The pattern of neuroprotection by borane−phosphine complexes PB1 and PB2 with different mitochondrial superoxide generators therefore correlates with the topology of superoxide release. Rotenone generates superoxide exclusively into the matrix, where the bioavailability of PB1 and PB2 would be expected to be low because of the negative charge of the carboxy group after cleavage of the ester bond by nonspecific cytoplasmic esterases (Figure 6A) and the negative mitochondrial membrane potential. Antimycin A and menadione lead to the production of superoxide in both the matrix and extramitochondrial spaces, with the latter being the likely site of action of these disulfide reductants. When superoxide is generated into the extramitochondrial space by redox cycling or complex III inhibition, the intracellular levels of the negatively charged phosphine ester metabolites is sufficient to reduce oxidized thiols. The intracellular charge of the phosphines therefore explains the difference between protection against complex I inhibitor-mediated neuronal death and death from complex III inhibition or redox cycling.
There are several caveats to this study. First, neuronal precursor cell line cells were used that have several phenotypic features of retinal ganglion cells but are not the same as primary retinal ganglion cells. Second, these are in vitro studies of a single homogeneous cell population, and the absence of other cells that normally reside in the milieu of retinal ganglion cell within the retina could confound the results. Third, the in vitro conditions involve incubation in the presence of room air and 5% CO2, which means that cells are exposed to higher oxygen concentrations than that of the RGC milieu under physiological conditions in the inner retina (14). Fourth, these are mitochondrial toxins, and although their major mode of action is on the electron transport chain, we cannot exclude other nonspecific effects.
The differences between these modes of mitochondria-associated oxidation have implications for the potential therapeutic use of borane−phosphine complexes as neuroprotectants in various diseases. RGCs exhibit increased levels of superoxide dependence on complex III after axotomy (6) and are subsequently protected after acute axotomy and culture by these borane−phosphine complex compounds (9). Since complex III generates superoxide toward the cytoplasm, we would expect that the borane−phosphine complex drugs act on disulfides generated via cytoplasmic superoxide. On the other hand, diseases like Leber’s hereditary optic neuropathy are due to mutations in mitochondrial DNA coding for components of complex I (15), and RGC death in this disease is likely signaled by increased levels of mitochondrially generated superoxide (7). PB1 and PB2 would theoretically not be effective in that or related diseases, such as Leigh syndrome and Parkinson’s disease. Modification of borane-protected reducing agents to achieve distribution in the mitochondrial matrix may be necessary for neuroprotection in such disorders.
Methods
Materials
RGC-5 cells were a generous gift of Neeraj Agarwal, Ph.D. Dulbecco’s modified Eagle’s medium (DMEM) and penicillin−streptomycin were from Mediatech, Inc. (Manassas, VA). Fetal bovine serum (FBS) was from Gemini Bio-Products (West Sacramento, CA). Staurosporine was from Alexis Biochemicals (San Diego, CA). Diazabicyclo-2,2,2-octane (DABCO) was from Acros Organics (Geel, Belgium). Antimycin A, rotenone, menadione, xanthine, xanthine oxidase, superoxide dismutase (SOD), poly(ethylene glycol)-conjugated superoxide dismutase (PEG−SOD), N,N-dimethylformamide, and TCEP were from Sigma-Aldrich (St. Louis, MO). Phosphate-buffered saline (PBS) was from Lonza Walkersville, Inc. (Walkersville, MD). Calcein-AM and propidium iodide were from Invitrogen (Carlsbad, CA). Dihydroethidium was from Anaspec (Fremont, CA). The novel borane-protected phosphines bis(3-propionic acid methyl ester)phenylphosphine borane complex (PB1) and (3-propionic acid methyl ester)diphenylphosphine borane complex (PB2) were synthesized as previously described (9).
Cell Culture
RGC-5 cells were cultured in DMEM containing 1 g/L glucose supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were passaged every 48−72 h when cells were approximately 60−75% confluent, replated at a 1:20 dilution in a 75 cm2 flask in 20 mL of cell culture media, and incubated at 37 °C in humidified 5% CO2. Experiments were performed in duplicate or triplicate.
Cell Treatment and Assessment of Viability
Cells were plated in 96-well plates at a density of approximately 60 cells/mm2 in 50 μL of media. Approximately 24 h after plating, cells were supplemented with an equal volume of media containing the METC-inhibitors with or without PB1 and PB2 to generate the final concentrations listed in the results. Control wells received media containing vehicle alone. Following 24 h of treatment, media was aspirated from wells using a 25-gauge needle, and cells were stained with calcein-AM (10 μg/mL) and propidium iodide (1 μg/mL) in PBS for 30 min. Staining solution was aspirated using a 25-gauge needle and replaced with PBS. Three randomly chosen fields per well were photographed on an Axiovert 135 microscope under epifluorescence with a Nikon D70s digital SLR camera (Nikon, Melville, NY) at a resolution of 3008 pixels × 2000 pixels and an exposure time of 1.6 s. Live (calcein-positive) and dead (propidium iodide-positive) cells were assessed using ImageJ software. Pictures were batch analyzed using a macro containing the following actions: subtract background, threshold, erode, erode, watershed, and analyze particles between the sizes of 1000 pixels and infinity. Automated counts were periodically checked against manual counts with good agreement. The absolute amount of death in the cell culture experiments varied depending on the passage number of the RGC-5 cells and small differences between batches of cryopreserved RGC-5 cells. Because of this, each experiment was analyzed with respect to its own set of controls.
Superoxide Measurement
Cell-Free
The superoxide scavenging ability of PB1 and PB2 were assessed in a cell-free system. In the presence of superoxide, dihydroethidium (HEt) is converted to an ethidium derivative that exhibits peak fluorescence in the red spectrum (excitation 480 nm; emission 586 nm). HEt (1 mM) was combined with PB1, PB2, or TCEP at concentrations of 1, 10, and 100 μM in the presence of 1 mM xanthine (X) and 0.05 U/mL xanthine oxidase (XO), a system known to generate both superoxide and hydrogen peroxide. Superoxide dismutase from bovine erythrocytes was tested as a known scavenger of superoxide at 1, 10, and 100 U/mL. Additionally, the borane group was removed from PB1 and PB2 by incubation at 60 °C in the presence of an equimolar amount of DABCO in N,N-dimethylformamide for 1 h under argon gas, followed by gradual cooling to room temperature. These deprotected compounds (dPB1 and dPB2) were also tested for scavenging ability at concentrations of 1, 10, and 100 μM. The reaction was initiated by addition of xanthine oxidase to wells, and fluorescence was monitored every 5 min for 30 min using a 1420 Victor 2 T Multilabel Counter (excitation 485 nm, emission 580 nm). The change in HEt fluorescence over 30 min was calculated for each well and compared with wells receiving no scavenging compound. Experiments were performed in triplicate, and results are representative of two or more experiments.
In Vitro
Superoxide levels were measured with HEt in cultured cells. Twenty-four hours after plating, RGC-5 cells in a 96-well plate were treated with 17.8 μM menadione. PB1 or PB2 was added to treatment wells to final concentrations of 100 nM, 1 μM, 10 μM, or 100 μM. Cells treated with PB1 or PB2 and menadione were compared with (1) cells treated with menadione alone, (2) cells treated with menadione and 300 U/mL PEG−SOD, and (3) untreated cells. All cells were treated with 3.2 μM HEt in medium 45 min after treatment with menadione, PB compounds, or PEG−SOD. Fluorescence was assessed 2 h after menadione treatment using a 1420 Victor 2 T Multilabel Counter (excitation 485 nm; emission 580 nm), with cells incubated at 37 °C in humidified 5% CO2 between readings. Conditions were performed in triplicate, and results are representative of two or more experiments.
Statistics
Viability was assessed as number of live cells per square millimeter and compared with untreated controls within each experiment. Means were compared using Student’s unpaired t-test.
NIH R21EY017970 and P30EY016665, Retina Research Foundation, and an unrestricted departmental grant from Research to Prevent Blindness, Inc.
L.A.L. conceived the study and designed the experiments. E.A.S., C.J.L., and A.F.T. performed experiments. E.A.S. wrote the initial draft of the manuscript, which was supplemented and revised with contributions from all authors.
Funding Statement
National Institutes of Health, United States
References
- Berkelaar M.; Clarke D. B.; Wang Y. C.; Bray G. M.; Aguayo A. J. (1994) Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. J. Neurosci. 14, 4368–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Valenzuela E.; Gorczyca W.; Darzynkiewicz Z.; Sharma S. C. (1994) Apoptosis in adult retinal ganglion cells after axotomy. J. Neurobiol. 25, 431–438. [DOI] [PubMed] [Google Scholar]
- Castagne V.; Lefevre K.; Natero R.; Clarke P. G.; Bedker D. A. (1999) An optimal redox status for the survival of axotomized ganglion cells in the developing retina. Neuroscience 93, 313–320. [DOI] [PubMed] [Google Scholar]
- Geiger L. K.; Kortuem K. R.; Alexejun C.; Levin L. A. (2002) Reduced redox state allows prolonged survival of axotomized neonatal retinal ganglion cells. Neuroscience 109, 635–642. [DOI] [PubMed] [Google Scholar]
- Swanson K. I.; Schlieve C. R.; Lieven C. J.; Levin L. A. (2005) Neuroprotective effect of sulfhydryl reduction in a rat optic nerve crush model. Invest. Ophthalmol. Visual Sci. 46, 3737–3741. [DOI] [PubMed] [Google Scholar]
- Lieven C. J.; Schlieve C. R.; Hoegger M. J.; Levin L. A. (2006) Retinal ganglion cell axotomy induces an increase in intracellular superoxide anion. Invest. Ophthalmol. Visual Sci. 47, 1477–1485. [DOI] [PubMed] [Google Scholar]
- Hoegger M. J.; Lieven C. J.; Levin L. A. (2008) Differential production of superoxide by neuronal mitochondria. BMC Neurosci. 9, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterboer A. L., Ribich J. R., Levin L. A. (2009) Identification of protein targets of novel neuroprotective sulfhydryl reducing drugs, presented at the Association for Research in Vision and Ophthalmology Annual Meeting, Fort Lauderdale, FL.
- Schlieve C. R.; Tam A.; Nilsson B. L.; Lieven C. J.; Raines R. T.; Levin L. A. (2006) Synthesis and characterization of a novel class of reducing agents that are highly neuroprotective for retinal ganglion cells. Exp. Eye Res. 83, 1252–1259. [DOI] [PubMed] [Google Scholar]
- Turrens J. F. (1997) Superoxide production by the mitochondrial respiratory chain. Biosci. Rep. 17, 3–8. [DOI] [PubMed] [Google Scholar]
- St-Pierre J.; Buckingham J. A.; Roebuck S. J.; Brand M. D. (2002) Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 277, 44784–44790. [DOI] [PubMed] [Google Scholar]
- Xu X.; Arriaga E. A. (2009) Qualitative determination of superoxide release at both sides of the mitochondrial inner membrane by capillary electrophoretic analysis of the oxidation products of triphenylphosphonium hydroethidine. Free Radical Biol. Med. 46, 905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meany D. L.; Thompson L.; Arriaga E. A. (2007) Simultaneously monitoring the superoxide in the mitochondrial matrix and extramitochondrial space by micellar electrokinetic chromatography with laser-induced fluorescence. Anal. Chem. 79, 4588–4594. [DOI] [PubMed] [Google Scholar]
- Ahmed J.; Braun R. D.; Dunn R. Jr.; Linsenmeier R. A. (1993) Oxygen distribution in the macaque retina. Invest. Ophthalmol. Visual Sci. 34, 516–521. [PubMed] [Google Scholar]
- Singh G.; Lott M. T.; Wallace D. C. (1989) A mitochondrial DNA mutation as a cause of Leber's hereditary optic neuropathy. N. Engl. J. Med. 320, 1300–1305. [DOI] [PubMed] [Google Scholar]
- Miwa S.; Brand M. D. (2005) The topology of superoxide production by complex III and glycerol 3-phosphate dehydrogenase in Drosophila mitochondria. Biochim. Biophys. Acta 1709, 214–219. [DOI] [PubMed] [Google Scholar]
- Muller F. L.; Liu Y.; Van Remmen H. (2004) Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 279, 49064–49073. [DOI] [PubMed] [Google Scholar]
- Miwa S.; St-Pierre J.; Partridge L.; Brand M. D. (2003) Superoxide and hydrogen peroxide production by Drosophila mitochondria. Free Radical Biol. Med. 35, 938–948. [DOI] [PubMed] [Google Scholar]
- Han D.; Antunes F.; Canali R.; Rettori D.; Cadenas E. (2003) Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 278, 5557–5563. [DOI] [PubMed] [Google Scholar]