

Abstract
We report that rapamycin, an allosteric inhibitor of certain but not all actions of the key cellular kinase mammalian target of rapamycin (mTOR), protects neurons from death in both cellular and animal toxin models of Parkinson's disease (PD). This protective action appears to be attributable to blocked translation of RTP801/REDD1/Ddit4, a protein that is induced in cell and animal models of PD and in affected neurons of PD patients and that causes neuron death by leading to dephosphorylation of the survival kinase Akt. In support of this mechanism, in PD models, rapamycin spares phosphorylation of Akt at a site critical for maintenance of its survival-promoting activity. The capacity of rapamycin to provide neuroprotection in PD models appears to arise from its selective suppression of some but not all actions of mTOR, as indicated by the contrasting finding that Torin1, a full catalytic mTOR inhibitor, is not protective and induces Akt dephosphorylation and neuron death.
Introduction
Parkinson's disease (PD) is characterized by neurodegeneration of specific populations of central and peripheral neurons, including those in the substantia nigra pars compacta (SNpc) and sympathetic ganglia (Fahn, 1998; Dauer and Przedborski, 2003; Marras and Lang, 2008). Current treatments principally ameliorate clinical manifestations of the disease rather than the underlying neuron degeneration and death. This reflects an incomplete understanding of the pathways that lead to neuron loss in PD (Levy et al., 2009).
A SAGE screen (Ryu et al., 2005) designed to uncover novel participants (and potential therapeutic targets) in PD-related neuron death revealed RTP801 (REDD1; Ddit4) as a gene that is highly induced in a cellular toxin model of PD. RTP801 protein and transcripts are induced by a variety of stresses relevant to PD, including oxidative stress, DNA damage, mitochondrial dysfunction, and endoplasmic reticulum stress (Ellisen et al., 2002; Shoshani et al., 2002; Wang et al., 2003; Schwarzer et al., 2005) as well as in various cellular PD toxin models (Malagelada et al., 2006). In addition, RTP801 is induced in dopaminergic SN neurons in a mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of PD and in dopaminergic SN neurons of PD patients (Malagelada et al., 2006).
RTP801 has variable effects on survival of non-neuronal cells but induces apoptotic death of all neuronal types tested to date (Shoshani et al., 2002; Malagelada et al., 2006, 2008). Significantly, RTP801 knockdown protects from death in cellular models of PD (Malagelada et al., 2006). RTP801 promotes neuron death by suppressing activation of mammalian target of rapamycin (mTOR) (Malagelada et al., 2006), a kinase that regulates key cellular functions (Fingar and Blenis, 2004; Swiech et al., 2008). This mTOR blockade in turn suppresses activation of Akt, a protein kinase with a major role in maintaining neuron survival (Dudek et al., 1997; Franke et al., 1997a,b), and it is this action that appears to underlie, at least in part, the proapoptotic actions of PD toxins and RTP801 (Malagelada et al., 2008). In consonance with this mechanism, SN neurons of PD patients possess substantially lower levels of phosphorylated (but not total) Akt (Malagelada et al., 2008).
Rapamycin is a macrolide that specifically inhibits certain mTOR actions by allosterically modulating access of a subset of substrates to the mTOR catalytic site (Sabers et al., 1995; Snyder et al., 1998; Pong and Zaleska, 2003). Thus, in contrast to RTP801, which apparently inhibits all mTOR actions by suppressing its activation by the Rheb GTPase (Brugarolas et al., 2004; Sofer et al., 2005), rapamycin blocks only a subset of mTOR activities (Lorenz and Heitman, 1995; Choo et al., 2008; Thoreen et al., 2009). One relevant consequence is that rapamycin suppresses mTOR-dependent translation of some classes of mRNAs but not others (Choo et al., 2008).
Here, we report that rapamycin blocks RTP801 induction in both in vitro and in vivo toxin models of PD. This appears to occur at the level of mTOR-controlled translation. Consistent with the role of RTP801 in mediating neuron death, rapamycin is also protective in such models. The contrasting actions of RTP801 and rapamycin in neuron death and survival appear to be attributable to their differential actions on mTOR and on regulating Akt phosphorylation.
Materials and Methods
Antibodies, plasmids, and materials
Anti-RTP801 antiserum was purchased from Millipore Bioscience Research Reagents or from Proteintech Group. Anti-ERK1 antibody was obtained from Santa Cruz Biotechnology. Anti-horseradish peroxidase secondary antibodies were obtained from Pierce. Antibodies against phospho-Ser473–Akt, phospho-Thr308–Akt, total Akt, phospho-Ser2448–mTOR, phospho-Ser235/236–S6, phospho-Thr37/46–4EBP1, phospho-Ser371–p70S6kinase, and p70S6kinase were obtained from Cell Signaling Technology. Tyrosine hydroxylase (TH) polyclonal antibody was purchased from Calbiochem EMD Biosciences. Donkey anti-rabbit or anti-mouse secondary antibodies conjugated with Alexa 488 or Alexa 568 were purchased from Invitrogen.
RTP801 constructs were generated as described previously (Malagelada et al., 2006). All newly made constructs were verified by DNA sequencing. 6-OHDA was purchased from Sigma or Tocris Bioscience. Rapamycin was purchased from Calbiochem or LC Laboratories for cell culture treatment or in vivo experiments, respectively. Cycloheximide was purchased from Calbiochem EMD Biosciences. FK506 was purchased from Axxora. pcDNA3 wild-type (WT) mTOR was a kind gift from Dr. Gary G. Chiang (Burnham Institute for Medical Research, La Jolla, CA) (Chiang and Abraham, 2005). Torin1 was a kind gift from Dr. D. M. Sabatini (Harvard University, Cambridge, MA).
Cell culture
PC12 cells were cultured and treated with nerve growth factor (NGF) as described previously (Greene and Tischler, 1976) For NGF treatment, the cells were cultured in RPMI 1640 medium (Cellgro) supplemented with 1% horse serum, penicillin/streptomycin, and 50 ng/ml recombinant human NGF (a kind gift from Genentech) for 8–10 d. Medium was changed every other day and immediately before treatments. 6-OHDA and 1-methyl-4-phenylpyridinium (MPP+) were prepared before use in 10 and 100 mm stocks, respectively, and diluted in medium to the indicated final concentrations. Neonatal rat superior cervical ganglion sympathetic neurons were cultured as described previously (Ryu et al., 2002; Malagelada et al., 2006, 2008). Treatments with 6-OHDA were performed at day 7 in vitro, and cell viability was assessed 24 h later as reported previously (Malagelada et al., 2006). Values represent the mean ± SEM of at least three different experiments. Evaluation of proportions of neurite-bearing cells was performed as described previously (Greene and Tischler, 1976) by counting the proportion of cells that have the neurites at least twice as long as the diameter of their soma.
Quantitative reverse transcription-PCR
Each sample of total RNA was isolated from neuronal PC12 cells by using TRI reagent (Molecular Research Center). cDNA was transcribed from total RNA with First-Strand cDNA Synthesis for Quantitative RT-PCR kit (Marlingen Biosciences). The primers used for quantitative-PCR amplification of RTP801 were 5′-GCTCTGGACCCCAGTCTAGT-3′ and 5′-GGGACAGTCCTTCAGTCCTT-3′.
Equal amounts of cDNA template were used for each quantitative PCR analysis of RTP801 and A-tubulin. Quantitative PCR was performed using a Cepheid SmartCycler following the specifications of the manufacturer. A-tubulin was used for normalization of RTP801 transcripts. cDNA was added to a 25 μl volume reaction mix containing Ready-to-Go Beads (GE Healthcare) or OmniMix HS master mix (Cepheid) and SYBR Green I (Invitrogen) together with appropriate primers at 0.2 μm each. Analyses of amplification curves of real-time fluorescence and of melting curves were performed as described previously (Troy et al., 2001).
RTP801 half-life
Cultures of neuronal PC12 cells were treated with 1 μm rapamycin for 1 h and then with 1 mm MPP+ for 8 h in the continued presence of rapamycin. The cultures were then treated with 1 μm cycloheximide for 0, 5, or 10 min and then harvested and subjected to Western immunoblotting for RTP801 and ERK1. The relative optical densities of the RTP801 signals were determined and normalized to that for ERK1. There were no evident effects of cycloheximide on ERK1 expression levels over the 5–10 min course of the experiment. The half-life of RTP801 protein under each condition was calculated from the slope derived by plotting the natural log of values for relative RTP801 expression versus time of cycloheximide treatment and fitting the data to a straight line using least-squares analysis.
Transfection
Neuronal PC12 cells were transfected with Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. At 48 h after transfection, cells were treated with 6-OHDA or MPP+. At 24 h after the treatments, viable transfected (as judged by expression of fluorophore) neuronal PC12 cells were scored by strip counting under an epifluorescence microscope (Malagelada et al., 2006).
In vivo MPTP treatments in mice
Rapamycin preparation.
Rapamycin (LC Laboratories) was dissolved in 0.2 ml of 100% ethanol and then diluted 100-fold with 40% propyleneglycol to obtain a final concentration of 0.75 mg/ml (Zhou et al., 2009).
Animals and MPTP regimens.
Ten-week-old, male, C57BL mice (Charles River Laboratories) were divided into four groups of 3–10 mice (MPTP/rapamycin, MPTP/vehicle, saline/rapamycin, and saline/vehicle) and were subjected to either an acute or a subacute MPTP regimen.
Acute regimen.
For additional details, see the study by Jackson-Lewis et al. (1995). Animals from each group received one intraperitoneal injection of MPTP-HCl (18 mg/kg free base suspended in saline; Sigma-Aldrich) or saline every 2 h for a total of four doses over an 8 h period in 1 d and were killed at the indicated time points after the last MPTP/saline injection. Rapamycin (7.5 mg/kg) (Zhou et al., 2009) or vehicle was administered intraperitoneally daily for 2 d before the first MPTP/saline injection and continued for 4 d after the last MPTP/saline injection. On the day of the MPTP/saline injections, rapamycin/vehicle was administrated 30 min before first MPTP/saline injection.
Subacute regimen.
For additional details, see the study by Tatton and Kish (1997). Animals from each group received one intraperitoneal injection of MPTP-HCl daily (30 mg/kg free base in saline) for 5 d and were killed at the indicated time points after the last MPTP injection. Rapamycin/vehicle was administered intraperitoneally daily, starting 2 d before the first MPTP/saline injection and continuing for 4 d after the last MPTP/saline injection. Rapamycin/vehicle was administrated 30 min before each MPTP/saline injection.
Quantification of dopamine neurodegeneration.
The total numbers of TH-positive (TH+) SNpc neurons were determined in the acute model at 7 d after the last MPTP or vehicle injection by unbiased stereology using the optical fractionator method (StereoInvestigator; MBF Bioscience). Quantification of the numbers of condensed chromatin SN cells in the subacute model were assessed 2 d after the last MPTP injection in the subacute regimen as described previously (Vila et al., 2001).
Western blot analysis.
Whole-cell extracts or mouse midbrain extracts were analyzed as described previously (Malagelada et al., 2006).
Statistics
All experiments were performed at least in triplicate, and results are reported as means ± SEM. Student's t test was performed as unpaired, two-tailed sets of arrays and presented as probability (p) values. ANOVA with Bonferroni's multiple comparison test or the Newman–Keuls test was performed to compare multiple groups with the control group.
Results
mTOR protects against 6-OHDA-induced death
Our past studies have indicated that PD toxins induce RTP801 and that RTP801 in turn causes neuron death by suppressing activation of mTOR (Malagelada et al., 2006, 2008). If this mechanism is correct, then restoring mTOR activity should prevent neuronal death induced by a PD-mimicking toxin such as 6-OHDA. Because 6-OHDA reduces but does not completely block mTOR activity (Malagelada et al., 2006), we reasoned that WT mTOR overexpression should provide a sufficient level of the activated kinase to confer neuroprotection. Accordingly, overexpression in neuronally differentiated PC12 cells of WT mTOR (Edinger et al., 2003) protected from 6-OHDA (Fig. 1 a). Conversely, overexpression of a kinase-inactive form of mTOR failed to rescue the cells from 6-OHDA (data not shown). Immunostaining for phosphorylated epitopes that are targets of mTOR or of its target, p70S6 kinase, confirmed that overexpression maintained mTOR kinase activity in presence of 6-OHDA (Fig. 1 b–d).
Figure 1.

mTOR overexpression protects from 6-OHDA. a, Neuronal PC12 cells were transfected with either enhanced green fluorescent protein (eGFP)/pcDNA3 (control) or eGFP/pcDNA3 WT mTOR and, after 48 h, were treated with or without 50 μm 6-OHDA. Replicate cultures were pretreated for 1 h with 1 μm rapamycin as indicated before 6-OHDA treatment and maintained with rapamycin for the duration of the experiment. One day later, transfected viable cells were scored under fluorescence microscopy. Values are expressed as mean ± SEM of at least three independent experiments in triplicate in each condition. ***p < 0.01 compared with cells transfected with eGFP/pcDNA3 empty vector; **p < 0.01 compared with cells transfected with eGFP/pcDNA3 empty vector exposed to 50 μm 6OHDA. b–d, Neuronal PC12 cells were transfected with eGFP/pcDNA3 or eGFP/pcDNA3 WT mTOR vector. After 48 h, cultures were treated with 50 μm 6-OHDA for 24 h. Cultures were then immunostained with antibodies to phopsho-Ser2448–mTOR, phospho-Ser235/236–S6, and phospho-Thr37/46–4EBP1, respectively, along with antiserum to GFP to visualize transfected GFP+ cells. Proportions of transfected cells positive for phospho-mTOR, phospho-S6, or phospho-4EBP1 were scored under fluorescence microscopy. Values represent means ± SEM for three independent experiments done in triplicate. *p < 0.05 compared with cells transfected with pcDNA3 empty vector treated with 6-OHDA. e, Neuronal PC12 cells were exposed to 250 nm or 1 μm Torin1 for 5 h. Cell extracts were then collected and subjected to SDS-PAGE and Western immunoblotting. Membranes were probed with the indicated immunoreagents and reprobed with an antibody for total Akt, as a loading control. M.W., Molecular weight; Ct, control.
To further assess the role of mTOR in neuronal survival, we treated cultures with 250 nm Torin1, a highly selective ATP-competitive inhibitor of all mTOR activities (Thoreen et al., 2009). This induced death of 22 ± 4% of the cells by 24 h (**p < 0.01 vs control untreated cells; result represents mean ± SEM of three independent experiments, done at least in triplicate). Immunoblot analysis of mTOR-dependent phosphorylations at 5 h of Torin1 treatment confirmed inhibition of mTOR activity at this time (Fig. 1 e). However, there was no inhibition at 24 h (data not shown), suggesting that Torin1 was either ineffective or unstable after more prolonged times. This may explain why Torin1 was not more effective in inducing death. Together, these findings indicate that activated mTOR provides neuroprotection, whereas mTOR inhibition induces neuronal death.
Rapamycin blocks RTP801 induction by PD toxins
If mTOR activity is required for neuron survival and this is inhibited by RTP801, then strategies that suppress RTP801 induction should be neuroprotective. Because there are often reciprocal control pathways between mTOR and its regulators (Huang and Manning, 2009), we assessed whether partial inhibition of mTOR function might affect RTP801 expression. We therefore tested the effect of rapamycin on RTP801 induction in 6-OHDA-treated PC12 cell cultures. As shown in Figure 2 a, we found inhibition at 0.1 and 1 μm (but not 20 nm) rapamycin. Western immunoblotting confirmed that phosphorylation of p70S6 kinase at Ser371, which is mTOR dependent and rapamycin sensitive (Burnett et al., 1998), was also suppressed (Fig. 2 a,b). Finally, rapamycin also blocked RTP801 induction in neuronal PC12 cells by a second PD-mimetic toxin, MPP+ (Fig. 2 c).
Figure 2.

Rapamycin blocks upregulation of RTP801 protein by PD mimetics. a, Neuronal PC12 cells were pretreated with or without 0.1 or 1 μm rapamycin (top) or with 20 nm rapamycin (bottom) and exposed to 100 μm 6-OHDA for 8 h. Cell extracts were subjected to SDS-PAGE and Western immunoblotting. Membranes were probed with the indicated immunoreagents and reprobed with antibodies for ERK1/2 or total Akt as loading controls. Right panels show quantification (means ± SEM) from three or more independent experiments. RTP801 relative expression (normalized to ERK1/2): *p < 0.05 versus untreated control (Ct) cultures; ++ p < 0.01, +++ p < 0.001 versus 6-OHDA-treated cultures. phospho-S6K relative expression (normalized to ERK1/2): **p < 0.01 versus untreated control cultures; + p < 0.05 versus 6-OHDA-treated cultures. b, Neuronal PC12 cells were pretreated with or without 1 μm rapamycin or 1 μm FK506 and then exposed to 100 μm 6-OHDA for 8 h. Cell extracts were subjected to SDS-PAGE and Western immunoblotting. Membranes were probed with the indicated immunoreagents and reprobed with antibodies for total S6K and ERK1. The vertical white line between lanes in b (bottom) indicates elimination of irrelevant intervening lanes in the blot. c, Neuronal PC12 cells were pretreated with or without 1 μm rapamycin and then exposed to 100 μm 6-OHDA or 500 μm MPP+ for 8 h. Cell extracts were subjected to SDS-PAGE and Western immunoblotting. Membranes were probed with the indicated immunoreagents and reprobed with an antibody for total ERK1 as loading control. Right panel shows quantification (means ± SEM; normalized to ERK1/2) from three or more independent experiments. RTP801 relative expression: *p < 0.05, **p < 0.01 versus untreated control cultures; ++ p < 0.01 versus 6-OHDA-treated cultures; ## p < 0.01 versus MPP+-treated cultures. d, Sympathetic neuron cultures were pretreated with or without 1 μm rapamycin and then exposed to 10 μm 6-OHDA for 16 h. Cell extracts were analyzed by Western immunoblotting with the indicated immunoreagents. Right panel shows quantification (means ± SEM; normalized to ERK1/2) from three or more independent experiments. RTP801 relative expression: *p < 0.05 versus untreated control (Ctl) cultures. The vertical white line between lanes indicates elimination of irrelevant intervening lanes in the blot.
Sympathetic neurons, which neuronal PC12 cells closely resemble, exhibit Lewy bodies and degenerate in PD (Marras and Lang, 2008; Orimo et al., 2008). Significantly, RTP801 is induced and mediates death of 6-OHDA-treated cultured sympathetic neurons (Malagelada et al., 2006, 2008). Rapamycin also suppresses RTP801 induction in these neurons (Fig. 2 d).
Rapamycin protects from PD toxins
Because RTP801 mediates death of neuronal PC12 cells and sympathetic neurons triggered by PD toxins, we next assessed whether rapamycin, which suppresses RTP801 induction, is protective. In parallel with effects on RTP801 induction, 100 nm and 1 μm, but not 20 nm, rapamycin protected neuronal PC12 cells from 6-OHDA (Figs. 1 a, 3 a). Rapamycin also effectively suppressed death induced by MPP+ (Fig. 3 b). Importantly, rapamycin protected not only cell bodies but also neurites (Fig. 3 c). Note that rapamycin alone slightly decreased the length of neurites (Costantini and Isacson, 2000; Zheng et al., 2002), a result most likely attributable to partial inhibition of mTOR by rapamycin and its consequent effects on Akt activity and protein synthesis. To extend our findings to neurons, sympathetic neurons were pretreated with rapamycin and then assessed for survival after 6-OHDA treatment. Here also, rapamycin protected from death (Fig. 3 d).
Figure 3.

Rapamycin protects cultured neuronal PC12 cells and sympathetic neurons from PD mimetics. a, Neuronal PC12 cells were pretreated with the indicated concentrations of rapamycin or FK506 for 1 h and then exposed to 100 μm 6-OHDA. Survival was measured 24 h later. Values are expressed as mean ± SEM of at least three independent experiments in triplicate in each condition. **p < 0.01 and ***p < 0.001 compared with untreated cultures exposed to 6-OHDA. b, Neuronal PC12 cells were pretreated with 1 μm rapamycin for 1 h and then exposed to no treatment [control (Ctl)], 100 μm 6-OHDA, or 200 μm MPP+ in the continued presence of rapamycin. Survival was measured 24 h later. Values are expressed as mean ± SEM of at least three independent experiments in triplicate in each condition. +++ p < 0.001 compared with untreated cultures; ***p < 0.001 compared with cultures treated with 6-OHDA alone; *p < 0.05 compared with cultures treated with MPP+ alone. c, Images of neuronal PC12 cells cultures treated with or without rapamycin and then exposed to the indicated PD mimetics. Note in the right that rapamycin diminishes neurite loss in neuronal PC12 cell cultures induced by PD mimetics. *p < 0.05 and ***p < 0.001 compared with untreated control cultures; ++ p < 0.01 compared with 6-OHDA-treated cultures alone; # p < 0.05 compared with MPP+ alone. d, Sympathetic neurons at 7 d in vitro were pretreated for 1 h with 20 nm, 0.1 μm, or 1 μm rapamycin and then exposed to 15 μm 6-OHDA. After 24 h, cultures were fixed and stained with nuclear dye Hoechst 33342. Surviving neurons (judged by the lack of chromatin condensation) were strip counted under fluorescence microscopy. Values are expressed as mean ± SEM of at least three independent experiments in triplicate in each condition. **p < 0.01 compared with untreated cultures exposed to 6-OHDA.
FK506 does not block RTP801 induction or protect from 6-OHDA
Rapamycin binds the immunophilin FK506-binding protein-12 (FKBP12), and this complex can mediate the interference of rapamycin with mTOR activities (Sabers et al., 1995). FK506, like rapamycin, is a macrolide that binds FKBP12 (Avramut and Achim, 2002). Although FK506 does not block mTOR, it and derivatives are reportedly neuroprotective (Snyder et al., 1998; Pong and Zaleska, 2003), thus raising the possibility that it is immunophilin interaction rather than mTOR inhibition that accounts for the protective actions of rapamycin in PD models. However, 100 μm FK506 did not block RTP801 induction by 6-OHDA (Fig. 2 b, right) nor did it protect against 6-OHDA (Fig. 3 a).
Rapamycin appears to suppress RTP801 translation
We next explored the mechanism by which rapamycin blocks RTP801 induction by PD toxins. There are three means by which this could occur: inhibition of transcription, increased turnover, and suppression of translation. Rapamycin did not inhibit induction of RTP801 transcripts by MPP+ and somewhat enhanced RTP801 mRNA levels (Fig. 4 a). We also assessed whether rapamycin regulates RTP801 protein by affecting its rate of turnover. RTP801 protein is unstable, with a half-life of 2–5 min in mouse embryo fibroblasts (Kimball et al., 2008). Neuronal PC12 cells were treated with or without MPP+ and/or rapamycin for 8 h and then with cycloheximide for 5–60 min. Relative RTP801 protein expression was then assessed by Western immunoblotting, and the data from the 5 and 10 min points were used to calculate RTP801 protein half-life under each condition (Fig. 4 b) (supplemental Table 1, available at www.jneurosci.org as supplemental material). MPP+ did not appreciably affect the half-life of RTP801 protein, and, in the presence of MPP+ and rapamycin, the half-life was somewhat increased rather than reduced (from 5–7 to ∼10 min). Consistent with the inhibition by RTP801 of mTOR activity and the short half-life of RTP801 protein, cycloheximide restored mTOR activity in cells treated with MPP+, as indicated by restoration of mTOR-dependent mTOR phosphorylation at Ser2448 (data not shown) (Kimball et al., 2008).
Figure 4.

Effects of rapamycin on RTP801 mRNA, RTP801 turnover, and Akt phosphorylation. a, Neuronal PC12 cells were pretreated with 1 μm rapamycin and then exposed to 1 mm MPP+ for 8 h. RNA was extracted, and reverse transcription-PCR was performed to quantify the number of copies of cDNA coding for RTP801 under the indicated conditions. Values represent mean ± SEM of at least four independent experiments. *p < 0.05 versus control (Ctl); **p < 0.01 versus control (Ctl). b, Neuronal PC12 cells were pretreated with 1 μm rapamycin and then exposed to 1 mm MPP+ for 8 h. Then they were treated with or without 1 μm cycloheximide (CHX) for 10 or 60 min. Cell extracts were collected immediately after and subjected to SDS-PAGE and Western immunoblotting. Membranes were probed with the indicated immunoreagents and reprobed with antibodies for total ERK1 and Akt. Densitometry values represent mean ± SEM of at least three independent experiments. *p < 0.05 versus control (−) and + p < 0.05 versus MPP+ alone. M.W., Molecular weight. c, Neuronal PC12 cells were pretreated with or without 1 μm rapamycin and exposed to 100 μm 6-OHDA for 16 h. Cell extracts were analyzed by SDS-PAGE and Western immunoblotting. Membranes were probed with anti-phospho-Thr308–Akt, anti-phospho-Ser473–Akt, and anti-RTP801 antibodies. Membranes were reprobed with antibody against total Akt as loading control. Films were scanned and densitometries (NIH ImageJ) were expressed as arbitrary units versus total Akt loading control for the two phospho-Akt residues (d) and RTP801 (e). Values represent mean ± SEM of at least three independent experiments in each condition. # p < 0.05 versus phospho-Ser473–Akt in control extracts (Ct); *p < 0.05 versus phospho-Thr308–Akt in control extracts (Ct); + p < 0.05 versus phospho-Thr308–Akt in 100 μm 6-OHDA-treated cells.
Together, these data indicate that rapamycin does not prevent RTP801 protein induction by either inhibiting RTP801 transcription or enhancing RTP801 turnover. Elimination of these possibilities thus indicates that rapamycin blocks RTP801 protein induction by inhibiting its translation. This is consistent with the capacity of rapamycin to suppress mTOR-dependent translation of a subset of cellular transcripts (Choo et al., 2008).
Rapamycin prevents 6-OHDA-promoted Akt dephosphorylation at Thr308
Our findings indicate that PD toxins and RTP801 trigger neuron death by blocking mTOR-dependent phosphorylation/activation of the survival kinase Akt (Malagelada et al., 2008). We therefore examined Akt phosphorylation in neuronal PC12 cells exposed to 6-OHDA and rapamycin. Rapamycin alone had little effect on Akt phosphorylation at Thr308 but depressed phosphorylation at Ser473, a site targeted by the mTOR complex mTORC2 (Sarbassov et al., 2006). As we reported previously, 6-OHDA repressed Akt phosphorylation at both Ser473 and Thr308 (Fig. 4 c,d). In contrast, rapamycin prevented 6-OHDA-promoted dephosphorylation of Akt at Thr308 (Fig. 4 c,d). Phosphorylation of this site, a target for the phosphatidylinositol 3-kinase (PI3K)–PDK1 pathway (Alessi et al., 1997), is sufficient to promote Akt activity (Jacinto et al., 2006) and cell survival. Such findings thus indicate that, in contrast to PD toxins and RTP801, rapamycin is not neurotoxic and is neuroprotective because it maintains Akt phosphorylation at Thr308.
Torin1 mimics RTP801 but not rapamycin
Our findings raise the hypothesis that RTP801 kills neurons because it fully blocks mTOR signaling, whereas rapamycin is neuroprotective because it blocks some mTOR actions (e.g., promoting RTP801 synthesis) but not others (e.g., maintaining Akt Thr308 phosphorylation). If this is correct, then Torin1, which blocks all actions of mTOR, should mimic RTP801 but not rapamycin. As reported above, Torin1 induces neuronal cell death. Consistent with this, Torin1, like RTP801 and PD toxins (Malagelada et al., 2008), but unlike rapamycin, suppresses Akt phosphorylation at both Thr308 and Ser473 (Fig. 1 e). Moreover, whereas Torin1 blocks RTP801 protein induction by 6-OHDA, it does not inhibit death promoted by 6-OHDA (data not shown). These findings thus further support the idea that rapamycin in neuroprotective because it selectively blocks some but not all mTOR actions.
Rapamycin does not block death induced by exogenous RTP801
Our findings suggest that rapamycin protects from PD toxins by blocking RTP801 induction. Alternatively, protection could arise from an action downstream or independent of RTP801. In this case, rapamycin should be neuroprotective even if RTP801 induction were not suppressed. To assess this, we transfected neuronal PC12 cells with RTP801, which is sufficient to promote their death, and evaluated protection by rapamycin 48 h later. Rapamycin at 1 μm did not protect from exogenous RTP801 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), even when readded to the cultures after 24 h (data not shown). These observations support the idea that rapamycin protects by suppressing RTP801 induction and does not block death downstream of RTP801.
Rapamycin blocks RTP801 induction and is neuroprotective in in vivo models of PD
RTP801 is induced in the mouse acute MPTP model (Malagelada et al., 2006). Based on the present in vitro findings, we assessed whether rapamycin blocks RTP801 induction and is neuroprotective in vivo. Rapamycin (7.5 mg/kg) (Zhou et al., 2009) was administered to mice intraperitoneally once a day, 2 d before MPTP. This was the highest dose that could be used without major toxicity when administered along with MPTP, apparently attributable to peripheral effects. Animals then received four MPTP injections at 2 h intervals. Rapamycin was continued on the day of treatment and for the next 4 d. Additional animals received vehicle alone, rapamycin alone, or MPTP alone. RTP801 levels increase in dopaminergic midbrain neurons by 24 h after acute MPTP treatment (Malagelada et al., 2006), and we therefore killed one group of animals at this time and examined RTP801 levels in ventral midbrain homogenates by Western immunoblotting. MPTP increased RTP801 protein levels in midbrains by an average of 55% compared with vehicle-treated animals (Fig. 5 a). Rapamycin had no effect on basal RTP801 but fully blocked the increase promoted by MPTP (Fig. 5 a).
Figure 5.
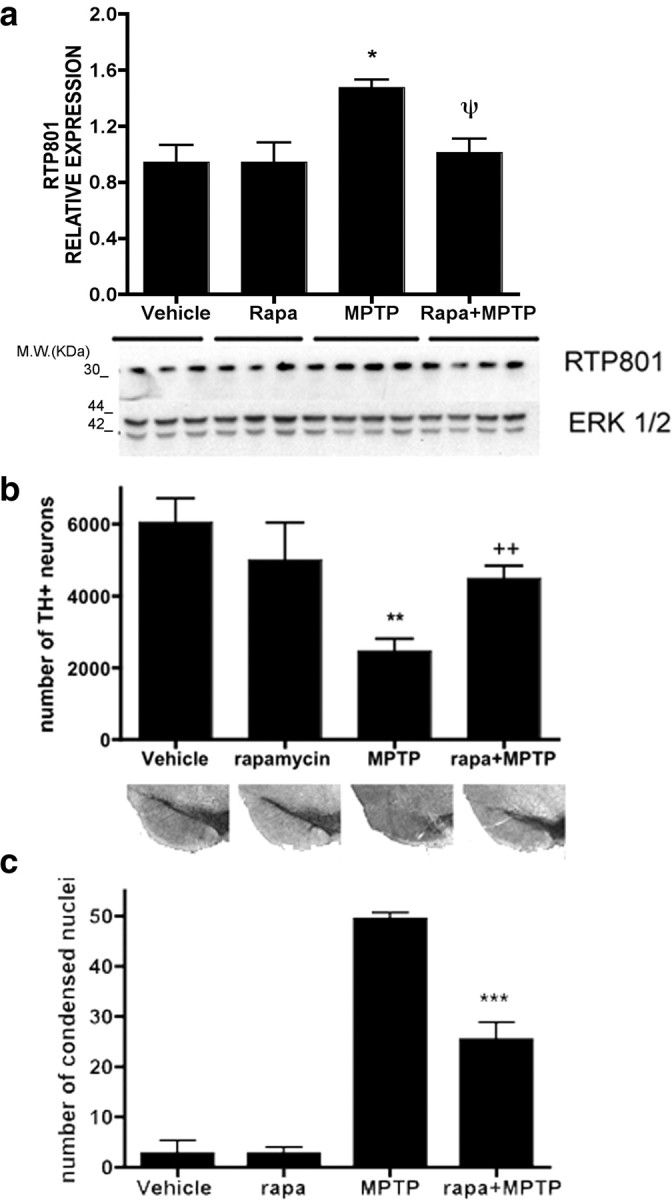
Rapamycin prevents RTP801 protein upregulation by MPTP in vivo and protects substantia nigral neurons from MPTP in living mice. a, Mice were injected with vehicle, rapamycin, MPTP, or rapamycin plus MPTP, as described in Materials and Methods for the acute regimen, and killed after 24 h. Ventral midbrains were dissected and homogenized in lysis buffer, and the homogenates were subjected to SDS-PAGE and Western immunoblotting. Membranes were probed with RTP801 antibody and reprobed with an antibody for total ERK1/2 as a loading control. Film resulting from the immunoblot was scanned, and relative densities of RTP801 bands were normalized in each case to the densities of the corresponding ERK1/2 signals. Values (in arbitrary units) for RTP801 are expressed as mean ± SEM for at least three mice per condition. *p < 0.01 versus mice injected with vehicle; ψ p < 0.01 versus mice injected with MPTP. M.W., Molecular weight. b, Mice subjected to the acute regimen as indicated were killed 7 d after the last MPTP injection. Midbrains were cryosectioned, immunostained with tyrosine hydroxylase antibody, and subjected to Nissl staining. Unbiased stereology was used to determine the total numbers of TH-positive neurons in the substantia nigra. Results represent mean ± SEM of total number of TH-positive cells per substantia nigra (right and left) for at least seven mice per each condition. **p < 0.001 versus mice injected with vehicle; ++ p < 0.01 versus mice injected with rapamycin plus MPTP. Bottom shows representative micrographs of SN stained with TH antibody in comparable sections for each condition. c, Mice subjected to the subacute MPTP regimen as indicated were killed 2 d after the last injection. Midbrains were cryosectioned, immunostained lightly with tyrosine hydroxylase antibody, and subjected to Nissl staining to visualize condensed nuclei (with fragmented chromatin). Results represent mean ± SEM of total number of apoptotic nuclei per midbrain for at least four mice per each condition. ***p < 0.001 versus mice injected with MPTP alone.
We also measured Akt phosphorylation in ventral midbrain homogenates obtained from animals subjected to the various treatments (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Although MPTP-insensitive glial cells and TH-negative (TH−) neurons in the ventral midbrain should contribute to the total and phospho-Akt signals, phosphorylation of Akt at Thr308 showed a tendency to decrease in response to MPTP, and there was a significant increase in phospho-Thr308–Akt in animals treated with MPTP plus rapamycin compared with those treated with MPTP alone. This apparent protection of phospho-Thr308–Akt by rapamycin is consistent with our in vitro findings.
Additional animals were killed 7 d after the last MPTP injection, and numbers of TH+ neurons in the SNpc were assessed by unbiased stereology. MPTP alone reduced the numbers of TH+ neurons by ∼60% (Fig. 5 b). Rapamycin substantially blocked this effect but had no significant effect on its own. Figure 5 b shows representative sections at comparable levels of the SNpc from each condition and illustrates the protection with rapamycin.
To assess the possibility that the differences in numbers of SNpc TH+ neurons among the various experimental groups were attributable to merely a loss of expression of the phenotypic marker TH rather than to neuron death, we also counted by unbiased stereology the total numbers of TH−/Nissl+ neurons in the SNpc (determined by staining with cresyl violet). This total number (which represents nondopaminergic neurons insensitive to MPTP plus MPTP-sensitive dopaminergic neurons that have lost TH expression) did not show a significant change among the various treatments (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), indicating that protection by rapamycin was attributable to effects on survival rather than on preservation of TH expression. In addition, analysis of control and treated striata confirmed that rapamycin did not affect the capacity of MPTP treatment to deplete levels of dopamine (DA) and its metabolites (data not shown) and that it did not interfere with MPTP metabolism or MPP+ uptake (striatal levels 90 min after one intraperitoneal injection of 30 mg/kg MPTP: 13.7 ± 1.0 μg/g for MPTP alone, 17.6 ± 1.3 μg/g for MPTP plus rapamycin; n = 3; p = 0.07 by Student's t test).
We additionally investigated the effects of rapamycin in the subacute MPTP model in which higher doses of MPTP are administered daily for 5 d, resulting in maximal induction of apoptotic profiles in the SN between 2 and 4 d after the last injection (Tatton and Kish, 1997). Rapamycin or vehicle was administered 30 min before MPTP. Mice were killed 2 d after the last MPTP injection, and SNpc apoptotic cells were assessed by quantitative stereology for numbers of condensed nuclei after Nissl staining. Rapamycin alone had no significant effect on numbers of condensed nuclei, whereas MPTP greatly increased this population (Fig. 5 c). In contrast, cotreatment with rapamycin reduced by half the numbers of condensed nuclei in MPTP-treated animals.
Discussion
RTP801 is induced in cell and animal models of PD and in dopaminergic SN neurons of PD patients. Moreover, RTP801 overexpression is sufficient to kill neurons (Shoshani et al., 2002; Malagelada et al., 2006, 2008), and interference with RTP801 expression protects from PD toxins (Malagelada et al., 2006). Here, we sought means to interfere with RTP801 induction and assessed whether and how this would provide neuroprotection in cell and animal PD models. We used sympathetic neurons and neuronal PC12 cells for mechanism studies because the former show Lewy body formation and functional impairment in PD (Marras and Lang, 2008; Orimo et al., 2008), whereas the latter closely model sympathetic neurons and their susceptibility to PD toxins and provide ample material for experimental analysis (Greene and Tischler, 1976; Malagelada and Greene, 2008).
The only described action for RTP801 is repression of mTOR activation through the TSC1/TSC2 tuberous sclerosis complex (Brugarolas et al., 2004; DeYoung et al., 2008). In line with this, RTP801 represses mTOR-dependent actions in neuronal cells such as phosphorylation of S6 kinase, 4EBP1, and Akt, and neuronal death caused by RTP801 or 6-OHDA requires TSC2 (Malagelada et al., 2006, 2008). Our findings that mTOR overexpression protects from 6-OHDA and that the mTOR inhibitor Torin1, like RTP801, induces death and blocks Akt phosphorylation at Ser473 and Thr308, further supporting a mechanism whereby RTP801 promotes neuron death by inhibiting mTOR and Akt signaling.
We observed that rapamycin suppresses RTP801 induction by PD toxins. RTP801 is an unstable protein (Kimball et al., 2008), and we found a half-life of 5–8 min in neuronal PC12 cells. Interference with either the transcription, translation, or protein stability of RTP801 would therefore cause its rapid depletion. Our data indicate that rapamycin does not reduce either the transcription or protein stability of RTP801 and thus favor a mechanism whereby rapamycin inhibits the translation of RTP801 transcripts. This is consistent with the well described actions of rapamycin on protein translation (Terada et al., 1994). Recent findings indicate that rapamycin can differentially inhibit mTOR-dependent translation promoted by p70S6 kinase versus cap-dependent translation regulated by 4EBP1 (Choo et al., 2008). p70S6 kinase appears to selectively control translation of mRNAs with structured 5′ untranslated regions (Fingar and Blenis, 2004). This would explain why rapamycin suppresses induction of endogenous RTP801 but not synthesis of exogenous RTP801 driven by a heterologous promoter.
RTP801 knockdown protects cultured neuronal cells from a variety of PD toxins (Malagelada et al., 2006). We therefore reasoned that, if rapamycin blocks RTP801 induction, it should also be neuroprotective. In consonance with this, rapamycin blocks death of neuronal PC12 cells and sympathetic neurons induced by 6-OHDA and MPP+ and of SN neurons in living mice in two different MPTP protocols. In contrast, rapamycin did not suppress death promoted by exogenous RTP801, ruling out a mechanism whereby rapamycin inhibits death downstream of RTP801 induction.
Schwarzer et al. (2005) reported on the basis of an array study that rapamycin reduces the basal level RTP801 transcripts in a prostate cell line. This contrasts with the present study in which rapamycin did not reduce either basal or induced levels of RTP801 message.
Rapamycin and RTP801 both inhibit mTOR signaling. This raises the issue as to why the former is neuroprotective whereas the latter promotes neuron death. The resolution appears to lie in the distinction in mechanisms and consequent actions of the two molecules. RTP801 acts upstream of mTOR to block its activation by the Rheb GTPase (Sofer et al., 2005; DeYoung et al., 2008). This suppresses most, if not all, mTOR activities. In contrast, rapamycin allosterically affects mTOR–substrate interactions and selectively blocks some mTOR actions but not others in a cell-type specific manner (Edinger et al., 2003; Choo et al., 2008; Thoreen et al., 2009). Relevant examples include rapamycin-resistant mTOR-dependent phosphorylation of Akt (Jacinto et al., 2004; Sarbassov et al., 2004) (Fig. 4) and cap-dependent mRNA translation (Choo et al., 2008; Choo and Blenis, 2009). These considerations suggest that rapamycin, unlike RTP801, spares mTOR-dependent activities in neurons needed for survival. This is consistent with findings that rapamycin does not impede neuron survival, dendritic growth and complexity, or action potentials (Rüegg et al., 2007). Additional support comes from our observation that Torin1, which, like RTP801 and in contrast to rapamycin, appears to block all actions of mTOR (Thoreen et al., 2009), promoted neuronal cell death.
An important aspect of the neuroprotective role of rapamycin in PD models appears to be its effects on Akt. Akt plays a major role in neuron survival (Franke et al., 1997a,b), and active Akt protects SN neurons from 6-OHDA in vivo (Ries et al., 2006) and neuronal PC12 cells from 6-OHDA and overexpressed RTP801 (Malagelada et al., 2008). Whereas Akt phosphorylation at both Ser473 and Thr308 provides maximum catalytic activity, phosphorylation at Thr308 (the site regulated by growth factors through PI3K signaling) is sufficient to activate the kinase and to maintain survival (Jacinto et al., 2006). Our studies indicate that one RTP801 action responsible for neuron death is inhibition of mTOR-dependent Akt phosphorylation at both Ser473 and Thr308 (Malagelada et al., 2008). This appears relevant to the pathophysiology of PD because DA neurons from PD patients have greatly reduced expression of phospho-Ser473 and phospho-Thr308–Akt but not of total Akt (Malagelada et al., 2008). In this context, it is significant that rapamycin blocks RTP801 protein induction and preserves Akt phosphorylation at Thr308, even in the presence of a PD toxin.
The capacity of rapamycin to preserve Thr308 phosphorylation may reflect the well described pathway in which mTOR-activated p70S6 kinase phosphorylates, and consequentially destabilizes, IRS-1, a scaffold protein involved in growth factor activation of PI3K and Akt (Shah et al., 2004). By blocking mTOR-dependent p70S6K activation (Chung et al., 1992; Price et al., 1992), rapamycin leads to elevated IRS-1 and enhances PI3K signaling and Akt phosphorylation at Thr308 (Haruta et al., 2000; Jacinto et al., 2006; Wan et al., 2007). In agreement with this, as in other systems (Hartley and Cooper, 2002; Tremblay et al., 2005), rapamycin maintained IRS-1 expression in our cultures, even in the presence of PD toxins (C. Malagelada, unpublished results).
Our findings highlight suppression of RTP801 induction as a likely mechanism by which rapamycin protects neurons from PD toxins. However, there are additional mechanisms by which rapamycin may confer neuroprotection. One is induction of autophagy. Autophagy has been suggested to be neuroprotective by enhancing clearance of harmful protein aggregates (Ravikumar et al., 2004, 2006; Sarkar and Rubinsztein, 2008; Sarkar et al., 2009). In particular, rapamycin protects in Huntington's disease models, and evidence suggests that this is attributable to stimulation of autophagy (Sarkar et al., 2009). Conversely, autophagy occurs in PD neurons (Anglade et al., 1997), and MPP+ induces autophagy in SH-SY5Y neuroblastoma cells (Zhu et al., 2007). Stimulation of macroautophagy also contributes to neuron death in an α-synuclein PD model (Xilouri et al., 2009). Thus, it is unclear whether or not rapamycin-stimulated autophagy would be protective in the context of PD. Additionally, it was reported that rapamycin protects macroautophagy-deficient fibroblasts from aggregated mutant huntingtin fragments by inhibiting mTOR-dependent protein synthesis (Wyttenbach et al., 2008). Such observations support the hypothesis that rapamycin confers neuroprotection by interfering with RTP801 translation.
In summary, rapamycin confers neuroprotection from PD toxins both in vitro and in vivo. Our data support a model in which rapamycin blocks translation of RTP801, thereby relieving RTP801 inhibition of mTOR and, consequently, of the survival-promoting kinase Akt (Fig. 6). The neuroprotective properties of rapamycin arise from its capacity to block some actions of mTOR (such as p70S6K activation) but not others (such as regulation of Akt Thr308 phosphorylation). Such findings, along with the elevation of RTP801 that occurs in human PD-affected neurons, support the idea of targeting RTP801 as a therapeutic strategy to suppress neuron death in PD.
Figure 6.

Scheme of hypothesized mechanisms by which neuron cell death and survival are regulated by mTOR and RTP801 signaling. a, Under basal conditions, RTP801 gene and protein expression are low. mTOR activity is unaffected and promotes rapamycin-sensitive and -insensitive translation as well as phosphorylation of Akt at Ser473 and Thr308. Cell survival is favored. b, In the presence of Torin1, all mTOR activities including Akt phosphorylation at Ser473 and Thr308 and rapamycin-sensitive and -insensitive translation are blocked. RTP801 expression is low. Cell death is favored because of inhibition of Akt phosphorylation/activation. c, In the presence of PD toxins, RTP801 gene and protein expression are induced. This leads to RTP801-dependent inhibition of mTOR activation and consequently to inhibition of Akt phosphorylation at Ser473 and Thr308. Cell death is favored. Although mTOR inhibition will affect RTP801 translation, the increased half-life of RTP801 protein in the presence of PD toxins and mutual feedback relationship between mTOR activity and RTP801 translation likely account for the sustained elevation of RTP801 levels in the presence of the toxins. d, Rapamycin does not affect induction of RTP801 transcripts by PD toxins but blocks mTOR-dependent RTP801 protein synthesis (most likely through inhibition of mTORC1-dependent p70S6 kinase activation) and phosphorylation of Akt at Ser473 (most likely through inhibition of mTORC2). Other activities of mTOR are left intact such as cap-dependent translation. Phosphorylation of Akt at Thr308 is also spared, leading to cell survival. This sparing of Thr308 phosphorylation may reflect inhibition by rapamycin of mTOR-dependent p70S6 kinase activation and of RTP801 synthesis. In addition, inhibition of p70S6 kinase activity may enhance Akt Thr308 phosphorylation by leading to stabilization of IRS-1 and to enhanced PI3K activity.
Footnotes
This work was supported in part by Department of Defense Grants DAMD 17-03-1, W81XWH-08-1-0465, and W81XWH-08-1-0522, National Institutes of Health/National Institute of Neurological Disorders and Stroke Grants AG21617, NS062180, NS064191, NS11766, NS38370, and NS042269, the Parkinson's Disease Foundation, the Thomas Hartman Foundation, Muscular Dystrophy Association/Wings Over Wall Street, and the American Parkinson's Disease Foundation. We thank Dr. Delphine Prou and Luís Parada for helpful advice, Dr. Gary Chiang for mTOR constructs, and Dr. D.M. Sabatini for providing Torin1.
References
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- Avramut M, Achim CL. Immunophilins and their ligands: insights into survival and growth of human neurons. Physiol Behav. 2002;77:463–468. doi: 10.1016/s0031-9384(02)00934-4. [DOI] [PubMed] [Google Scholar]
- Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG., Jr Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–2904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci U S A. 1998;95:1432–1437. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang GG, Abraham RT. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J Biol Chem. 2005;280:25485–25490. doi: 10.1074/jbc.M501707200. [DOI] [PubMed] [Google Scholar]
- Choo AY, Blenis J. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle. 2009;8:567–572. doi: 10.4161/cc.8.4.7659. [DOI] [PubMed] [Google Scholar]
- Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J, Kuo CJ, Crabtree GR, Blenis J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell. 1992;69:1227–1236. doi: 10.1016/0092-8674(92)90643-q. [DOI] [PubMed] [Google Scholar]
- Costantini LC, Isacson O. Immunophilin ligands and GDNF enhance neurite branching or elongation from developing dopamine neurons in culture. Exp Neurol. 2000;164:60–70. doi: 10.1006/exnr.2000.7417. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008;22:239–251. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Edinger AL, Linardic CM, Chiang GG, Thompson CB, Abraham RT. Differential effects of rapamycin on mammalian target of rapamycin signaling functions in mammalian cells. Cancer Res. 2003;63:8451–8460. [PubMed] [Google Scholar]
- Ellisen LW, Ramsayer KD, Johannessen CM, Yang A, Beppu H, Minda K, Oliner JD, McKeon F, Haber DA. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol Cell. 2002;10:995–1005. doi: 10.1016/s1097-2765(02)00706-2. [DOI] [PubMed] [Google Scholar]
- Fahn S. Medical treatment of Parkinson's disease. J Neurol. 1998;245:P15–P24. doi: 10.1007/pl00007742. [DOI] [PubMed] [Google Scholar]
- Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23:3151–3171. doi: 10.1038/sj.onc.1207542. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997a;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997b;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley D, Cooper GM. Role of mTOR in the degradation of IRS-1: regulation of PP2A activity. J Cell Biochem. 2002;85:304–314. doi: 10.1002/jcb.10135. [DOI] [PubMed] [Google Scholar]
- Haruta T, Uno T, Kawahara J, Takano A, Egawa K, Sharma PM, Olefsky JM, Kobayashi M. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol Endocrinol. 2000;14:783–794. doi: 10.1210/mend.14.6.0446. [DOI] [PubMed] [Google Scholar]
- Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37:217–222. doi: 10.1042/BST0370217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Jakowec M, Burke RE, Przedborski S. Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neurodegeneration. 1995;4:257–269. doi: 10.1016/1055-8330(95)90015-2. [DOI] [PubMed] [Google Scholar]
- Kimball SR, Do AN, Kutzler L, Cavener DR, Jefferson LS. Rapid turnover of the mTOR complex 1 (mTORC1) repressor REDD1 and activation of mTORC1 signaling following inhibition of protein synthesis. J Biol Chem. 2008;283:3465–3475. doi: 10.1074/jbc.M706643200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy OA, Malagelada C, Greene LA. Cell death pathways in Parkinson's disease: proximal triggers, distal effectors, and final steps. Apoptosis. 2009;14:478–500. doi: 10.1007/s10495-008-0309-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz MC, Heitman J. TOR mutations confer rapamycin resistance by preventing interaction with FKBP12-rapamycin. J Biol Chem. 1995;270:27531–27537. doi: 10.1074/jbc.270.46.27531. [DOI] [PubMed] [Google Scholar]
- Malagelada C, Greene LA. PC12 cells as a model for Parkinson's disease research. In: Przedborski S, Nass R, editors. Parkinson's disease: molecular and therapeutic insights from experimental models. Amsterdam: Elsevier; 2008. pp. 375–389. [Google Scholar]
- Malagelada C, Ryu EJ, Biswas SC, Jackson-Lewis V, Greene LA. RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson's disease by a mechanism involving mammalian target of rapamycin inactivation. J Neurosci. 2006;26:9996–10005. doi: 10.1523/JNEUROSCI.3292-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malagelada C, Jin ZH, Greene LA. RTP801 is induced in Parkinson's disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J Neurosci. 2008;28:14363–14371. doi: 10.1523/JNEUROSCI.3928-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marras C, Lang A. Invited article: changing concepts in Parkinson disease: moving beyond the decade of the brain. Neurology. 2008;70:1996–2003. doi: 10.1212/01.wnl.0000312515.52545.51. [DOI] [PubMed] [Google Scholar]
- Orimo S, Uchihara T, Nakamura A, Mori F, Kakita A, Wakabayashi K, Takahashi H. Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson's disease. Brain. 2008;131:642–650. doi: 10.1093/brain/awm302. [DOI] [PubMed] [Google Scholar]
- Pong K, Zaleska MM. Therapeutic implications for immunophilin ligands in the treatment of neurodegenerative diseases. Curr Drug Targets CNS Neurol Disord. 2003;2:349–356. doi: 10.2174/1568007033482652. [DOI] [PubMed] [Google Scholar]
- Price DJ, Grove JR, Calvo V, Avruch J, Bierer BE. Rapamycin-induced inhibition of the 70-kilodalton S6 protein kinase. Science. 1992;257:973–977. doi: 10.1126/science.1380182. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Berger Z, Vacher C, O'Kane CJ, Rubinsztein DC. Rapamycin pre-treatment protects against apoptosis. Hum Mol Genet. 2006;15:1209–1216. doi: 10.1093/hmg/ddl036. [DOI] [PubMed] [Google Scholar]
- Ries V, Henchcliffe C, Kareva T, Rzhetskaya M, Bland R, During MJ, Kholodilov N, Burke RE. Oncoprotein Akt/PKB induces trophic effects in murine models of Parkinson's disease. Proc Natl Acad Sci U S A. 2006;103:18757–18762. doi: 10.1073/pnas.0606401103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüegg S, Baybis M, Juul H, Dichter M, Crino PB. Effects of rapamycin on gene expression, morphology, and electrophysiological properties of rat hippocampal neurons. Epilepsy Res. 2007;77:85–92. doi: 10.1016/j.eplepsyres.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci. 2002;22:10690–10698. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu EJ, Angelastro JM, Greene LA. Analysis of gene expression changes in a cellular model of Parkinson disease. Neurobiol Dis. 2005;18:54–74. doi: 10.1016/j.nbd.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, Abraham RT. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem. 1995;270:815–822. doi: 10.1074/jbc.270.2.815. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Rubinsztein DC. Small molecule enhancers of autophagy for neurodegenerative diseases. Mol Biosyst. 2008;4:895–901. doi: 10.1039/b804606a. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009;16:46–56. doi: 10.1038/cdd.2008.110. [DOI] [PubMed] [Google Scholar]
- Schwarzer R, Tondera D, Arnold W, Giese K, Klippel A, Kaufmann J. REDD1 integrates hypoxia-mediated survival signaling downstream of phosphatidylinositol 3-kinase. Oncogene. 2005;24:1138–1149. doi: 10.1038/sj.onc.1208236. [DOI] [PubMed] [Google Scholar]
- Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14:1650–1656. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Shoshani T, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S, Moshel Y, Elbaz S, Budanov A, Chajut A, Kalinski H, Kamer I, Rozen A, Mor O, Keshet E, Leshkowitz D, Einat P, Skaliter R, Feinstein E. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol Cell Biol. 2002;22:2283–2293. doi: 10.1128/MCB.22.7.2283-2293.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder SH, Sabatini DM, Lai MM, Steiner JP, Hamilton GS, Suzdak PD. Neural actions of immunophilin ligands. Trends Pharmacol Sci. 1998;19:21–26. doi: 10.1016/s0165-6147(97)01146-2. [DOI] [PubMed] [Google Scholar]
- Sofer A, Lei K, Johannessen CM, Ellisen LW. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol Cell Biol. 2005;25:5834–5845. doi: 10.1128/MCB.25.14.5834-5845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiech L, Perycz M, Malik A, Jaworski J. Role of mTOR in physiology and pathology of the nervous system. Biochim Biophys Acta. 2008;1784:116–132. doi: 10.1016/j.bbapap.2007.08.015. [DOI] [PubMed] [Google Scholar]
- Tatton NA, Kish SJ. In situ detection of apoptotic nuclei in the substantia nigra compacta of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice using terminal deoxynucleotidyl transferase labelling and acridine orange staining. Neuroscience. 1997;77:1037–1048. doi: 10.1016/s0306-4522(96)00545-3. [DOI] [PubMed] [Google Scholar]
- Terada N, Patel HR, Takase K, Kohno K, Nairn AC, Gelfand EW. Rapamycin selectively inhibits translation of mRNAs encoding elongation factors and ribosomal proteins. Proc Natl Acad Sci U S A. 1994;91:11477–11481. doi: 10.1073/pnas.91.24.11477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay F, Gagnon A, Veilleux A, Sorisky A, Marette A. Activation of the mammalian target of rapamycin pathway acutely inhibits insulin signaling to Akt and glucose transport in 3T3-L1 and human adipocytes. Endocrinology. 2005;146:1328–1337. doi: 10.1210/en.2004-0777. [DOI] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Hohl JB, Angelastro JM, Greene LA, Shelanski ML. Death in the balance: alternative participation of the caspase-2 and -9 pathways in neuronal death induced by nerve growth factor deprivation. J Neurosci. 2001;21:5007–5016. doi: 10.1523/JNEUROSCI.21-14-05007.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila M, Jackson-Lewis V, Vukosavic S, Djaldetti R, Liberatore G, Offen D, Korsmeyer SJ, Przedborski S. Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Proc Natl Acad Sci U S A. 2001;98:2837–2842. doi: 10.1073/pnas.051633998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932–1940. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
- Wang Z, Malone MH, Thomenius MJ, Zhong F, Xu F, Distelhorst CW. Dexamethasone-induced gene 2 (dig2) is a novel pro-survival stress gene induced rapidly by diverse apoptotic signals. J Biol Chem. 2003;278:27053–27058. doi: 10.1074/jbc.M303723200. [DOI] [PubMed] [Google Scholar]
- Wyttenbach A, Hands S, King MA, Lipkow K, Tolkovsky AM. Amelioration of protein misfolding disease by rapamycin: translation or autophagy? Autophagy. 2008;4:542–545. doi: 10.4161/auto.6059. [DOI] [PubMed] [Google Scholar]
- Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE. 2009;4:e5515. doi: 10.1371/journal.pone.0005515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng WH, Kar S, Quirion R. FKHRL1 and its homologs are new targets of nerve growth factor Trk receptor signaling. J Neurochem. 2002;80:1049–1061. doi: 10.1046/j.0022-3042.2002.00783.x. [DOI] [PubMed] [Google Scholar]
- Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, Sinton C, Powell CM, Parada LF. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci. 2009;29:1773–1783. doi: 10.1523/JNEUROSCI.5685-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol. 2007;170:75–86. doi: 10.2353/ajpath.2007.060524. [DOI] [PMC free article] [PubMed] [Google Scholar]