

Summary
The Ser/Thr kinase mTOR is a central regulator of anabolism, growth and proliferation. We investigated the effects of Toxoplasma gondii on host mTOR signaling. Toxoplasma invasion of multiple cell types rapidly induced sustained mTOR activation that was restricted to infected cells, as determined by rapamycin-sensitive phosphorylation of ribosomal protein S6; however, phosphorylation of the growth-associated mTOR substrates 4E-BP1 and S6K1 was not detected. Infected cells still phosphorylated S6K1 and 4E-BP1 in response to insulin, although the S6K1 response was blunted. Parasite-induced S6 phosphorylation was independent of S6K1 and did not require activation of canonical mTOR-inducing pathways mediated by PI3K–Akt and ERK. Host mTOR was localized in a vesicular pattern surrounding the parasitophorous vacuole, suggesting potential activation by phosphatidic acid in the vacuolar membrane. In spite of a failure to phosphorylate 4E-BP1 and S6K1, intracellular T. gondii triggered host cell cycle progression in an mTOR-dependent manner and progression of infected cells displayed increased sensitivity to rapamycin. Moreover, normal cell growth was maintained during parasite-induced cell cycle progression, as indicated by total cellular S6 levels. The Toxoplasma-infected cell provides a unique example of non-canonical mTOR activation supporting growth that is independent of signaling through either S6K1 or 4E-BP1.
Introduction
The Ser/Thr kinase mammalian-target-of-rapamycin (mTOR) plays a central role in coordinating global cellular metabolic and growth responses to environmental conditions with respect to nutritional status, growth factor signaling and stress (Fingar and Blenis, 2004; Hay and Sonenberg, 2004; Wullschleger et al., 2006). A key function of mTOR is the promotion of cellular growth and proliferation. Dysregulation of mTOR is a frequent occurrence in neoplasms, and excessive stimulation of this pathway leads to defective control of cell survival and proliferation (Petroulakis et al., 2006). Conversely, rapamycin, a specific inhibitor of mTOR, prevents or delays mitogenic responses in certain cell types (Jayaraman and Marks, 1993; Mourani et al., 2004; Slavik et al., 2001; Yeager et al., 2008), and is an effective anti-tumor agent in certain settings (Carraway and Hidalgo, 2004; Kurmasheva et al., 2006; LoPiccolo et al., 2008; Thomas et al., 2006). An even broader role for mTOR as a critical control point for cellular growth is indicated by studies of mTOR-deficient cells that have revealed rapamycin-insensitive growth-promoting functions for this protein (Murakami et al., 2004; Wang et al., 2005).
A number of potential downstream effectors of mTOR have been identified, including the S6 kinases (S6K1 and S6K2) as well as several proteins associated with the translational initiation complex (Tee and Blenis, 2005). The latter include the mTOR substrate 4E-BP1, an inhibitory factor whose phosphorylation leads to its dissociation from eIF4E and the subsequent recruitment to the complex of eIF4G, which competes with 4E-BP1 for binding to a shared site on eIF4E. The binding of eIF4G, together with its associated RNA helicase, eIF4A, results in the formation of the eIF4F complex, which serves to promote global translation and also modulates the translation of specific mRNAs in a manner that favors anabolism, cell growth and proliferation (Bilanges et al., 2007; Dmitriev et al., 2003; Ramirez-Valle et al., 2008; Yoon et al., 2007). An additional potential effector is the ribosomal protein S6, which is a substrate for S6K1 and S6K2, although its role in translation and cell growth is not well understood (Ruvinsky and Meyuhas, 2006). In addition to these growth-promoting effects on the translational apparatus, mTOR activation also enhances transcription of rRNA, tRNA and ribosomal protein genes (Wullschleger et al., 2006).
These various effects of mTOR might be envisaged as acting in concert to promote a unified program of growth and cell cycle progression. Alternatively, distinct sets of mTOR effectors may act independently to govern distinct functions. The latter view is supported by recent studies of S6K-deficient mice. It was observed that S6K1-deficient myoblasts are defective with respect to proliferation-associated cell growth; however this kinase is not required for mTOR-dependent control of either cell proliferation (Ohanna et al., 2005), global protein translation or protein turnover in these cells (Mieulet et al., 2007). Further support for a specific linkage between S6K1 and cell growth comes from the observation that pancreatic β-cells have a reduced size in S6K1-deficient mice (Pende et al., 2000). Other cell types were unaffected, however, implying the likely existence of additional pathways to maintain cell growth. The nature of such pathways and their connection to mTOR are unresolved questions.
The notion that cell growth and cell cycle progression may each be governed by distinct mTOR effectors suggests that subsets of effectors might be regulated independently downstream of mTOR. However, there is as yet little evidence for such selective regulation of downstream elements of the pathway.
Intracellular parasites represent a relatively untapped resource for the investigation of regulatory mechanisms in mammalian cells. While cellular responses to parasitism may involve host defense-related functions, such as TLR signaling and stress responses, it is becoming increasingly clear that they also reflect sophisticated parasite adaptations that target and modify various aspects of host cell signaling. In particular, the apicomplexan parasite Toxoplasma gondii, which invades most mammalian cells, has been shown to generate novel modifications of several host cell signaling pathways. For example, infection of fibroblasts by T. gondii leads to initiation of the NF-κB signaling pathway (activation of IKKβ and degradation of IκB) but the pathway is blocked due to incomplete phosphorylation and failed nuclear translocation of p65/RelA (Shapira et al., 2005). In addition, intracelluar Toxoplasma can export a secretory kinase termed ROP16 to the host cytoplasm and nucleus, resulting in activation of host Stat3, suppression of host IL-12 production and enhanced parasite virulence (Saeij et al., 2007). In addition to ROP16, several other functionally uncharacterized putative kinases and phosphatases are contained in the specialized parasite secretory vesicles known as rhoptries and may potentially modulate host cell signaling (Ravindran and Boothroyd, 2008).
The mTOR pathway represents a potential target for manipulation by T. gondii, as the regulation of the anabolic / catabolic balance of the host cell may have a substantial impact on the availability of host-derived nutrients to support parasite expansion. We now report that the mTOR pathway is functionally activated upon infection in a manner that leads to an unusual dissociation between mTOR and its effectors, resulting in a physiological demonstration of mTOR-dependent cell growth independent of signaling through S6K1 and 4E-BP1.
Results
Stable activation of host mTOR in Toxoplasma – infected cells
To assess activation of host cell mTOR after infection by T. gondii, we examined the phosphorylation of ribosomal protein S6 by immunoblot analysis. Initial studies were conducted in fibroblasts deprived of serum in order to minimize basal mTOR activity. As demonstrated in Fig. 1, an increase in phosphorylated S6 (pS6) was observed within 2 hours of infection in either BALB/c 3T3 murine fibroblasts (3T3)(Fig. 1A) or human foreskin fibroblasts (HFF)(Fig. 1B). The induced level of pS6 was maintained for at least 24 hours and was not related to the number of parasites in the cell (as determined by YFP content using YFP-expressing T. gondii). A similar elevation of pS6 was observed in other cultured cells, including HeLa cells and murine peritoneal exudate macrophages (Fig. 1C). Parasite induction of host pS6 was comparable in intensity to the response to insulin. Densitometric analysis of band intensity indicated that the fold increase in pS6 upon infection was 7.1 ± 2.7 for 3T3 (n = 9 independent experiments), 3.3 ± 0.6 for HeLa (n = 6), and 5.5 ± 1.3 for HFFs (n = 4). The increase in pS6 was abolished by treatment with 200 ng/ml rapamycin (Fig. 1D), confirming that pS6 generation reflected activation of host cell mTOR. Rapamycin at 20 ng/ml was equally effective (Fig. 2E). The effect of rapamycin was not due to an inhibition of parasite survival or growth (Figs 1D, 6D). No reactivity was detected in lysates of extracellular parasites (Fig. 1A). Furthermore, immunostaining of pS6 revealed a generalized staining of host cytoplasm specific to infected cells, confirming that a host cell pathway had been activated (Fig. 1E). The fact that immunostaining is confined to infected cells indicates that T. gondii-mediated activation of host cell mTOR is a direct consequence of parasite invasion, rather than an indirect result of the secretion of diffusible mediators within the culture, or of incidental contact between parasites and fibroblasts.
Figure 1. T. gondii induces host cell mTOR activation.

(A) 3T3 cells were deprived of serum for one day and then infected or not with YFP-RH at m.o.i of 8 for the indicated times. Lysates of host cells or of free T.gondii tachyzoites (T) were subjected to immunoblotting. (B) HFFs were deprived of serum for one day and then infected at the indicated m.o.i for 2, 4, 8, or 24 h. Protein extracts were prepared for immunoblotting. (C) Murine peritoneal exudate macrophages or HeLa cells were infected at the indicated m.o.i for 24 h in the absence of serum. Uninfected HeLa cells were stimulated with insulin (Ins) for 30 min prior to harvest. The loading control is total Akt for macrophages and total S6 for HeLa. (D) 3T3 cells were infected overnight at m.o.i. of 4 in the absence of serum. Cells were treated as indicated with insulin for the final 2 h and with rapamycin (Rap, 200 ng/ml) for the final 30 min of culture. (E) 3T3 cells were infected with YFP-RH (green) overnight in the absence of serum, and then subjected to pS6 (S235/236) immunostaining (red). Brightfield (BF) and fluorescent images of the same field are displayed. The data in the figure are representative of either two or three independent experiments.
Figure 2. Characterization of mTOR-activating signals in infected cells.

(A) 3T3 cells or HeLa cells were infected at the indicated m.o.i for 24 h prior to Western blot analysis. Uninfected cells were treated with insulin for 30 min (Ins). (B) 3T3 cells were serum-starved overnight and then infected or not at m.o.i of 8 for the indicated times. The lower panel displays the band intensity for pErk1/2 (dashed) and pS6 (solid), relative to 0 h values. The values were normalized to the intensity of total S6. The open and closed symbols represent data from two experiments. (D) 3T3 cells were infected or not for 22 h and then treated for 2 h with LY294002 (LY), Akti-1/2 (AKTi), PD98059 (PD) or rapamycin (Rap, 200 ng/ml). (E) 3T3 or HeLa cells were infected at m.o.i. of 4 for 24 h and then treated with either medium (C), LY294002 (LY), wortmannin (Wort) or 20 ng/ml rapamycin (Rap) for 2 h prior to harvest. U = uninfected. The data are representative of either two (A,C,E) or three (B,D) independent experiments. (A) HFFs were infected with YFP-RH for 4 h, after which plates were rinsed to remove free parasites. To vary amino acid supply, the medium was replaced with DMEM diluted with HBSS to the indicated percentage DMEM and culture continued for 24 h prior to harvest for Western blot analysis.
Figure 6. 4E-BP1 phosphorylation is not altered by Toxoplasma infection.
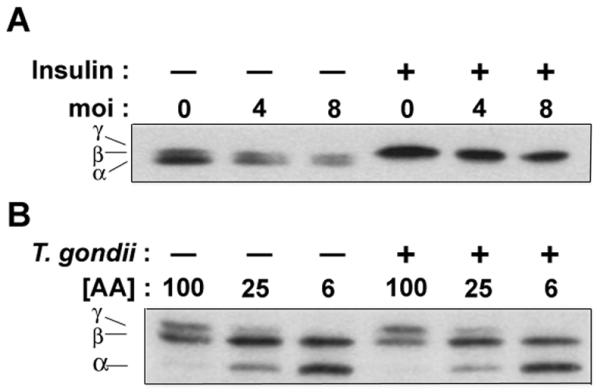
(A) 3T3 cells were infected with T.gondii at the indicated m.o.i for 23.5 h followed by treatment with insulin for 30 min and Western blot analysis. The positions of the α, β, and γ species, representing states of progressively increased phosphorylation (Khaleghpour et al., 1999), are indicated. (B) Mouse embryonic fibroblasts were infected with T.gondii at m.o.i of 8 for 24 h. Amino acid concentration ([AA]) was controlled by dilution of DMEM to the indicated percentages in HBSS. The data in the figure are representative of two independent experiments.
Toxoplasma-induced mTOR is independent of canonical mTOR-regulatory signals
At least three pathways leading to mTOR activation have been described, mediated by either Akt, Erk, or amino acids (principally leucine). Akt activation by class I phosphatidylinositol 3-kinase (PI3K) results in an inactivating phosphorylation of tuberin (TSC2), which functions as a GTPase-activating protein suppressing Rheb, a member of the Ras GTPase superfamily that associates with mTOR and is required for its activation (Bai et al., 2007; Li et al., 2004). In addition to its potential direct activation of mTOR, Rheb may indirectly stimulate mTOR through its ability to bind and activate phospholipase D (PLD)(Sun et al., 2008). PLD is required for mTOR activation in several settings (Foster, 2007) and acts by converting phosphatidylcholine to phosphatidic acid, which can bind directly to mTOR (Veverka et al., 2008). The Erk connection to mTOR activation is similar to that of Akt, as it depends on Erk2-dependent phosphorylation of TSC2 (Arvisais et al., 2006; Ma et al., 2005; Rolfe et al., 2005). The amino acid-dependent pathway, less well characterized, is also dependent on Rheb, as well as the Rag GTPase, but it is independent of TSC2 and is mediated by Vps34, a class III PI3K (Avruch et al., 2008; Gulati et al., 2008; Sancak et al., 2008).
To investigate these pathways as potential mediators of mTOR activation by Toxoplasma, we examined infected cells with respect to the activation of these signals and their contribution to mTOR activity. Akt phosphorylation did not correlate with mTOR activation in T. gondii-infected cells. While Akt activation did occur in HFF, it was delayed relative to pS6 accumulation (Fig. 1B). Furthermore Akt activation was absent in both 3T3 and HeLa cells, and phosphorylation of the Akt substrate GSK-3β was not detected (Fig. 2A). An examination of shorter infection times in 3T3 cells failed to reveal a transient activation of the kinase (Fig. 2B). Nevertheless, both basal and infection-induced mTOR activity were strongly dependent on basal levels of Akt activity in 3T3 cells (Fig. 2C). Inhibitors of PI3-kinase, wortmannin and LY294002, were also inhibitory of both basal and infection-induced mTOR activity in both 3T3 and HeLa cells (Fig. 2D), although this result may be less informative as these compounds can also inhibit mTOR. Overall, these data indicate that, while the PI3K - Akt pathway may play an important permissive role for mTOR activation, the stimulation of mTOR activity in response to infection is not mediated via this pathway.
With respect to Erk activity, a mild stimulation was detected in some experiments at 24 hours post-infection in 3T3 (Fig. 2B) or HeLa cells (data not shown), as well as a transient elevation at 2 hours in 3T3 (Fig. 2B). A biphasic response of Erk activity to T. gondii infection has been previously reported in HFF (Molestina et al., 2008). However, the kinetics of this variable response did not correlate well with the consistent and more uniform increase in pS6 accumulation (Fig. 2B, lower panel). Inhibition of Erk activity with PD98059, an inhibitor of the Erk-activating kinase MKK1, resulted in a partial diminution of both basal and parasite-induced mTOR activity in 3T3 cells (Fig. 2C). Therefore, while MEK/Erk signaling may partially contribute to mTOR function in infected cells, the role of this pathway, as with PI3K - Akt, appears to be permissive and does not provide an explanation for parasite-induced mTOR activation.
We considered the possibility that T. gondii might amplify signaling through the pathway mediating amino acid control of mTOR. In this case, infected cells might be expected to show reduced sensitivity of mTOR activity to amino acid depletion. As shown in Fig. 2E, this is not the case: parasite-induced pS6 accumulation was strongly dependent on amino acids in a dose-dependent manner. In addition, we have observed no effect on pS6 accumulation following siRNA-mediated knockdown of Vps34 in infected cells (data not shown). Therefore it is unlikely that this pathway contributes to parasite-mediated mTOR activation.
Association of mTOR with the parasitophorous vacuole
Since our results do not support a major involvement of the canonical pathways in driving the host mTOR response to Toxoplasma infection, it was necessary to search for additional clues to the mechanism of this response. A notable feature of Toxoplasma is the ability to export parasite-encoded proteins to the surface of the parasitophorous vacuole, and there is evidence suggesting that such proteins can perform a signaling function (Ravindran and Boothroyd, 2008; Saeij et al., 2007). In addition, there is evidence suggesting that the vacuolar membrane is enriched in phosphatidic acid, an important mediator of mTOR activation (Charron and Sibley, 2002). Several different kinds of host vesicles and organelles have been shown to accumulate in the vicinity of the vacuole (Coppens et al., 2006; Martin et al., 2007; Sinai et al., 1997). Since mTOR activity, at least when driven by amino acid signaling, has recently been shown to involve mTOR association with vesicular compartments (Sancak et al., 2008), we considered the possibility that vesicular mTOR might interact with the vacuole as part of an activating mechanism. We therefore investigated the localization of mTOR in infected HeLa cells. As shown in Fig. 3A-C, infected cells uniformly displayed punctate mTOR staining that was preferentially localized to a region surrounding the parasitophorous vacuole. In many infected cells, it was possible to observe a distinct ring of punctate stain approximately coinciding with the vacuolar membrane (Fig. 3B). In comparison, uninfected cells displayed a variable pattern of punctate mTOR staining that was frequently perinuclear (Fig. 3D), as expected for mTOR in amino acid-replete cells (Sancak et al., 2008). In comparison, pS6 was distributed diffusely through the cytoplasm of infected cells as expected (Fig. 3E). Western blot analysis showed that infection did not alter the total level of mTOR (data not shown). These data support the possibility of a functional interaction between mTOR-bearing vesicles and the parasitophorous vacuole.
Figure 3. Localization of mTOR in T. gondii-infected cells.

HeLa cells were infected overnight with YFP-RH (green) and stained for mTOR (A-D) or pS6 (E) using a Cy5-conjugated secondary antibody. Panels A-D display the Cy5 signal in inverted grayscale. In panel E, the red Cy5 signal is merged with a grayscale phase contrast image. Panel B represents the boxed region in panel A, rotated clockwise. Arrowheads indicate mTOR-stained puncta closely adjoined to the parasitophorous vacuole. (C) Illustration of the mTOR-staining pattern in a cell containing two vacuoles. Note that mTOR-stained puncta are associated with both vacuoles. (D) Uninfected culture. (E) Arrowheads indicate infected cells displaying uniform cytoplasmic pS6 stain. Scale bars: 10 μm (A, D, E); 5 μm (B); 6 μm (C).
Activation of mTOR in infected cells is dissociated from downstream events
Activation of mTOR by mitogens and growth factors leads to phosphorylation of S6K1 by mTOR at the hydrophobic motif site T389 within the catalytic domain, a critical event in S6K1 activation (Volarevic and Thomas, 2001). Surprisingly, infection of 3T3 cells resulted in only a transient phosphorylation of S6K1 at T389 at 2 to 4 hours post-infection, even though high levels of pS6 are maintained for 24h hours (Fig. 4A). Transiently induced S6K1 activity cannot account for the sustained elevation of pS6, as the latter is completely reversed by exposure to rapamycin for 1.5 hours (Fig. 1C). Therefore the results imply that while Toxoplasma-activated mTOR may initially interact with its canonical substrate S6K1, this linkage appears to be disrupted after the first several hours of infection. We therefore considered the possibility that at these later time points the infected cell might actively suppress S6K1 activity. To test this possibility, we asked whether overnight infection of 3T3 cells would affect the subsequent activation of S6K1 in response to treatment with insulin for 30 minutes. As shown in Fig. 4B, activation of S6K1 was substantially reduced in the infected culture. Very similar results were obtained with HeLa cells (Fig. 4B) and HFF (data not shown). In cells infected at an moi of 8, the insulin-induced intensity of the pS6K1 band was reduced by 56 percent for 3T3 cells and 51 percent for HeLa. The inhibition of signaling to S6K1 was not the consequence of a general loss of insulin responsiveness, as insulin-stimulated phosphorylation of 4E-BP1 was unimpaired in infected cells (Fig. 6A). Therefore Toxoplasma infection appears to generate a suppressive activity that interrupts signal transduction between mTOR and S6K1.
Figure 4. Absence of sustained S6K1 activation in T.gondii-infected cells.

(A) 3T3 cells were deprived of serum for one day and then infected or not with YFP-RH at m.o.i of 8 for the indicated times prior to harvest and Western blot analysis. The asterisk indicates a novel band of unknown identity found consistently and uniquely in infected cells. (B) 3T3 or HeLa cells were infected at the indicated m.o.i for 23.5 h followed by treatment with insulin (Ins) for 30 min. The data are representative of two (A) or three (B) independent experiments.
The dissociation of mTOR and S6K1 activation suggested that S6 phosphorylation in infected cells is likely to be S6K1-independent. To test this prediction, we analyzed S6 phosphorylation in 3T3 cells treated with S6K1 siRNA. As shown in Fig. 5, treatment with specific siRNA led to a reduction in both total and phosphorylated S6K1, but had no effect on the Toxoplasma-induced level of pS6. Notably, S6K1 knockdown also had no effect on pS6 elevation in response to insulin, in either infected or uninfected cultures. Therefore an mTOR-dependent, S6K1-independent mechanism of S6 phosphorylation is present both in infected and in insulin-stimulated cells.
Figure 5. T. gondii-induced S6 phosphorylation is independent of S6K1.
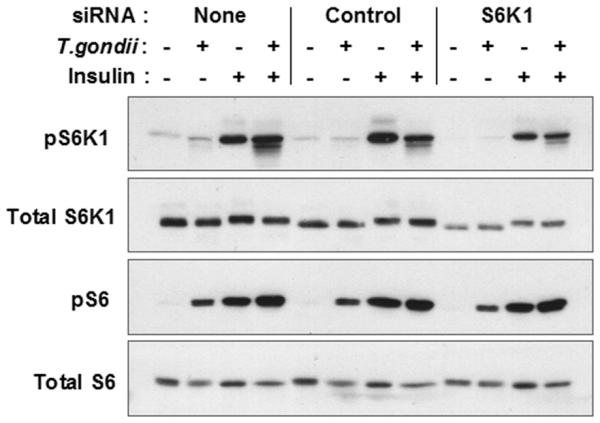
3T3 cells were transfected with the indicated oligoribonucleotides and then infected with YFP-RH for 23.5 h, followed by stimulation with insulin for 30 min. Lysates were prepared for Western blot analysis. The data are representative of two independent experiments.
In addition to S6K1, a second substrate of mTOR that is phosphorylated in response to growth signals is 4E-BP1. Phosphorylation of 4E-BP1 is hierarchical, with phosphorylation at two N-terminal sites preceding phosphorylation at Ser64 and Ser69. These late phosphorylations are associated with the conversion of the faster-migrating α and β isoforms to the slower migrating γ isoform in SDS-PAGE, and also with the dissociation of 4E-BP1 from eIF4E, for which Ser64 phosphorylation appears to be particularly important (Mothe-Satney et al., 2000a; Wang et al., 2005). We assessed 4E-BP1 phosphorylation in T. gondii - infected 3T3 cells. As shown in Fig. 6A, Toxoplasma infection failed to generate detectable levels of γ isoform. In contrast to S6K1, no transient phosphorylation was detected at early time points (data not shown). Therefore the activation of mTOR in infected cells is dissociated from phosphorylation of both S6K1 and 4E-BP1. However, unlike S6K1, whose activation by insulin is partly suppressed by infection, 4E-BP1 was efficiently converted to the γ isoform by insulin in both infected and uninfected cells (Fig. 6A). Similar results were obtained with HeLa cells (data not shown). Amino acid-dependent phosphorylation of 4E-BP1, which was readily observable in serum-starved MEF cells, was similarly unaffected by Toxoplasma infection (Fig. 6B). These results imply a selectivity of the suppressive function of T. gondii within the mTOR pathway.
Toxoplasma promotes mTOR-dependent cell cycle progression and cell growth
The mTOR pathway functions to enhance cell growth and cell cycle progression. We therefore asked whether, in spite of the failure to phosphorylate S6K1 and 4E-BP1, activated mTOR in infected cells could still promote these functions. Quiescent 3T3 cells were infected and assessed for cell cycle progression in the presence or absence of rapamycin. In order to specifically detect cell-autonomous effects of the parasite, the YFP-transgenic strain of T. gondii was employed, so that cells of varying infection level in the same culture could be compared. The ethanol fixation step required for DNA content analysis limits the sensitivity of YFP detection. By comparison of ethanol-treated samples with aliquots fixed with paraformaldehyde alone, in which the signal from single parasites is readily detected, it was estimated that the YFP-positive gate (‘highly infected cells’) in ethanol-treated samples includes cells with eight or more parasites (Fig. 7A), and should therefore include nearly all cells that were productively infected for the entire culture interval. Similar results were obtained in experiments in which the effect of ethanol on parasite enumeration was less pronounced (data not shown). As shown in Fig. 7B and C, infection was a highly efficient inducer of cell cycle entry, leading to the accumulation of 82 percent YFP-positive cells in S phase by 20 hours, compared to < 10 percent in control uninfected cultures. The YFP-negative fraction of the infected culture (‘minimally infected cells’, including uninfected and lightly-infected cells) showed only minor progression. The finding of T. gondii - induced S-phase entry in quiescent fibroblasts is confirmatory of similar results recently obtained by two other groups (Brunet et al., 2008; Molestina et al., 2008); however the data presented here demonstrate for the first time that this is predominantly due to a cell-autonomous effect of the parasite on host cell signaling.
Figure 7. Infection promotes mTOR-dependent cell cycle progression.

3T3 cells were deprived of serum beginning one day prior to infection with YFP-RH at m.o.i. of 2. the cells were cultured for 20 or 30 h in the presence or absence of 200 ng/ml rapamycin (Rap) and then fixed with 0.5% paraformaldehyde (PFA) for 1h. PFA-only aliquots were reserved for parasite enumeration (A, upper panel; D) and the remainder of the samples were treated with ethanol, stained with propidium iodide (PI) and analyzed by flow cytometry. (A) Estimation of parasite content in PI-stained samples. The upper panel displays a YFP histogram for ethanol-treated, PI-stained cells from a culture infected for 30 h (solid line). The shaded area represents the gate defined as YFP-positive (‘highly infected cells’, 57% of the total). An uninfected culture is shown for comparison (dashed line). The lower panel displays a PFA-only aliquot of the same sample. The upper 57% of the distribution is shown to contain cells with at least 8 parasites/cell. Bars indicate fractions containing uninfected cells (U), singly infected cells (1) and cells with 1 - 8 parasites (1-8). (B) Histograms display DNA content for samples cultured for the indicated times. Cells were either from uninfected cultures (shaded), the YFP-negative (‘minimally infected’) fraction of infected cultures (dashed), or the YFP-positive (‘highly infected’) fraction (solid, unshaded). Rap, rapamycin-treated. (C) Data from panel B was analyzed with respect to cell cycle phase distribution and median DNA content (normalized to the mean PI intensity of cells in G1, with G1 DNA content = 2n). (D) The parasite content of total infected cells in PFA-only aliquots was analyzed. YFP intensity was normalized to the mean intensity of free parasites in the same sample. Data in the figure represent the mean ± s.e.m. of triplicates and are representative of two independent experiments.
To determine whether activated mTOR in infected cells was responsible for parasite-induced cell cycle progression, we assessed the effect of rapamycin on cell cycle status. As shown in Fig. 7B, C, treatment with rapamycin severely retarded T. gondii-induced cell cycle progression. At 20 hours, rapamycin restored the G1 and S-phase frequencies of the highly infected fraction nearly to baseline levels. The effect of the drug was even more pronounced when examined with respect to median DNA content (Fig. 7C). At 30 hours, rapamycin-treated infected cells had begun to enter S phase, but their progress was markedly inhibited, as revealed by median DNA content (Fig. 7C). The effect of rapamycin was not due to any alteration of parasite proliferation by the drug (Fig. 7D). These data imply that the level of mTOR activation in infected cells is sufficient to support S phase entry, and that mTOR-mediated promotion of cell cycle progression can occur even in the absence of elevated levels of S6K1 and 4E-BP1 phosphorylation.
We next asked whether the mTOR - dependent cell cycle - promoting effect of T. gondii was redundant with the effects of serum. Quiescent serum-deprived 3T3 cells were briefly infected and then replated at subconfluent density in the presence of serum to initiate cell cycle entry. As shown in Fig. 8, cell cycle progression in the highly infected fraction was enhanced relative to the minimally infected fraction in the same culture. After 24 hours, 19% of minimally infected cells were in G1 and 48% in G2, whereas only 5% of highly infected cells were in G1 and 68% were in G2 (Fig. 8B). Treatment with rapamycin reduced the G2 frequency to 12% for minimally infected and 10% for highly infected cells. Therefore, as evident from Fig. 8B, the effect of the drug was more pronounced in the highly infected relative to the minimally infected fraction, and even more so in comparison with the uninfected culture. We draw two inferences from these findings. First, T. gondii-induced cell cycle progression is nonredundant with serum-induced signals. Second, the heightened rapamycin sensitivity of infected cells suggests that the elevation of mTOR activity in these cells contributes to the observed acceleration of cell cycle progression.
Figure 8. T. gondii-induced cell cycle progression is non-redundant with serum.

3T3 cells were deprived of serum beginning one day prior to infection with YFP-RH at m.o.i. of 4. After 4 h cells were trypsinized and replated at low density in DMEM containing 10% serum with or without 200 ng/ml rapamycin (Rap). Cell cycle analysis was carried out by flow cytometry, using PI-stained and PFA-only aliquots to define minimally-infected and highly-infected gates as for Fig. 6. (A) Histograms display DNA content for uninfected control cultures (thin lines), minimally-infected fractions of infected cultures (dashed lines) and highly-infected fractions (thick lines). (B) Data from panel A was analyzed for cell cycle phase distribution. Data represent the mean ± s.e.m. of triplicates and are representative of two independent experiments.
We next asked whether T. gondii could generate rapamycin-sensitive cell cycle progression in transformed cells whose growth was normally less mTOR - dependent than that of 3T3. The cell cycle status of asynchronously proliferating HeLa cells displayed minimal sensitivity to rapamycin (Fig. 9) The behavior of uninfected cultures was indistinguishable from the minimally infected fraction of infected cultures (data not shown). After 24 hours, highly infected HeLa cells displayed significantly reduced G1 frequency (36 ± 0.6% vs. 52 ± 0.7% for minimally infected) and increased G2 frequency (31 ± 0.6% vs 16 ± 0.3%). This effect of the parasite was sensitive to rapamycin: the gain in median DNA content in the highly infected compared to minimally infected fraction was reduced by 60% in the presence of the drug (Fig. 9C). Therefore, in both primary and transformed cells, infection led to an alteration of cell cycle progression, and the parasite effect was more mTOR-dependent than the progression of uninfected cells.
Figure 9. T. gondii infection leads to increased mTOR-dependence of cell cycle progression in a transformed cell line.

Proliferating HeLa cells were infected with YFP-RH in the presence or absence of 200 ng/ml rapamycin (Rap) for 24 h. Cell cycle analysis was carried out by flow cytometry, using PI-stained and PFA-only aliquots to define minimally-infected and highly-infected gates as for Fig. 6. (A) Histograms display DNA content for uninfected control cultures (0 h), minimally-infected fractions of infected cultures (24 h, dashed lines) and highly-infected fractions (24 h, solid lines). (B) Data from panel A was analyzed for cell cycle phase distribution. (C) Data from panel A was analyzed with respect to median DNA content. PI values were normalized to the mean PI intensity of cells in G1 (G1 DNA content = 2n). Data in the figure represent the mean ± s.e.m. of triplicates and are representative of two independent experiments.
For both 3T3 and HeLa cells, an alternative interpretation of our data is that the accumulation of highly infected cells in G2 (Figs. 8 and 9) represents G2 arrest rather than enhanced cell cycle transit. A recent report by Brunet et al. presents evidence for such G2 arrest after Toxoplasma infection of either human dermal fibroblasts or BeWo choriocarcinoma cells (Brunet et al., 2008), while two studies of infected HFFs suggest either arrest in S/G2 (Molestina et al., 2008) or impaired host cell cytokinesis (Walker et al., 2008). However, when we followed the proliferation of infected 3T3 cells using the vital dye DDAO-SE, we observed no difference in cell division between infected cells and cells from an uninfected culture (Fig 10A). A very similar result was obtained comparing the highly-infected and minimally-infected fractions of DDAO-SE-labeled HeLa cells (Fig. 10B). The behavior of minimally-infected cells was indistinguishable from that of parallel uninfected cultures (data not shown). For HeLa, only G2 cells were assessed, in order to minimize the impact of cell cycle stage-dependent variation in DDAO-SE intensity. To further confirm these findings, mitosis of infected subconfluent HeLa cells was observed by timelapse microscopy. During the 2.5 hour period of observation, 6 of 81 infected cells entered mitosis (in comparison with 2 of 85 uninfected cells), implying that the frequency of M phase in the infected population was at least comparable to that of uninfected cells. This finding is illustrated by the timelapse images in Fig. 10C, which displays three mitoses of infected cells occurring simultaneously in one field. Formation of a metaphase plate in infected cells is illustrated in Fig. 10D. It therefore is likely that our cell cycle data reflect parasite-mediated enhanced progression rather than G2 arrest. The reason for the difference between our results and those of other investigators is unclear, but may relate to the use of different cell lines.
Figure 10. T. gondii does not prevent host cell mitosis.

(A) 3T3 cells were labeled with DDAO-SE and then seeded at 1.6 × 104 cells/cm2 in the presence of serum to permit proliferation. After one day, cells were infected with YFP-RH at m.o.i. of 4 and then collected after 4 h or 28h for PFA fixation and flow cytometric analysis. DDAO-SE intensity is inversely related to cell proliferation. The histogram displays total cells at 4 h (shaded), the uninfected fraction of the infected culture (dashed) and the infected fraction (solid). The data are representative of two separate experiments. (B) HeLa cells were labeled with DDAO-SE, infected with YFP-RH at m.o.i. of 0.5, and then replated at low density and harvested after one or two days. Cell cycle analysis was carried out by flow cytometry, using PI-stained and PFA-only aliquots to define minimally-infected and highly-infected gates as for Fig. 6. The histogram displays the DDAO-SE intensity of G2 cells in either the minimally infected (dashed) or highly infected (solid) fractions in infected cultures. The data are representative of two separate experiments. (C) HeLa cells infected overnight with YFP-RH (yellow) were imaged at 15 min intervals. The displayed field was chosen to illustrate the occurrence of mitosis in three infected cells (arrows). (D) Mouse embryonic fibroblasts were infected with YFP-RH (green) for 24 h. The cells were processed for immunofluorescence analysis to stain tubulin (red) and mounted with DAPI (blue). Brightfield DIC and fluorescent images of the same field are displayed.
We next asked whether T. gondii - induced cell cycle progression is accompanied by normal regulation of cell growth, in spite of the absence of S6K1 and 4E-BP1 phosphorylation. Cellular ribosome content is a sensitive indicator of cell growth in mitogen-treated fibroblasts, as the increase in ribosome content between the quiescent and growing states exceeds the increase in cellular volume and protein content (Becker et al., 1971; Tushinski and Warner, 1982). Since direct measurement of host cell volume in our experiments is complicated by the varying contribution of the parasitophorous vacuole, we chose to assess host cell ribosome content (as indicated by the level of total ribosomal protein S6) as a measure of host cell growth. We compared quiescent serum-starved 3T3 cells infected with T. gondii to similar quiescent cultures that were not infected but were instead replated at low density in serum in order to maximally induce cell growth and proliferation. Immunoblot analysis confirmed that phosphorylation of S6K1 and 4E-BP1 was increased by serum treatment, whereas these events did not occur in the infected, serum-deprived cultures (Fig. 11A). Nevertheless, total cellular S6 content steadily increased as a function of cell cycle stage under both stimuli, and the increase in infected cells was at least as great as that in serum-stimulated cultures (Fig. 11D). This result implies that sustained signaling through S6K1 and 4E-BP1 is not required for the mTOR-dependent growth response in infected cells.
Figure 11. T. gondii-induced cell cycle progression is accompanied by normal host cell growth.

3T3 cells were deprived of serum for one day. Cells were then either trypsinized and replated at one-half original cell density in the presence of serum, or infected with YFP-RH at m.o.i. of 1 in the absence of serum (without replating), or harvested (G0). Cells treated with serum or infected were cultured for 30 h. and then harvested. Aliquots of harvested cells were processed for either Western blot analysis or cell cycle analysis, using PI-stained and PFA-only aliquots to define minimally-infected and highly-infected gates as for Fig. 6. Prior to PI staining, cells were stained with anti-total S6 antibody. (A) Western blot analysis of duplicate samples of either G0, serum-treated or infected cells. (B) Definition of gates used for S6 content analysis. The lower panel represents the highly infected fraction of an infected culture. Shaded segments of the DNA histograms represent the gates used to define G0/G1, early S, mid S and late S fractions. (C) Representative staining of total S6 (solid line) compared to a control with secondary antibody alone (dashed line). The data represent total cells from a G0 sample. (D) The intensity of total S6 staining is displayed for the indicated cell cycle fractions, as defined in panel B. Data represent mean ± s.e.m. for 3 - 6 replicates. The two panels represent two independent experiments (the left panel corresponds to the data shown in panels A - C).
Discussion
Studies of mTOR signaling have largely focused on the effects of microenvironmental signals such as growth factors and nutrients. The current study provides the first evidence that an intracellular parasite can activate mTOR and that the parasite-dependent initiation of host cell S-phase entry occurs via this pathway. It is conceivable that parasite-induced cell cycle progression requires only basal levels of mTOR function, acting in concert with an unidentified parasite-derived signal. However, the correlation we observe between increased mTOR activity and increased rapamycin-sensitivity of cell cycle progression in infected cells argues that stimulation of mTOR is likely to be of functional importance. This correlation was observed for both 3T3 and HeLa cells. These data, as well as the many studies documenting the cell-dependence of rapamycin sensitivity (Noh et al., 2004; Slavik et al., 2001; Zeiser et al., 2008), imply the existence in host cells of a regulated balance between mTOR-dependent and mTOR-independent proliferative pathways, and suggest that the presence of intracellular Toxoplasma tips this balance towards mTOR-dependence. While several mechanisms could produce such an effect, including downregulation of an mTOR-independent pathway, the simplest explanation of our results is that T. gondii-induced mTOR activity is responsible for the increased mTOR-dependence of host cell cycle transit.
A remarkable aspect of our data is that parasite-induced cell cycle progression and cell growth, while mTOR-dependent, take place in the absence of either sustained activation of S6K1 or phosphorylation of 4E-BP1. Ohanna et al. have described independence of mTOR-dependent cell cycle progression from S6K1 for cultured myoblasts from S6K1-deficient mice. While mTOR-mediated S6 phosphorylation remained intact in S6K1-deficient myoblasts, as in our studies with Toxoplasma-infected cells, this phosphorylation did not account for the mTOR-dependent proliferation, which remained unchanged in S6K1/S6K2 double knockout cells that no longer phosphorylated S6 (Ohanna et al., 2005). It therefore seems likely that mTOR-dependent cell cycle progression can proceed via a pathway, as yet uncharacterized, that is independent of both S6K1 and S6. Our study of parasite-infected cells demonstrates, in addition, that such S6K1-independent progression can take place through mTOR signaling that does not involve phosphorylation of 4E-BP1. Consistent with this finding, transfection with non-phosphorylatable mutants of 4E-BP1 had no effect on proliferation in S6K-deficient myoblasts (Ohanna et al., 2005). However, the roles of S6K1 and 4E-BP1 in growth may be cell-dependent. S6K1-deficient mice display a selective defect in pancreatic β-cell size (Pende et al, 2000), and forced expression of hypophosphorylated 4E-BP1 leads to marked inhibition of global translation in HEK293 cells, in a manner directly related to the ability of the mutant proteins to bind eIF4E (Mothe-Satney et al., 2000b). Ongoing studies in our laboratory are directed to examining the ability of mTOR to enhance global translation in parasitized cells in spite of the persistence of hypophosphorylated forms of 4E-BP1. Broad upregulation of host cell translation is indicated by preliminary results showing recruitment of host ribosomal proteins to polysomes in infected cells (data not shown). Alternatively, mTOR function in these cells may be focused to translational enhancement of specific mRNAs. A recent comparison of the alterations in host cell transcriptome and proteome after T. gondii-parasitized cells detected many instances of potential translational regulation in response to infection (Nelson et al., 2008).
Dissociation between mTOR and its effectors, S6K1 and eIF4E, is a highly unusual occurrence. In myotube cultures, amino acid starvation is reported to partly reduce S6K1 activity, while leaving intact mTOR-dependent S6 phosphorylation (Talvas et al., 2006). In our experiments, mTOR-mediated S6 phosphorylation is strictly amino acid-dependent. We are not aware of any previous study in which upregulation of pS6 levels via mTOR is not accompanied by phosphorylation of S6K1 and 4E-BP1. It is possible that this unique pattern represents an adaptation of the parasite. A recent study indicates that phosphorylation of S6 may serve to protect cells from apoptosis mediated by TRAIL, a death receptor ligand expressed by cytotoxic T cells and of potential importance to the control of T. gondii infection (Jeon et al., 2008). On the other hand, both S6K1 and 4E-BP1 have recently been implicated in the mediation of mTOR-dependent effects on cytoskeletal organization (Liu et al., 2008). There is evidence that Toxoplasma actively modifies the host cell cytoskeleton (Coppens et al., 2006) and may therefore need to limit interfering effects derived from host mTOR. The pattern of mTOR-dependent signaling we have observed might represent a parasite-initiated mechanism to optimize host cell survival and structural remodeling.
How might Toxoplasma achieve uncoupling of mTOR activation from phosphorylation of S6K1 and 4E-BP1? Several investigators have demonstrated that these substrates share a N-terminal TOS (TOR Signaling) motif that is essential for efficient phosphorylation by mTOR, and that this motif does not mediate interaction directly with mTOR but rather with the mTOR-binding protein, Raptor (Schalm and Blenis, 2002). One mechanism that could explain our findings is that T. gondii invasion generates a factor that reduces the availability of the TOS-interacting site in the N-terminal portion of Raptor. Under this mechanism, other sites on the Raptor scaffold would remain available for interaction with other potential mTOR substrates, including the kinase responsible for the pS6 generation in infected cells. An obvious candidate for such a kinase is S6K2. Our attempts to assess S6K2 activity have so far been inconclusive (data not shown). A potential difficulty with this mechanism is that S6K2 also contains a TOS motif (Schalm and Blenis, 2002); however, a role for this motif, or of interaction with Raptor, has not been established for this kinase. In the case of at least one other Raptor-binding (and TOS-motif-containing) protein, phospholipase D2, it has been demonstrated that binding can occur to the C-terminal region of Raptor, even when the N-terminal region is simultaneously occupied with S6K1 or 4E-BP1 (Ha et al., 2006). A second difficulty with this mechanism is that it does not account for the ability of insulin to efficiently induce 4E-BP1 phosphorylation in infected cells. However, insulin or serum-induced phosphorylation of 4E-BP1, in contrast to that of S6K1, has been reported to occur in some instances via a rapamycin-insensitive mechanism (Choo et al., 2008; Wang et al., 2005), and we have noted a partial rapamycin-resistance for the generation of the γ band of 4E-BP1 by insulin in 3T3 cells (data not shown). Finally, the possibility must be considered that the absence of phosphorylated S6K1 and 4E-BP1 reflects the parasite-induced activation of specific phosphatase activity rather than a restriction of mTOR function. The phosphatase PP2A has been proposed to act on both S6K1 and 4E-BP1 (Lin and Lawrence, 1997; Peterson et al., 1999).
A further question raised by our results is how Toxoplasma invasion induces S6 phosphorylation. Based on the exquisite sensitivity both to rapamycin and to amino acid withdrawal, as well as dependence on basal Akt activity, we have concluded that the immediate cause of pS6 generation is mTOR activation, although we cannot absolutely rule out the contribution of unknown kinases with similar regulatory features. Our data do not clearly identify a mechanism for host mTOR activation. Canonical pathways that act through Akt or Erk activation, or alternatively via amino acids and Vps34, do not sufficiently account for mTOR activity in infected cells. While basal Akt function was required for mTOR activity, we observed no correlation between mTOR and Akt activation and indeed could detect no elevation of Akt phosphorylation at either Thr308 or Ser473 in infected 3T3 cells. Similarly, we observed a partial dependence of S6 phosphorylation on basal Erk function, but only a modest and variable elevation of Erk activity that did not correlate with pS6 accumulation.
A possible explanation for these negative findings is that T. gondii initiates host mTOR signaling at a point downstream of Akt and Erk. A candidate for this point of intervention is suggested by the observation that host phosphatidic acid, an important downstream mediator of mTOR signaling from several stimuli, becomes highly enriched in a membrane that surrounds the parasite and likely represents the parasitophorous vacuolar membrane (Charron and Sibley, 2002). It is plausible that phosphatidic acid concentrated at this membrane could directly associate with host mTOR and facilitate its activation. Consistent with this hypothesis, we observed that host mTOR was localized to a region surrounding the vacuole, and often to puncta that appeared to line the vacuolar membrane. The hypothesis accounts not only for the independence of the parasite-mediated mTOR response from Akt and Erk stimulation, but also for the dependence of this response on basal Akt and Erk. Activation of mTOR by either exogenous phosphatidic acid or overexpression of phospholipase D1 requires PI3K activity, although Akt activation is not elevated, implying a dependence on basal Akt function (Foster, 2007). Basal Akt signals might enhance mTOR activation by maintaining adequate GTP loading of mTOR-associated Rheb. Amino acid signaling may contribute to the localization of mTOR to Rheb-bearing vesicles (Sancak et al., 2008). Parasite-mediated colocalization of these vesicles to phosphatidic acid at the vacuolar surface may then lead to the induction of mTOR activity.
Alternative mechanisms may link mTOR activation to the trafficking of mTOR-bearing vesicles to the parasitophorous vacuole. One possibility is that the concentration of these vesicles in the vicinity of the vacuole is sufficient to increase mTOR activity, perhaps by facilitating interaction between mTOR-bearing vesicles with vesicles bearing an mTOR-activating factor. Alternatively, parasite-imposed trafficking of these vesicles might alter their maturation in a manner facilitating mTOR activation. Finally, it is possible that vesicular mTOR is activated by signals initiated by a parasite-encoded factor. Toxoplasma is capable of exporting parasite-encoded proteins either to the surface of the parasitophorous vacuole, where contact with host cytosol is possible, or alternatively into host cytosol and nuclei (Ravindran and Boothroyd, 2008). The parasite secretory organelles known as rhoptries contain a number of proteins that can potentially function as kinases or phosphatases that might initiate signal transduction cascades in the host cell, and there is evidence for the export of signaling kinases to the vacuolar surface (Molestina and Sinai, 2005). The rhoptries are thought to be capable of releasing their contents at a very early stage of parasite invasion; nevertheless, at least one rhoptry product, ROP16, is able to generate effects on host cell signaling at least 18 hours post-infection (Saeij et al., 2007), consistent with the kinetics we have observed for S6 phosphorylation. Alternatively, the initiating factor may be a parasite moiety that engages a host cell surface receptor. During the process of attachment and invasion of host cells, Toxoplasma secretes a number of proteins that can bind the host cell membrane, and whose effects on host cells are yet to be characterized (Soldati et al., 2001).
The potential role of the host mTOR pathway in Toxoplasma pathogenesis will require further investigation. We have already alluded to a potential function of pS6 under apoptotic stress, and also to the possibility that the parasite might modify the translational regulation of specific mRNA, for example encoding mediators of host defense. An additional possibility arises from our recent finding that host cell autophagy becomes supportive of parasite growth when ambient amino acid concentration is reduced to physiological levels (Wang et al., 2009). This observation gave rise to the hypothesis that Toxoplasma derives nutritive benefit by driving host amino acids through a ‘futile cycle’ of protein synthesis and degradation (Orlofsky, 2009), resulting in increased amino acid flux through host lysosomes that can be captured by the parasitophorous vacuole (Coppens et al., 2006). It will be of interest to determine the impact of host mTOR impairment under conditions of nutritional stress. The nutritional demand from the parasitophorous vacuole is expected to increase exponentially with time, and therefore the critical pathogenetic role of mTOR under this hypothesis might only occur during the late stages of intracellular residence.
Another noteworthy aspect of our results is the demonstration of the ability of T. gondii - infected cells to progress through an apparently normal complete cell cycle, including cytokinesis. This has not been observed previously, and several recent studies, using cell lines different from the ones studied here, suggest that the cell cycle of infected cells is impaired or incomplete (Brunet et al., 2008; Molestina et al., 2008; Walker et al., 2008). It is possible that parasite interference in the late stages of host cell mitosis is a cell-dependent phenomenon. It will be interesting to determine whether the parasite-dependent reorganization of host microtubules is similar in cell lines that either do or do not progress through mitosis after infection. It may be also be significant that the cell lines in which we observed mitosis (3T3 and HeLa) failed to activate Akt upon infection; in comparison, Akt was activated in infected HFFs, in which defective mitosis has been reported (Walker et al., 2008). Further investigation will be needed to determine the basis for alternative outcomes with respect to host cell cycle completion in Toxoplasma-infected cells.
In summary, this study identifies several novel features of Toxoplasma - host cell interaction: the stimulation of host mTOR; the uncoupling of mTOR signaling from the growth-promoting effectors, S6K1 and eIF4E; and the ability of mTOR to support both cell growth and cell cycle progression in the absence of signaling through these two effectors. Each of these effects points to an area of further investigation that may enhance our understanding of both parasite-host cell coadaptation and the mechanisms underlying the coordination of growth and proliferation in mammalian cells. In light of these findings, it will be of particular interest to assess the impact of T. gondii on host cell translational regulation, and this area is a focus of ongoing studies.
Experimental Procedures
Parasite and mammalian cell culture
YFP-expressing Toxoplasma gondii RH strain (YFP-RH; a kind gift of Dr. B. Striepen, Univ. of Georgia) was maintained in primary human foreskin fibroblasts (HFF). After cell lysis, parasites were recovered by centrifugation and resuspended in Dulbecco's minimal essential medium (DMEM, Invitrogen). All cell culture was performed in DMEM containing (except as indicated) 10% fetal calf serum (HyClone) in the absence of antibiotics at 5% CO2. BALB/c 3T3 cells were obtained from Dr. R. Baserga (Thomas Jefferson Univ.). HeLa cells were obtained from the late Dr. M. Horwitz (Albert Einstein Coll. of Med.). Peritoneal exudate macrophages were obtained as peritoneal lavage from mice injected intraperitoneally four days earlier with 1 ml of 3% thioglycolate broth. Cells were pooled from at least three mice and stored as frozen aliquots. Cells were infected 24 hours after plating in multiwell dishes. Prior to infection, one well was harvested for cell counting to determine multiplicity of infection (m.o.i.). Initial experiments were conducted using cells deprived of serum beginning one day prior to infection. Subsequently, it was observed that similar parasite-dependent signals were obtained when serum was removed at the time of infection. This procedure was routinely adopted (except as indicated), as it was less stressful to host cells. Where indicated, cells were treated with insulin (200 nM; Invitrogen), LY294002 (10 uM; LC Labs), Akti-1/2 (10 uM; EMD Biosciences), PD98059 (20 uM; LC Labs), wortmannin (200 nM; EMD Biosciences) or the indicated concentrations of rapamycin (LC labs).
siRNA transfection
Cells were transfected with either nonspecific oligoribonucleotides or S6K1 RNAi (ON-TARGETplus SMARTpool, L-040893-00, Dharmacon), using lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Cells were replated 24 hours after transfection, infected for 24 hours and then harvested for Western blot analysis.
Immunoblot analysis
Cells were lysed in RIPA buffer (1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS in PBS) supplemented with protease inhibitor and phosphatase inhibitor cocktails (Sigma). Protein concentration was determined by the microbicinchoninic acid assay (Pierce). Ten or twenty micrograms of protein extracts were resolved by SDS-PAGE, followed by transfer to PVDF membranes (Millipore). Membranes were blocked with 5% non-fat milk in TPBS (0.5% Tween-20 in PBS) and subsequently probed with primary antibody in TPBS containing 1% bovine serum albumin (BSA). After overnight incubation at 4°C, blots were washed with TPBS, incubated with horseradish peroxidase-conjugated secondary antibody (KPL) in 5% milk in TPBS for 1 hour at room temperature, and imaged using enhanced chemiluminescence (ECL, Pierce) followed by exposure to X-ray film. Primary antibodies from Cell Signaling include: anti-pAkt (S473) (9271), anti-total Akt (9272), anti-pGsk3β(S9) (9336),anti-pERK1/2 (T202/Y204) (9101), anti-pS6k1 (T389) (9234), anti-total S6K1 (9202), anti-pS6 (S235/236) (2211), anti-total S6 (2217), anti-4E-BP1 (9644). Other primary antibodies include anti-actin (Abcam) and anti-YFP (Becton Dickinson). Band intensities were analyzed using ImageJ.
Immunofluorescence and timelapse imaging
Cells were seeded onto coverslips (Fisher scientific) in a 24-well plate. For analysis, cells were fixed with 4% buffered paraformaldehyde (PFA) and permeabilized with 0.1% Triton X-100. After blocking with 10% fetal bovine serum in PBS, the samples were incubated at 4°C overnight with anti-pS6 (S235/236) diluted in 1% BSA in PBS. Cells were then washed and incubated with Cy5-conjugated anti-rabbit IgG (Jackson ImmunoResearch) for 1 h at room temperature. After extensive washing, the samples were dried and mounted with GOLDEN anti-fade reagent (Invitrogen). Images were collected on a fluorescence microscope (Olympus 1X81) in the Albert Einstein College of Medicine Analytical Imaging Facility. For timelapse imaging, cells were grown in coverslip-thickness chamber slides (Nunc) and infected overnight with YFP-RH at m.o.i. of 4. The medium was replaced with medium containing 20 uM HEPES (pH 7.5) and timelapse images were collected at 15 minute intervals on a Zeiss AxioObserver fluorescence microscope.
Flow cytometry
For vital dye labeling, plates were rinsed with Hank's balanced salt solution (HBSS) and the cells were incubated with 1 uM DDAO-SE (CellTrace Far Red, Invitrogen) in HBSS for 15 minutes and then washed with medium. Cells were harvested with trypsin and single-cell suspensions were prepared by repeated trituration on ice. After centrifugation, cells were suspended in 500 μl cold PBS, followed by addition 500 ul 1% PFA in PBS. After a one hour incubation on ice, cells were washed with PBS containing 0.5% BSA. Aliquots (‘PFA-only’) were reserved for assessment of parasite content in the absence of ethanol treatment. The remainder of the sample was centrifuged and the pellet suspended by the addition of 70% ethanol (-20°C) in a dropwise manner while vortexing. For determination of S6 content, cells were then washed several times with 0.5% BSA in PBS and incubated with 100 ul anti-S6 (1:100 in BSA-PBS) for one hour at room temperature. The cells were then washed, incubated with Cy5-conjugated anti-rabbit IgG for 30 minutes, and washed extensively. Prior to flow cytometry, all samples (except the ‘PFA only’ aliquots) were washed with PBS containing 0.5% BSA, and incubated with 50 μg/ml propidium iodide and 50 μg/ml RNase A for 30 min at 37°C. Flow cytometry was performed on a FacsCalibur (Becton Dickinson). Data were analyzed using FCSExpress (De Novo Software), except for cell cycle analysis, which was performed in FlowJo (TreeStar) using a Watson pragmatic model in which the G2/G1 ratio, as well as the G1 and G2 coefficients of variation, were constrained. Prior to cell cycle analysis, the contribution of parasite DNA to PI intensity was corrected by using compensation software in FCSExpress to remove the dependence of PI intensity on YFP intensity in G1 phase cells. Compensation samples were used to correct for minor spillover from the PI signal (FL3) to Cy5 (FL4).
Acknowledgements
The authors wish to acknowledge Drs. Jonathan Backer, Renato Baserga, Umadas Maitra, Boris Striepen, and Jonathan Warner for valuable suggestions and reagents. We also thank Ms. Yanfen Ma for assistance with parasite preparation. This study was supported by funds from National Institutes of Health grant AI-55358 to A.O and AI-39454 to L.M.W., and by the Flow Cytometry Core of the Center for AIDS Research (AI-51519).
References
- Arvisais EW, Romanelli A, Hou XY, Davis JS. AKT-independent phosphorylation of TSC2 and activation of mTOR and ribosomal protein S6 kinase signaling by prostaglandin F2 alpha. J Biol Chem. 2006;281:26904–26913. doi: 10.1074/jbc.M605371200. [DOI] [PubMed] [Google Scholar]
- Avruch J, Long X, Ortiz-Vega S, Rapley J, Papageorgiou A, Dai N. Amino Acid Regulation of TOR Complex 1. Am J Physiol Endocrinol Metab. 2008 doi: 10.1152/ajpendo.90645.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XC, Ma DZ, Liu AL, Shen XY, Wang QJM, Liu YJ, Jiang Y. Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science. 2007;318:977–980. doi: 10.1126/science.1147379. [DOI] [PubMed] [Google Scholar]
- Becker H, Stanners CP, Kudlow JE. Control of macromolecular synthesis in proliferating and resting Syrian hamster cells in monolayer culture. II. Ribosome complement in resting and early G1 cells. J Cell Physiol. 1971;77:43–50. doi: 10.1002/jcp.1040770106. [DOI] [PubMed] [Google Scholar]
- Bilanges B, Argonza-Barrett R, Kolesnichenko M, Skinner C, Nair M, Chen M, Stokoe D. Tuberous sclerosis complex proteins 1 and 2 control serum-dependent translation in a TOP-dependent and -independent manner. Mol Cell Biol. 2007;27:5746–5764. doi: 10.1128/MCB.02136-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet J, Pfaff AW, Abidi A, Unoki M, Nakamura Y, Guinard M, Klein JP, Candolfi E, Mousli M. Toxoplasma gondii exploits UHRF1 and induces host cell cycle arrest at G2 to enable its proliferation. Cell Microbiol. 2008;10:908–920. doi: 10.1111/j.1462-5822.2007.01093.x. [DOI] [PubMed] [Google Scholar]
- Carraway H, Hidalgo M. New targets for therapy in breast cancer - Mammalian target of rapamycin (mTOR) antagonists. Breast Cancer Res. 2004;6:219–224. doi: 10.1186/bcr927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron AJ, Sibley LD. Host cells: mobilizable lipid resources for the intracellular parasite Toxoplasma gondii. J Cell Sci. 2002;115:3049–3059. doi: 10.1242/jcs.115.15.3049. [DOI] [PubMed] [Google Scholar]
- Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppens I, Dunn JD, Romano JD, Pypaert M, Zhang H, Boothroyd JC, Joiner KA. Toxoplasma gondii sequesters lysosomes from mammalian hosts in the vacuolar space. Cell. 2006;125:261–274. doi: 10.1016/j.cell.2006.01.056. [DOI] [PubMed] [Google Scholar]
- Dmitriev SE, Terenin IM, Dunaevsky YE, Merrick WC, Shatsky IN. Assembly of 48S translation initiation complexes from purified components with mRNAs that have some base pairing within their 5 ′ untranslated regions. Mol Cell Biol. 2003;23:8925–8933. doi: 10.1128/MCB.23.24.8925-8933.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DA. Regulation of mTOR by phosphatidic acid? Cancer Res. 2007;67:1–4. doi: 10.1158/0008-5472.CAN-06-3016. [DOI] [PubMed] [Google Scholar]
- Gulati P, Gaspers LD, Dann SG, Joaquin M, Nobukuni T, Natt F, Kozma SC, Thomas AP, Thomas G. Amino acids activate mTOR Complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab. 2008;7:456–465. doi: 10.1016/j.cmet.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha SH, Kim DH, Kim IS, Kim JH, Lee MN, Lee HJ, Kim JH, Jang SK, Suh PG, Ryu SH. PLD2 forms a functional complex with mTOR/raptor to transduce mitogenic signals. Cell Signal. 2006;18:2283–2291. doi: 10.1016/j.cellsig.2006.05.021. [DOI] [PubMed] [Google Scholar]
- Jayaraman T, Marks AR. Rapamycin-Fkbp12 Blocks Proliferation, Induces Differentiation, and Inhibits Cdc2 Kinase-Activity in A Myogenic Cell-Line. J Biol Chem. 1993;268:25385–25388. [PubMed] [Google Scholar]
- Jeon YJ, Kim IK, Hong SH, Nan H, Kim HJ, Lee HJ, Masuda ES, Meyuhas O, Oh BH, Jung YK. Ribosomal protein S6 is a selective mediator of TRAIL-apoptotic signaling. Oncogene. 2008;27:4344–4352. doi: 10.1038/onc.2008.73. [DOI] [PubMed] [Google Scholar]
- Khaleghpour K, Pyronnet S, Gingras AC, Sonenberg N. Translational homeostasis: Eukaryotic translation initiation factor 4E control of 4E-binding protein 1 and p70 S6 kinase activities. Mol Cell Biol. 1999;19:4302–4310. doi: 10.1128/mcb.19.6.4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurmasheva RT, Huang S, Houghton PJ. Predicted mechanisms of resistance to mTOR inhibitors. Br J Cancer. 2006;95:955–960. doi: 10.1038/sj.bjc.6603353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Corradetti MN, Inoki K, Guan KL. TSC2: filling the GAP in the mTOR signaling pathway. Trends Biochem Sci. 2004;29:32–38. doi: 10.1016/j.tibs.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Lin TA, Lawrence JC. Control of PHAS-I phosphorylation in 3T3-L1 adipocytes: Effects of inhibiting protein phosphatases and the p70(S6K) signalling pathway. Diabetologia. 1997;40:S18–S24. doi: 10.1007/s001250051391. [DOI] [PubMed] [Google Scholar]
- Liu L, Chen L, Chung J, Huang S. Rapamycin inhibits F-actin reorganization and phosphorylation of focal adhesion proteins. Oncogene. 2008;27:4998–5010. doi: 10.1038/onc.2008.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist Updates. 2008;11:32–50. doi: 10.1016/j.drup.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Chen ZB, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk: Implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- Martin AM, Liu T, Lynn BC, Sinai AP. The Toxoplasma gondii parasitophorous vacuole membrane: Transactions across the border. J Eukaryot Microbiol. 2007;54:25–28. doi: 10.1111/j.1550-7408.2006.00230.x. [DOI] [PubMed] [Google Scholar]
- Mieulet V, Roceri M, Espeillac C, Sotiropoulos A, Ohanna M, Oorschot V, Klumperman J, Sandri M, Pende M. S6 kinase inactivation impairs growth and translational target phosphorylation in muscle cells maintaining proper regulation of protein turnover. Am J Physiol. 2007;293:C712–C722. doi: 10.1152/ajpcell.00499.2006. [DOI] [PubMed] [Google Scholar]
- Molestina RE, Sinai AP. Detection of a novel parasite kinase activity at the Toxoplasma gondii parasitophorous vacuole membrane capable of phosphorylating host I kappa B alpha. Cell Microbiol. 2005;7:351–362. doi: 10.1111/j.1462-5822.2004.00463.x. [DOI] [PubMed] [Google Scholar]
- Molestina RE, El Guendy N, Sinai AP. Infection with Toxoplasma gondii results in dysregulation of the host cell cycle. Cell Microbiol. 2008;10:1153–1165. doi: 10.1111/j.1462-5822.2008.01117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothe-Satney I, Brunn GJ, McMahon LP, Capaldo CT, Abrahams RT, Lawrence JC. Mammalian target of rapamycin-dependent phosphorylation of PHAS-I in four (S/T)P sites detected by phospho-specific antibodies. J Biol Chem. 2000a;275:33836–33843. doi: 10.1074/jbc.M006005200. [DOI] [PubMed] [Google Scholar]
- Mothe-Satney I, Yang DQ, Fadden P, Haystead TAJ, Lawrence JC. Multiple mechanisms control phosphorylation of PHAS-I in five (S/T)P sites that govern translational repression. Mol Cell Biol. 2000b;20:3558–3567. doi: 10.1128/mcb.20.10.3558-3567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourani PM, Garl PJ, Wenzlau JM, Carpenter TC, Stenmark KR, Weiser-Evans MCM. Unique, highly proliferative growth phenotype expressed by embryonic and neointimal smooth muscle cells is driven by constitutive Akt, mTOR, and p70S6K signaling and is actively repressed by PTEN. Circulation. 2004;109:1299–1306. doi: 10.1161/01.CIR.0000118462.22970.BE. [DOI] [PubMed] [Google Scholar]
- Murakami M, Ichisaka T, Maeda M, Oshiro N, Hara K, Edenhofer F, Kiyama H, Yonezawa K, Yamanaka S. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol Cell Biol. 2004;24:6710–6718. doi: 10.1128/MCB.24.15.6710-6718.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MM, Jones AR, Carmen JC, Sinai AP, Burchmore R, Wastling JA. Modulation of the host cell proteome by the intracellular apicomplexan parasite Toxoplasma gondii. Infect Immun. 2008;76:828–844. doi: 10.1128/IAI.01115-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh WC, Mondesire WH, Peng JY, Jian WG, Zhang HX, Dong JJ, Mills GB, Hung MC, Meric-Bernstam F. Determinants of rapamycin sensitivity in breast cancer cells. Clin Cancer Res. 2004;10:1013–1023. doi: 10.1158/1078-0432.ccr-03-0043. [DOI] [PubMed] [Google Scholar]
- Ohanna M, Sobering AK, Lapointe T, Lorenzo L, Praud C, Petroulakis E, Sonenberg N, Kelly PA, Sotiropoulos A, Pende M. Atrophy of S6K1(−/−) skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control. Nature Cell Biol. 2005;7:286–294. doi: 10.1038/ncb1231. [DOI] [PubMed] [Google Scholar]
- Orlofsky A. Toxoplasma - induced autophagy: a window into nutritional futile cycles in mammalian cells? Autophagy. 2009 doi: 10.4161/auto.5.3.7807. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pende M, Kozma SC, Jaquet M, Oorschot V, Burcelin R, Marchand-Brustel Y, Klumperman J, Thorens B, Thomas G. Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature. 2000;408:994–997. doi: 10.1038/35050135. [DOI] [PubMed] [Google Scholar]
- Peterson RT, Desai BN, Hardwick JS, Schreiber SL. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycin-associated protein. Proc Natl Acad Sci U S A. 1999;96:4438–4442. doi: 10.1073/pnas.96.8.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroulakis E, Mamane Y, Le Bacquer O, Shahbazian D, Sonenberg N. mTOR signaling: implications for cancer and anticancer therapy. Br J Cancer. 2006;94:195–199. doi: 10.1038/sj.bjc.6602902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Valle F, Braunstein S, Zavadil J, Formenti SC, Schneider RJ. eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J Cell Biol. 2008;181:293–307. doi: 10.1083/jcb.200710215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindran S, Boothroyd JC. Secretion of proteins into host cells by Apicomplexan parasites. Traffic. 2008;9:647–656. doi: 10.1111/j.1600-0854.2008.00723.x. [DOI] [PubMed] [Google Scholar]
- Rolfe M, Mcleod LE, Pratt PF, Proud CG. Activation of protein synthesis in cardiomyocytes by the hypertrophic agent phenylephrine requires the activation of ERK and involves phosphorylation of tuberous sclerosis complex 2 (TSC2) Biochem J. 2005;388:973–984. doi: 10.1042/BJ20041888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvinsky I, Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci. 2006;31:342–348. doi: 10.1016/j.tibs.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich-Rain M, Nir T, Dor Y, Zisman P, Meyuhas O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005;19:2199–2211. doi: 10.1101/gad.351605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeij JPJ, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature. 2007;445:324–327. doi: 10.1038/nature05395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalm SS, Blenis J. Identification of a conserved motif required for mTOR signaling. Curr Biol. 2002;12:632–639. doi: 10.1016/s0960-9822(02)00762-5. [DOI] [PubMed] [Google Scholar]
- Shapira S, Harb OS, Margarit J, Matrajt M, Han J, Hoffmann A, Freedman B, May MJ, Roos DS, Hunter CA. Initiation and termination of NF-kappa B signaling by the intracellular protozoan parasite Toxoplasma gondii. J Cell Sci. 2005;118:3501–3508. doi: 10.1242/jcs.02428. [DOI] [PubMed] [Google Scholar]
- Sinai AP, Webster P, Joiner KA. Association of host cell endoplasmic reticulum and mitochondria with the Toxoplasma gondii parasitophorous vacuole membrane: a high affinity interaction. J Cell Sci. 1997;110:2117–2128. doi: 10.1242/jcs.110.17.2117. [DOI] [PubMed] [Google Scholar]
- Slavik JM, Lim DG, Burakoff SJ, Hafler DA. Uncoupling p70(s6) kinase activation and proliferation: Rapamycin-resistant proliferation of human CD8(+) T lymphocytes. J Immunol. 2001;166:3201–3209. doi: 10.4049/jimmunol.166.5.3201. [DOI] [PubMed] [Google Scholar]
- Soldati D, Dubremetz JF, Lebrun M. Microneme proteins: structural and functional requirements to promote adhesion and invasion by the apicomplexan parasite Toxoplasma gondii. Int J Parasitol. 2001;31:1293–1302. doi: 10.1016/s0020-7519(01)00257-0. [DOI] [PubMed] [Google Scholar]
- Sun Y, Fang Y, Yoon MS, Zhang C, Roccio M, Zwartkruis FJ, Armstrong M, Brown HA, Chen J. Phospholipase D1 is an effector of Rheb in the mTOR pathway. Proc Natl Acad Sci U S A. 2008;105:8286–8291. doi: 10.1073/pnas.0712268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talvas J, Obled A, Fafournoux P, Mordier S. Regulation of protein synthesis by leucine starvation involves distinct mechanisms in mouse C2C12 myoblasts and myotubes. J Nutr. 2006;136:1466–1471. doi: 10.1093/jn/136.6.1466. [DOI] [PubMed] [Google Scholar]
- Tee AR, Blenis J. mTOR, translational control and human disease. Sem Cell Dev Biol. 2005;16:29–37. doi: 10.1016/j.semcdb.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Thomas GV, Tran C, Mellinghoff IK, Welsbie DS, Chan E, Fueger B, Czernin J, Sawyers CL. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12:122–127. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- Tushinski RJ, Warner JR. Ribosomal proteins are synthesized preferentially in cells commencing growth. J Cell Physiol. 1982;112:128–135. doi: 10.1002/jcp.1041120119. [DOI] [PubMed] [Google Scholar]
- Veverka V, Crabbe T, Bird I, Lennie G, Muskett FW, Taylor RJ, Carr MD. Structural characterization of the interaction of mTOR with phosphatidic acid and a novel class of inhibitor: compelling evidence for a central role of the FRB domain in small molecule-mediated regulation of mTOR. Oncogene. 2008;27:585–595. doi: 10.1038/sj.onc.1210693. [DOI] [PubMed] [Google Scholar]
- Volarevic S, Thomas G. Role of S6 phosphorylation and S6 kinase in cell growth. Prog Nucleic Acid Res Mol Biol. 2001;65:101–127. doi: 10.1016/s0079-6603(00)65003-1. 65. [DOI] [PubMed] [Google Scholar]
- Walker ME, Hjort EE, Smith SS, Tripathi A, Hornick JE, Hinchcliffe EH, Archer W, Hager KM. Toxoplasma gondii actively remodels the microtubule network in host cells. Microbes Infect. 2008;10:1440–1449. doi: 10.1016/j.micinf.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XM, Beugnet A, Murakami M, Yamanaka S, Proud CG. Distinct signaling events downstream of mTOR cooperate to mediate the effects of amino acids and insulin on initiation factor 4E-binding proteins. Mol Cell Biol. 2005;25:2558–2572. doi: 10.1128/MCB.25.7.2558-2572.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Weiss LM, Orlofsky A. Host Cell Autophagy Is Induced by Toxoplasma gondii and Contributes to Parasite Growth. J Biol Chem. 2009;284:1694–1701. doi: 10.1074/jbc.M807890200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager N, Brewer C, Cai KQ, Xu XX, Di Cristofano A. Mammalian target of rapamycin is the key effector of phosphatidylinositol-3-OH-initiated proliferative signals in the thyroid follicular epithelium. Cancer Res. 2008;68:444–449. doi: 10.1158/0008-5472.CAN-07-3030. [DOI] [PubMed] [Google Scholar]
- Yoon S, Lee MY, Park SW, Moon JS, Koh YK, Ahn YH, Park BW, Kim KS. Up-regulation of Acetyl-CoA carboxylase alpha and fatty acid synthase by human epidermal growth factor receptor 2 at the translational level in breast cancer cells. J Biol Chem. 2007;282:26122–26131. doi: 10.1074/jbc.M702854200. [DOI] [PubMed] [Google Scholar]
- Zeiser R, Leveson-Gower DB, Zambricki EA, Kambham N, Beilhack A, Loh J, Hou JZ, Negrin RS. Differential impact of mammalian target of rapamycin inhibition on CD4(+)CD25(+)Foxp3(+) regulatory T cells compared with conventional CD4(+) T cells. Blood. 2008;111:453–462. doi: 10.1182/blood-2007-06-094482. [DOI] [PMC free article] [PubMed] [Google Scholar]