

Abstract
A strategy for preparing high relaxivity, metabolically stable peptide-based MR contrast agents is described.
The chemical and topological diversity of peptides offer tremendous possibilities to identify new diagnostic imaging compounds. Peptides have been widely used to target an imaging probe to a specific protein or receptor and thereby provide greater specificity. Typically an imaging reporting moiety (e.g. positron emitter, gamma emitter, paramagnetic ion, near infrared fluorophore) is conjugated to the peptide. The site of conjugation, the linker, and the type of imaging reporter all play a role in determining biological activity and pharmacokinetics.1, 2 For peptide-based magnetic resonance imaging (MRI) contrast agents, an additional factor is detection sensitivity of the imaging probe.3 Multiple copies of the MR active reporter, typically a gadolinium complex, are required to provide robust image contrast.
An additional major challenge to creating new drugs from peptides is peptide degradation by endogenous peptidases. There are numerous medicinal chemistry approaches to improve peptide stability, biological activity, and/or bioavailability that increase in complexity from modified peptides to pseudopeptides to small molecule peptidomimetics.4, 5 In this report, we explore the potential of using the imaging reporter to block peptidase activity.
We,6–9 and others,10–14 have been interested in developing gadolinium-based peptide-targeted MR imaging agents. Compared to other modalities, MRI provides a favorable combination of high spatial resolution, depth penetration, and lack of ionizing radiation. Unlike nanoparticles, these relatively small molecules can rapidly reach targets in extravascular spaces and can be readily excreted through the kidneys to reduce/avoid long-term gadolinium retention and toxicity. On the other hand, extravasation into the kidneys and liver exposes these compounds to a range of peptidases.
There is some flexibility as to where and how the gadolinium chelates are conjugated to the peptide. Conjugation is possible at the N- or C-terminus and/or within the peptide structure.6 We recently reported some fibrin-specific peptides conjugated with one or four [Gd(DTPA)]2− moieties.8 The construct with highest affinity had two peptides linked via their N-terminus to a GdDTPA tetramer, i.e. PepN-Gd4-NPep, termed EP-1084 (Cmpd 1 in Scheme 1). 1 showed good fibrin affinity and specificity, high relaxivity, and positive uptake in an in vivo venous thrombus model. However, subsequent in vivo studies revealed that this compound was being metabolized over the course of the study, and that the Gd-containing metabolites no longer bound fibrin. This communication describes the effort to improve the metabolic stability of this class of compounds while maintaining their biological and relaxometric activity. We demonstrate that the Gd-chelate moieties can be used to block peptidase activity in addition to their role in MRI signal generation. Parts of this work have been communicated previously in conference abstracts.15, 16
Scheme 1.

Compounds used in this study.
Scheme 1 shows the compounds described in this communication which used 3 similar peptides that differ only at the C-terminus. The L-Asp to D-Asp substitution did not affect fibrin affinity, but D-amino acids are known to sometimes provide metabolic stability.4 The Leu to biphenylalanine (Bip) substitution resulted in higher fibrin affinity8 and it was hoped that the unnatural amino acid would improve stability. Peptides are linked to the Gd-chelates via an oxime or amide bond.
A typical in vitro assay for metabolism is to incubate the compound with tissue homogenate or liver microsomes. Rat liver homogenate consists of a mixture of proteases and other enzymes and represents a harsh challenge for compound stability. The half-life of 1 in liver homogenate at 37 °C was 10.8 min, which was consistent with observed instability in vivo. The peptides alone, with or without the Bip or D-Asp substitutions, were completely metabolized within the time taken for measurement, t1/2 < 2 min. The bulky hydrophilic GdDTPA tetramer at the N-terminus appeared to block metabolism. This was supported by studies with the single peptide analog 2 (Gd4-NPep). The half-life for this compound was similar to 1 (10.0 min, Table 1) suggesting that blockade of the C-terminus might be required for enhanced stability. Compound 3 (Bpc-NPep-Gd4) was synthesized with the Gd tetramer conjugated directly to the C-terminus and the N-terminus capped with biphenyl carboxamide (Bpc), a group used to block exopeptidase activity. However this compound also had a similar half-life in rat liver homogenate (9 min). With compound 4, we took this strategy further and capped the C- and N-termini with a GdDTPA dimeric unit (Gd2A-NPep-Gd2A) resulting in a 6 to 7-fold increase in metabolic stability. This suggests that while the D-amino acid and unnatural Bip in 2 and the Bpc in 3 may increase stability, these substitutions are not as effective as using the metal chelate with this specific peptide.
Table 1.
Effect of peptide-chelate architecture on metabolic stability, as assessed by half-life (t1/2) in rat liver homogenate, and affinity to DD(E) (soluble fibrin fragment).
| Compound | Architecture | t1/2[a] [min] | Ki[a] [μM] | Compound |
|---|---|---|---|---|
| 1 (EP-1084) | PepN-Gd4-NPep | 10.8 | 1.1 | 1 (EP-1084) |
| 2 (EP-1086) | Gd4-NPep | 10.0 | 4.7 | 2 (EP-1086) |
| 3 | NPep-Gd4 | 9.0 | 9.1 | 3 |
| 4 | Gd2-NPep-Gd2 | 65.2 | 12.0 | 4 |
| 5 (EP-1242) | Gd2-NPep-Gd2 | >70 | 4.7 | 5 (EP-1242) |
Uncertainties estimated at ±10%.
While metabolic stability was improved with compound 4, unfortunately its fibrin affinity was significantly reduced. Table 1 also shows inhibition constants, Ki, for each compound to inhibit the binding of a fluorescent peptide to the soluble fibrin fragment DD(E). A lower Ki value indicates higher fibrin affinity. The Ki values for 1 and 2 are comparable to the binding dissociation constants to insoluble fibrin reported previously (for 1, Ki (DD(E)) = 1.1 μM, Kd (fibrin) = 1.9 μM). To improve the fibrin affinity of metabolically stable 4, we made the Leu to Bip substitution used in 1 to give compound 5 (Gd2B-Pep-Gd2B), which resulted in a 2–3 fold increase in fibrin affinity, while maintaining the metabolic stability. The other modifications from 4 to 5 were practical: the m-bis-(aminomethyl)-benzene linker is much less expensive than the para analog; the bis(GdDTPA) moiety Gd2B prepared from diethylene triamine gave better yields than Gd2A prepared from diaminopropionic acid. Although the affinity of 5 was still less than 1, it was a simpler and more cost-effective molecule that only utilized one peptide per compound. Compound 5 (aka EP-1242) subsequently showed efficacy in a guinea pig model of arterial thrombosis.17
Based on its favorable fibrin affinity and metatabolic stability properties, we sought to further characterize the MR properties of 5. Nuclear magnetic relaxation dispersion (NMRD) profiles of 5 in either pH 7.4 Tris buffered saline (TBS), 30 μM human fibrinogen/TBS solution, human plasma, or 30 μM gelled human fibrin in TBS are shown in Figure 1. The large enhancement in relaxivity going from TBS to fibrin and the peak in relaxivity at ca. 30 MHz are consistent with binding to fibrin and a reduction in the rotational correlation time (τR). There was little relaxivity enhancement in fibrinogen solution suggesting 5 does not bind/binds weakly to fibrinogen. Some enhanced relaxivity in plasma suggests some weak binding to plasma proteins. Relaxivity in Figure 1 is plotted on a per molecule basis to demonstrate the increased detection sensitivity of 5 compared to commercial [Gd(DTPA)]2−.
Figure 1.
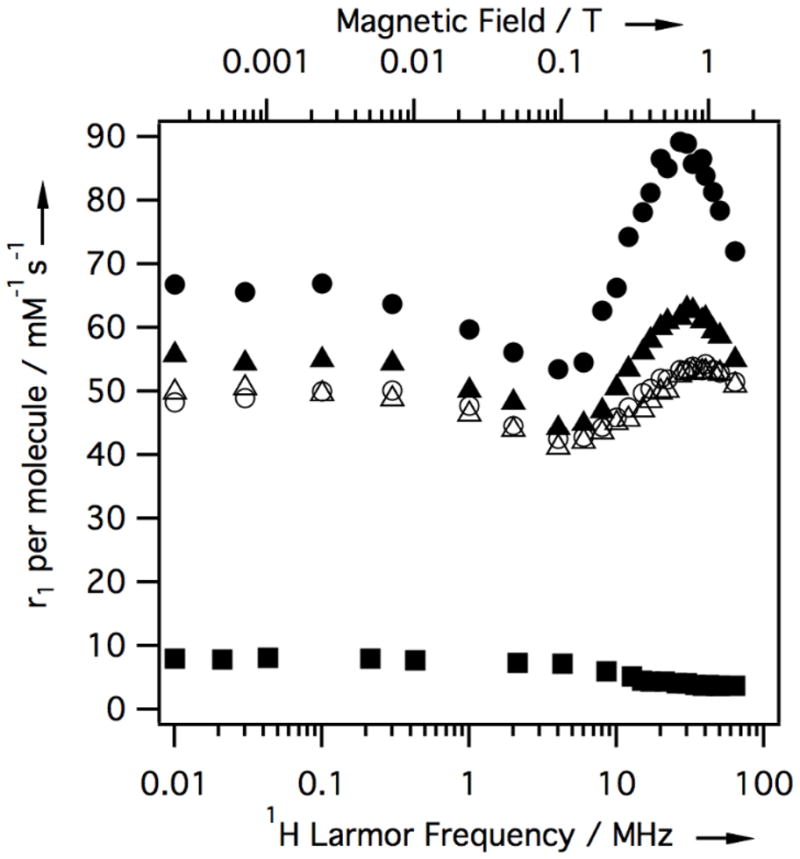
NMRD showing field- and medium-dependent relaxivity of 5 at 35 °C in TBS buffer (open triangles), 30 μM human fibringogen/TBS (open circles), human plasma (filled triangles), and 30 μM fibrin gel (filled circles). NMRD of [Gd(DTPA)]2− in plasma (filled squares) shown for reference.
Figure 2 shows the dual effect of fibrin targeting and relaxation enhancement on thrombus imaging. Two 500 μL solutions were prepared, each containing 30 μM compound 5, in fibrinogen enriched (10 mg/mL) human plasma. One sample was clotted by addition of 4 μL of a 2 M CaCl2 solution and 2 μL of human thrombin (0.6 units). After clotting, the clot was allowed to retract by gentle agitation with an Eppendorf pipet tip. Relative to 5 in plasma (Fig 2A), the signal intensity in the clot (Fig 2B) is higher because of increased concentration of 5 due to binding and also because of higher relaxivity. The concentration of 5 in the serum is depleted resulting in lower signal intensity.
Figure 2.

T1-weighted MR images of 5 in unclotted (A) and clotted (B) human plasma from a 1.5T clinical scanner (General Electric) with a 6 cm surface coil, using a spoiled gradient echo sequence (TE/TR/α: 3/39/40°)
Additional NMRD at 5, 15, 25, and 35 °C for 5 in TBS or in fibrin gel, and variable temperature O-17 solvent relaxation studies (T1, T2) for 5 in TBS were performed to better understand the underlying dynamics influencing relaxation. Figure 3 shows the results of these studies with solid lines as fits to the data. The high field NMRD and O-17 data were analyzed as described previously18 using the usual Solomon Bloembergen Morgan equations with two-site exchange.19 The data were well described with an isotropic model of rotation, and τR of this peptide-gadolinium tetramer is substantially increased compared to [Gd(DTPA)]2− (390 ps vs 44 ps20 at 37 °C) accounting for the increased relaxivity of 5 compared to [Gd(DTPA)]2−. Not surprisingly, the water exchange rate and parameters for electronic relaxation at the backbone modified Gd(DTPA)(H2O) moiety in 5 are very similar to those of [Gd(DTPA)]2− and related derivatives.18, 20
Figure 3.
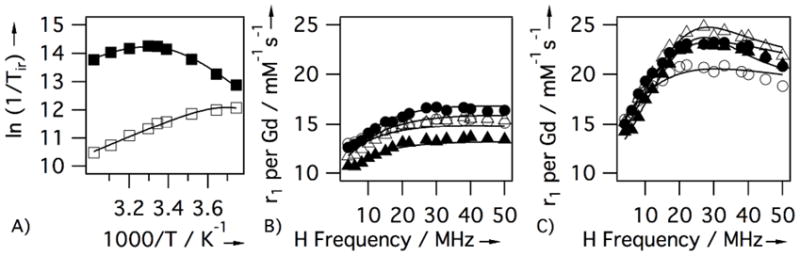
Relaxometric analysis of 5: A) VT O-17 NMR (1/T1 open squares, 1/T2 filled squares) at 7.05T in TBS. VT NMRD in TBS (B) and fibrin gel (C) at 5° (open circles), 15° (filled circles), 25° (open triangles), and 35°C (filled triangles\). Solid lines are fits to data.
The lack of temperature dependence on the relaxivity of fibrin-bound 5 implies that there is substantial internal motion limiting relaxivity. If fibrin binding caused true immobilization, then relaxivity should decrease with decreasing temperature because relaxation would be limited by slow water exchange. The NMRD data of the fibrin-bound 5 was modeled with the Lipari-Szabo formalism where two correlation times describe rotational diffusion: a slow, global correlation time (τR) for the protein-bound compound and a shorter local correlation time for (τl) for internal motion.18, 21 These are weighted by an order parameter, 1 ≥ F2 ≥ 0 where F2=1 represents isotropic global motion and F2=0 represents local motion decoupled from the slow global motion. The NMRD data was well described by adjusting the rotational parameters without changing the electronic relaxation and water exchange parameters from those determined in TBS. Best fits were obtained when the global τR was very long, >20 ns. However F2 was quite small (0.08) indicating that relaxivity of fibrin-bound 5 was limited by internal. As temperature is decreased, internal motion is reduced but this positive effect on relaxivity is offset by a decrease in water exchange rate. The overall effect is a near temperature independence on relaxivity.
The NMRD analysis indicates that relaxivity at 37 °C is not likely to be improved by changing the Gd chelate to one with faster water exchange kinetics since relaxivity at 37 °C is limited by fast internal motion and not water exchange. However reducing internal motion should have a major impact on increasing relaxivity at 1.5T (the field at which the vast majority of clinical MRI scanners operate). Nonetheless, the relaxivity observed for 5 is still quite high: 18 mM−1s−1 per Gd and 72 per molecule bound to fibrin at 1.5T. This is 4–5 fold higher than GdDTPA on a per Gd basis and 18 fold higher per molecule, providing the sensitivity to detect thrombi in vivo.
A similar Gd2-Pep-Gd2 design was used in EP-2104R,9, 22 which has been used to detect thrombus by MRI in animal models and in human clinical trials. 22 EP-2104R uses a different peptide than the compounds in this report, but its metabolic stability may arise in part because of blocking both the C- and N- termini with Gd chelates.
In summary, the metabolic stability of a fibrin-targeted peptide is greatly increased when the Gd chelates are positioned at both the C- and N-termini to block exopeptidase activity. Using multiple Gd chelates also results in increased molecular relaxivity, although NMRD analysis suggests that relaxivity can be even further increased by reducing internal motion. Using more than one site of chelate conjugation may represent a general strategy to increase metabolic stability of peptide-targeted imaging agents.
Experimental
General procedures for synthesis of the protected peptides
Compounds 1 and 2 were prepared as described previously.8 Peptides were synthesized on an automated peptide synthesizer, Advanced ChemTech ACT-348 Ω. Standard Fmoc chemistry was used to elongate the peptide on the resin. The linear peptide containing Cys(ACM) residuals on resin was cyclized with a solution of Tl(TFA)3 (2.2 eq.) in DMF. The cyclic peptide was cleaved from the resin by treating several times with 1% TFA/DCM (10–15 mL/g resin) for 5 min.
Synthesis of tetrakis(DTPA)-3-peptide
The protected cyclic peptide (3-peptide) was prepared by using Fmoc-Asp(OtBu)-SUSRIN resin (0.54 mmol/g). Molecular weight for C88H125N11O20S2: 1721.13. MS (ESI) m/z: calcd: 1722.1 (M+H)+; found: 1722.8. 3-peptide (202 mg, 117 μmol) and tetra(DTPE)-diamine8 (122 mg, 39 μmol) were dissolved in dichloromethane (25 mL) and DMF (25 mL). DIPEA was added dropwise until the pH measured 9, and DIC (15 mg, 117 μmol) and HOBt (21 mg, 137 μmol) were added to the mixture. The mixture was stirred at room temperature overnight. The solvents were removed under reduced pressure. The crude was purified by RP-HPLC with a C4 column (1%TFA/H2O/CH3CN) to provide tetrakis(DTPE)-3-peptide as a white solid (72.6 mg, 15 μmol, 12.9% yield). Molecular weight for C240H397N31O65S2: 4821.02. MS (ESI) m/z: calcd: 1608.0 [(M+3H)/3]3+; found: 1608.3. Tetrakis(DTPE)-3-peptide (72.6 mg, 15 μmol) was dissolved in dichloromethane (2.0 mL) and anisole (2.0 mL) and the solution was stirred at 4 °C for 10 min. To the solution was added concentrated HCl solution (2.0 mL) dropwise. The mixture was stirred at 4 12 °C for 4 h and then to the mixture was added water. The mixture was extracted four times with ether. The aqueous solution was lyophilized to give a crude product that was purified by using RP-HPLC with a C18 column (eluents: 0.1% TFA/H2O/CH3CN). The fractions containing the pure product were pooled and lyophilized to give tetrakis(DTPA)-3-peptide as a white foam (21.9 mg, 6.4 μmol, 42.7% yield). Molecular weight for C140H197N31O65S2: 3418.38. MS (ESI) m/z: calcd: 1710.2 [(M+2H)/2]2+; found: 1710.8.
Synthesis of tetrakis(DTPA)-4-peptide
The protected cyclic peptide (4-peptide) was synthesized by using 1,4-bis-(aminomethyl)-benzene trityl NovaSyn TGT resin (0.32 mmol/g). Molecular weight for C83H127N13O18S2: 1657.89. MS (ESI) m/z: calcd: 830.6 [(M+2H)/2]2+; found: 831.0. To a solution of 4-peptide (187 mg, 0.113 mmol) in DMF (40 mL) and was added a solution of bis(DTPE)-acid-B23 (508 mg, 0.338 mmol) in dichloromethane (40 mL), HOBt (60.4 mg, 0.395 mmol), DIPEA (102 mg, 0.789 mmol) and then DIC (42.7 mg, 0.338 mmol). The mixture was stirred at room temperature overnight. The solvents were removed under reduced pressure to give a pale-yellow oil, which was purified by using RP-HPLC with C4 column (eluents: 0.1% TFA/H2O/CH3CN) to give tetrakis(DTPE)-4-peptide as a white solid (210 mg, 0.045 mmol, 40.1% yield): Molecular weight for C229H383N29O64S2: 4630.78. MS (ESI) m/z: calcd: 1544.6 [(M+3H)/3]3+; found: 1545.2. Tetrakis(DTPE)-4-peptide (210 mg, 0.045 mmol) was deprotected using dichloromethane (8.0 mL), anisole (8.0 mL) and concentrated HCl solution (8.0 mL) to obtein tetrakis(DTPA)-4-peptide as a white foam (55 mg, 0.017 mmol, 38% yield). Molecular weight for C129H183N29O64S2: 3228.14. MS (ESI) m/z: calcd: 1615.1 [(M+2H)/2]2+; found: 1615.3.
Synthesis of tetrakis(DTPA)-5-peptide
The protected cyclic peptide (5-peptide) was synthesized by using 1,3-bis-(aminomethyl)-benzene trityl NovaSyn TGT resin (0.59 mmol/g). Molecular weight for C92H129N13O18S2: 1769.21. MS (ESI) m/z: calcd: 885.61 [(M+2H)/2]2+; found: 885.8. 5-peptide (1.527 g, 0.863 mmol) and bis(DTPE)-acid-A23 (2.964 g, 1.90 mmol) were dissolved in dichloromethane (100 mL) and DMF (100 mL). DIPEA (about 0.76 mL) was added dropwise until the pH measured 9, and DIC (240 mg, 1.90 mmol) and HOBt (330 mg, 2.16 mmol) were added to the mixture. The mixture was stirred at room temperature for two hours. The reaction is monitored using LC/MS. If needed, additional pre-activated bis(DTPE)-acid-A (with DIC, DIPEA, and HOBt) was added in one portion, and this was repeated two hours later. After the reaction was completed, solvents were removed under reduced pressure and the crude product was purified by a RP-HPLC to give tetrakis(DTPE)-5-peptide as a pale yellow oil (807 mg, 0.166 mmol, 19.3% yield). Molecular weight for C244H399N31O64S2: 4851.84. MS (ESI) m/z: calcd: 1618.3 [(M+3H)/3]3+; found 1620.2. Tetrakis(DTPE)-5-peptide (357 mg, 0.074 mmol) was deprotected using dichloromethane (12 mL), anisole (12 mL) and concentrated HCl solution (12 mL) to obtain tetrakis(DTPA)-5-peptide as a white foam (103 mg, 0.030 mmol, 40.2% yield). Molecular weight for C144H199N31O64S2: 3452.42. MS (ESI) m/z: calcd: 1727.2 [(M+2H)/2]2+; found: 1727.6.
General procedure for final compound preparation
All final compounds 3, 4, and 5 were each prepared by reacting the respective tetrakis(DTPA)-peptide with gadolinium chloride in situ. Each tetrakis(DTPA)-peptide was dissolved in a small volume of distilled, deionized water (3 mL), and the pH was adjusted to 6.5 with NaOH. The exact ligand concentration of the solution was determined by photometric titration with standardized gadolinium chloride in 0.02 M xylenol orange (pH 4.9, acetate buffer, monitor at 572 nm). There is a marked increase in absorbance once the endpoint has been reached. Four equivalents of GdCl3 6H2O was added to the tetrakis(DTPA)-peptide solution and the pH adjusted to 6.5 by the addition of NaOH to give an aqueous solution of the final compound 3, 4, or 5. The exact concentration was determined by ICP-MS and contained no excess gadolinium as detected by xylenol orange, nor measurable amounts of underchelated product by photometric titration.
Rat liver homogenate assay for metabolic stability
Rat liver homogenate was prepared from livers isolated from male Sprague Dawley rats.24 Freshly prepared rat liver homogenate (630 μL) was placed in a glass test tube and incubated at 37 °C in a water bath for 4 minutes. To the rat liver homogenate at 37 °C was added 70 μL of a 1 mM solution of test compound. At time points 0, 5, 15, 30, and 60 minutes, a 100 μL aliquot of the reaction mixture was removed and mixed with 100 μL of methanol in a microfuge tube to quench the reaction. The quenched reaction mixture is centrifuged for 3 minutes at 10,000 rpm to pellet the precipitated protein. The supernatant is analyzed by LC-MS to quantify the amount of test compound remaining by comparing the area of the single ion MS peak to that of a series of standards.
DD(E) binding assay
The soluble fibrin fragment DD(E) was prepared as previously described.25 DD(E) used in this study contained subunits of 61 kD and 72kD, assigned to Fragments E1 and E2 and present in a roughly 1:1 ratio, and 180 kD (Fragment DD). Peptide conjugate binding to DD(E) was measured by a fluorescence polarization (FP) assay. The anisotropy (robs) of 0.1 μM fluorescein labeled peptide (Fluor-Aca-LPCDYYGTCLD, where Aca=aminocaproic acid) binding to DD(E) (0 – 20 μM) was measured in TBS. The data were fit to a single site model, equation 1, to obtain the dissociation constant, Kd, for the DD(E)·(fluorescent peptide, “Fl”) complex and reference values for rbd, the anisotropy of “Fl” when fully bound to DD(E) and rfr, the anisotropy in solution. The Kd for the fluorescent peptide binding to DD(E) was 1.3 ± 0.4 μM.
| (1) |
Binding of peptide-chelate conjugates 1, 2, 3, 4, and 5 to DD(E) was measured by fluorescent peptide displacement. DD(E) (1.5 μM) and Fl (1 μM) and competing peptide conjugate (0.1 – 50 μM) were mixed in TBS·Ca. Sample robs (100 μL, n = 3 wells) was measured in a 96 well microplate (Costar, Cat. No. 3915), using a Tecan Polarian FP microplate reader (ex = 485 nm; em = 535 nm). In the presence of inhibitor, an apparent dissociation constant for the fluorescent probe, Kdapp, is determined, eq 2. The inhibition constant, Ki, is related to Kdapp by eq 3 where Kd is the true dissociation constant of the fluorescent probe measured in the absence of inhibitor. Ki values were obtained by least squares fitting of the data as described elsewhere.26
| (2) |
| (3) |
Relaxivity determination
Relaxivities were determined using a a field cycling relaxometer at NY Medical College over the frequency range 0.01 to 50 MHz. Relaxivity was determined for 5 in either pH 7.4 Tris (50 mM) buffered saline (TBS), human plasma, 10 mg/mL (30 μM) fibrinogen in TBS, or 30 μM fibrin gel in TBS. The fibrin gel samples were prepared by first mixing appropriate amounts of fibrinogen stock solution (15 – 20 mg/mL), 5 stock, and TBS to a total volume of 400 μL. To this solution was added 4 μL of a 2 M CaCl2 solution and 2 μL of human thrombin (0.6 units), the resultant solution was vigorously mixed for 3 seconds and then incubated for one hour at 37 °C to allow for complete polymerization of the fibrinogen. Twenty-two data point 1/T1 dispersions were recorded for either a 100 μM 5 solution in TBS, 50 μM 5 in 30 μM fibrin gel, 50 μM 5 in 30 μM fibrinogen solution, 100 μM 5 in human plasma, human plasma alone, 30 μM fibrinogen solution alone, or 30 μM fibrin gel without compound. There are known to be 2 equivalent binding sites on fibrin for these peptides,8 so the NMRD in fibrin gel was recorded under conditions where the [binding sites] > [5]. Based on the measured binding constant, under these conditions 5 is 81% bound to fibrin. Compound concentration was determined from ICP-MS analysis of total Gd content and dividing by the number of Gd/molecule. Relaxivity was computed by subtracting the relaxation rate of the medium (TBS, plasma, fibrinogen, or fibrin gel/TBS) from the relaxation rate of the Gd solution at each field strength and dividing the difference by the gadolinium concentration in millimolar.
Variable temperature O-17 NMR
H217O transverse relaxation rates were determined for a TBS buffer solution in the presence and absence of 11.75 mmolal 5 as a function of temperature (−6 to 92 °C) on a Varian Unity 300 NMR operating at 40.6 MHz. Probe temperatures were determined from ethylene glycol or methanol chemical shift calibration curves. T2 was determined by a CPMG pulse sequence and T1 by inversion recovery. Data were analyzed as previously described.18
Table 2.
Parameters (and their temperature dependence in parentheses) obtained from modeling variable temperature 1H NMRD and H217O relaxation data for 5 in TBS or in 30 μM fibrin gel (FBN).
| τm37[a] (ΔH‡) | τR37 (ΔER‡) | τv37 | Δ2 ×10−18 | F2 | τloc37 (ΔEl‡) | |
|---|---|---|---|---|---|---|
| TBS | 124±9 (45.7±1.4) | 389±4 (24.9±0.4) | 18.0 ±1.7 | 10.2 ±0.5 | 1 | NA |
| FBN | 124[b] (45.7[b]) | >20,000 (NA) | 18[b] | 10.2[b] | 0.08 ±0.02 | 720±20 (30±2) |
units: correleation times τ have units of ps except τm which is ns; activation energies have units kJ·mol−1; Δ2 has units of s2; F2 is dimensionless.
values fixed to those obtained previously for 5 in absence of fibrin.
NA=not applicable.
Notes and references
- 1.Kim YS, He Z, Hsieh WY, Liu S. Bioconjug Chem. 2007;18:929–936. doi: 10.1021/bc0603182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu S. Chem Soc Rev. 2004;33:445–461. doi: 10.1039/b309961j. [DOI] [PubMed] [Google Scholar]
- 3.Caravan P. Acc Chem Res. 2009;42:851–862. doi: 10.1021/ar800220p. [DOI] [PubMed] [Google Scholar]
- 4.Adessi C, Soto C. Curr Med Chem. 2002;9:963–978. doi: 10.2174/0929867024606731. [DOI] [PubMed] [Google Scholar]
- 5.Sawyer TK. In: Peptide-Based Drug Design. Taylor MD, Amidon GL, editors. American Chemical Society; Washington: 1995. p. 387. [Google Scholar]
- 6.Caravan P, Das B, Deng Q, Dumas S, Jacques V, Koerner SK, Kolodziej A, Looby RJ, Sun WC, Zhang Z. Chem Commun. 2009:430–432. doi: 10.1039/b819098d. [DOI] [PubMed] [Google Scholar]
- 7.Caravan P, Das B, Dumas S, Epstein FH, Helm PA, Jacques V, Koerner S, Kolodziej A, Shen L, Sun WC, Zhang Z. Angew Chem Int Ed Engl. 2007;46:8171–8173. doi: 10.1002/anie.200700700. [DOI] [PubMed] [Google Scholar]
- 8.Nair S, Kolodziej AF, Bhole G, Greenfield MT, McMurry TJ, Caravan P. Angew Chem Int Ed Engl. 2008;47:4918–4921. doi: 10.1002/anie.200800563. [DOI] [PubMed] [Google Scholar]
- 9.Overoye-Chan K, Koerner S, Looby RJ, Kolodziej AF, Zech SG, Deng Q, Chasse JM, McMurry TJ, Caravan P. J Am Chem Soc. 2008;130:6025–6039. doi: 10.1021/ja800834y. [DOI] [PubMed] [Google Scholar]
- 10.Amirbekian V, Aguinaldo JG, Amirbekian S, Hyafil F, Vucic E, Sirol M, Weinreb DB, Le Greneur S, Lancelot E, Corot C, Fisher EA, Galis ZS, Fayad ZA. Radiology. 2009;251:429–438. doi: 10.1148/radiol.2511080539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Leon-Rodriguez LM, Ortiz A, Weiner AL, Zhang S, Kovacs Z, Kodadek T, Sherry AD. J Am Chem Soc. 2002;124:3514–3515. doi: 10.1021/ja025511v. [DOI] [PubMed] [Google Scholar]
- 12.Morisco A, Accardo A, Gianolio E, Tesauro D, Benedetti E, Morelli G. J Pept Sci. 2009;15:242–250. doi: 10.1002/psc.1087. [DOI] [PubMed] [Google Scholar]
- 13.Poduslo JF, Wengenack TM, Curran GL, Wisniewski T, Sigurdsson EM, Macura SI, Borowski BJ, Jack CR., Jr Neurobiol Dis. 2002;11:315–329. doi: 10.1006/nbdi.2002.0550. [DOI] [PubMed] [Google Scholar]
- 14.Ye F, Jeong EK, Jia Z, Yang T, Parker D, Lu ZR. Bioconjug Chem. 2008;19:2300–2303. doi: 10.1021/bc800211r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caravan P, Kolodziej AF, Greenwood JM, Witte S, Looby RJ, Zhang Z, Spiller M, McMurry TJ, Weisskoff RM, Graham PB. Proc. 10th ISMRM Scientific Sessions; Honolulu, HI, USA. 2002. p. 217. [Google Scholar]
- 16.Zhang Z, Nair S, Wang X, Kolodziej AF, Case A, Caravan P, Greenfield MT, Graham PB, McMurry TJ. Proc. 4th Soc. Mol. Imaging; Cologne, Germany. 2005. [Google Scholar]
- 17.Sirol M, Aguinaldo JG, Graham PB, Weisskoff R, Lauffer R, Mizsei G, Chereshnev I, Fallon JT, Reis E, Fuster V, Toussaint JF, Fayad ZA. Atherosclerosis. 2005;182:79–85. doi: 10.1016/j.atherosclerosis.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Caravan P, Parigi G, Chasse JM, Cloutier NJ, Ellison JJ, Lauffer RB, Luchinat C, McDermid SA, Spiller M, McMurry TJ. Inorg Chem. 2007;46:6632–6639. doi: 10.1021/ic700686k. [DOI] [PubMed] [Google Scholar]
- 19.Bertini I, Luchinat C, Parigi G. Solution NMR of Paramagnetic Molecules. Elsevier; Amsterdam: 2001. [Google Scholar]
- 20.Powell DH, Ni Dhubhghaill OM, Pubanz D, Helm L, Lebedev YS, Schlaepfer W, Merbach AE. J Am Chem Soc. 1996;118:9333–9346. [Google Scholar]
- 21.Lipari G, Szabo A. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 22.Spuentrup E, Botnar RM, Wiethoff AJ, Ibrahim T, Kelle S, Katoh M, Ozgun M, Nagel E, Vymazal J, Graham PB, Gunther RW, Maintz D. Eur Radiol. 2008;18:1995–2005. doi: 10.1007/s00330-008-0965-2. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Z, Amedio J, Caravan P, Dumas S, Kolodziej A, McMurry TJ. Peptide-based multimeric targeted contrast agents. 7,238,341 B2. US. Issued July 3, 2007.
- 24.Graham J, Harris JR. In: Cell Biology Protocols. Harris JR, Graham J, Rickwood D, editors. Wiley and Sons; New York: 2008. p. 88. [Google Scholar]
- 25.Moskowitz KA, Budzynski AZ. Biochemistry. 1994;33:12937–12944. doi: 10.1021/bi00248a001. [DOI] [PubMed] [Google Scholar]
- 26.Feng S, Kasahara C, Rickles RJ, Schreiber SL. Proc Natl Acad Sci U S A. 1995;92:12408–12415. doi: 10.1073/pnas.92.26.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]