

Abstract
Today’s generally accepted view of the self–nonself discrimination was voiced by Miller1 in 2004 in a thought-provoking essay. In spite of its popularity, this position has its limitations, which are analyzed here with a view toward establishing an interactive discussion that hopefully will culminate in agreed upon decisive experiments. The inadequacies of Miller’s view of the self–nonself discrimination and their resolution under the associative recognition of antigen model are analyzed.
Keywords: self-nonself, tolerance, T cell, processing
The conflicting views of the self–nonself discrimination as depicted by Miller1,2 and ourselves3–6 lack a consensus on the ground rules for an interactive discussion. The one thing agreed upon is that the subject is important. The basic differences between the two positions relate to the meaning of the term ‘self–nonself discrimination’ and to whether, once defined, it can be accounted for by a unitary mechanism. The Miller position is that the term ‘self–nonself discrimination’ is a collection of failsafe mechanisms or singularities called into play by what he refers to as ‘circumstances’1 and others refer to as ‘context’7. Our position is that there is a single evolutionarily selected mechanism.8
BACKGROUND
Delineating the problem
If the germline-selected recognitive (paratopic) repertoire (so-called ‘innate’ immune system) were adequate to protect vertebrates against the pathogenic universe, then the somatically derived paratopic repertoire (so-called ‘adaptive’ immune system) would have been unselectable. Vertebrates with defective ‘adaptive’ systems (RAG−, class II MHC−, athymic and so on) but intact ‘innate’ systems die of infection. The difference is that the somatically derived (‘adaptive’) paratopic repertoire recognizes epitopes that the germline-selected (‘innate’) paratopic repertoire cannot see.9 That portion unique to the somatically derived repertoire is what must be dealt with as the self–nonself discrimination because it is the deciding factor in determining the mechanism. How does this problem arise?
The ‘adaptive’ immune system functions by coupling a somatically generated and encoded, large and random (with respect to recognition of self or nonself) paratopic repertoire to a handful of biodestructive and ridding effector functions that are shared with the ‘innate’ repertoire. Any biodestructive and ridding protective mechanism must have a way to distinguish self (not-to-be-ridded) from nonself (to-be-ridded). The ‘innate’ repertoire, being germline selected, distinguishes nonself from the self-of-the-species, whereas the ‘adaptive’ repertoire, being somatically sorted, distinguishes nonself from the self-of-the-individual (that is, what is self for one individual is nonself for another). It might be well to stress that antigen receptors (paratopes) recognize epitopes, not antigens that are collections of linked epitopes. The immune system responds to antigens using a collection of paratopes associated with their recognition of epitopes that are linked on an antigen. This is referred to as associative recognition of antigen (ARA).
Although I have always viewed the sorting of the repertoire as the mechanism of the self–nonself discrimination, this position has been constantly challenged by substituting the ‘tolerance–immunity’ or ‘unresponsiveness–responsiveness’ discrimination, which is designed to include events at the level of the regulation of effector class. This is a black box approach and has no where to go because the requirements for an effective effector output are antithetical to any process that can distinguish self from nonself as classes.
The sorting of the repertoire is both necessary and sufficient to make a self–nonself discrimination. The selection pressure on this first decision mechanism is to reduce the frequency of autoimmunity to an evolutionarily acceptable level. Once a paratopic specificity is put under the control of the second decision mechanism regulating the effector class, the response is geared to destroy and rid the target independent of whether it is self or nonself. The selective pressure on this second decision step is to destroy and rid the target without attacking innocent bystanders (that is, to reduce immunopathology to an evolutionarily acceptable level). The necessity to make a self–nonself discrimination is the sole selection pressure for the specificity of the paratope. The necessity to adequately couple the paratope to the appropriate effector mechanism is the sole selective pressure to regulate the range and magnitude of the ridding response. As the ligand for the paratope is an epitope (not an antigen), the sorting of the repertoire involves the purging of anti-self paratopes consequent to recognition, epitope by epitope, whereas the regulation of effector class involves the selection for a convergent set of anti-nonself paratopes, antigen by antigen, because the effector response must be independent and coherent for each nonself antigen.4,10
The discussion of the self–nonself discrimination can then be reasonably defined by the epitope-specific interactions required for the sorting of the repertoire.
The cells and their ligands
For this analysis we will need some terms and abbreviations.
The cell that is born responsive to antigen will be referred to as an initial state or i cell. The i cell expresses no effector function; it receives and responds to signals that direct it along one of the two possible pathways, inactivation or activation. The pathway initiated by activation eventually results in an effector or e cell. The e cell when triggered sends signals, unlike the i cell, which receives signals.
The ligands for the three classes of i cell, iB, iTc (cytotoxic T) and iTh (helper), are distinct. The iB cell recognizes shape patches on extracellular and surface-expressed antigens. It does not encounter the intracellular antigens of viable cells. As a consequence, these intracellular antigens are defined by the B cell as nonself. The ligand for the iTc cell is protein processed to peptide (P) presented by MHC-encoded class I restricting elements (RIs), symbolized as (P-RI). As all cells express RI, most host antigens are processed and presented to iTc as self-(Ps-RI) by the cell of origin. The ligand for the iTh cell is processed peptide presented by MHC-encoded class II restricting elements (RIIs), symbolized as (P-RII). As few cell types process protein to RII, the iTh has a special relationship with host components that necessitates a more detailed analysis (see later).
The associative recognition of antigen model
This model for the self–nonself discrimination has been described with increasing sophistication over many years.3,6,11–16 It has also been attacked with increasing sophistication over many years, thus driving its evolution by interactive selection. While it will be referred to here, I will not take up space to describe it once more. When introduced for comparison with competing models, the differences should be self-explanatory.
THE SELF–NONSELF DISCRIMINATION AS SEEN BY MILLER
Miller1 postulates that the sorting of the repertoire of T cells involves two distinct mechanisms, one intrathymic, the other in the periphery. As it is not possible to define self as anything encoded in the genome, he argues validly that attributes such as the following must be evaluated to define self.
-
(a)
Stage of development of the individual;
-
(b)
stage of maturity of the lymphocyte;
-
(c)
site of encounter: thymus or extrathymic tissues;
-
(d)
type of cells presenting epitopes;
-
(f)
‘danger’.
THE STAGE OF DEVELOPMENT OF THE INDIVIDUAL
Miller1 rules out developmental time as a factor in the self–nonself discrimination by the following reasoning:
‘Implicit in this hypothesis is the requirement for prenatal generation of the entire immune repertoire. This is not the case, since lymphocyte differentiation continues throughout postnatal life, and somatic mutation generates new B-cell specificities after antigenic stimulation, thus the key factor in determining whether tolerance or immunity results cannot be the developmental stage of the individual.’
Miller’s dismissal of a role for developmental time in establishing ‘tolerance’ is misplaced. He rejects the 1948 Burnet version of the developmental time hypothesis, not the later updated version.3,15,17,18
I have reviewed the history of the developmental time hypothesis19 so that here I will concentrate on the argument used by Miller.
In 1955, Jerne20 proposed that the entire repertoire was expressed as secreted antibody at a developmental time when all self and no nonself was present. The anti-self was subtracted from the repertoire leaving anti-nonself, which was somehow replicated by interaction with nonself. Talmage21 and Burnet22 in 1957, realizing the fallacy of Jerne’s assumption of a self-replicating protein, put the antibody on cells. Burnet, however, simply assimilated Jerne’s assumption that Miller describes as ‘the prenatal generation of the entire repertoire.’ For Jerne, this assumption was a logical necessity; for Burnet it was gratuitous.
The fact that lymphocyte production occurs throughout life does not disprove the developmental time model because the persistence of self maintains a steady-state inactivation of anti-self. Further, ‘prenatal generation of the entire immune repertoire’ is not disproven by whether or not ‘lymphocyte differentiation continues throughout postnatal life.’ If the size of the repertoire of functionally distinct specificities was less than the number of prenatal lymphocytes produced, then their continued production would only generate redundancy. In any case, even these considerations are an aside because whether or not the entire paratopic repertoire is expressed prenatally is irrelevant. What is relevant is that the entire ‘self’ be expressed as an appropriate ligand while the developmental time window is open (that is, when there is an insufficiency of effector T-helper (eTh)) and persist after it closes (that is, when there is a sufficiency of eTh). It is the sufficiency or insufficiency of eTh that determines whether the immune system is responsive (‘mature’) or unresponsive (‘immature’). Birth may be a convenient marker for the immunologist; it is an approximate marker for the immune system that has been selected to be prepared for the appearance of nonself.
The sorting of the antigenic universe into self and nonself as a function of developmental time and as a prerequisite to the sorting of the repertoire (the self–nonself discrimination) is, in my mind, a default postulate.
STAGE OF MATURITY OF THE LYMPHOCYTE
Lederberg23 in 1959, using the same argument as Miller, rejected the Jerne/Burnet version of the developmental time model. In its place, he proposed that the responsive cells are born inactivatable-only (‘tolerizable-only’) and after a period of failure to encounter antigen they differentiate to activatable-only (‘inducible-only’). The state of ‘inducible-only’ is essentially equivalent to that of an effector. Miller cites experimental studies to demonstrate that peripheral lymphocytes from neonates have the potential to respond to nonself. This is not surprising, but are they inducible-only? Under the ARA model, peripheral T cells are tolerizable and inducible. If they were inducible-only, then the mechanism of peripheral tolerance would have to be suppressive. Miller leaves us in suspense. Instead he asks ‘what about immature T-cells in the thymus?’ I take this to mean, are they tolerizable-only?
Before considering that question, the Lederberg model needs clarification. Under his model, the immune system responds using the cells that have differentiated antigen-independently to inducible-only. Therefore, the initiation of the Lederberg pathway must occur when the developmental time window is open and all self is present. The Lederberg model requires a developmental time window. His model is embedded in the developmental time ARA model, which it neither replaces nor contradicts. Today this model can only be applied to the T helper as discussed.earlier.8 In 1996, Miller and Basten2 took a purely Lederbergean position when they argued that ‘one key factor in determining the nature of the response, whether tolerance or immunity, is, therefore, not the developmental stage of the individual but rather the state of differentiation of the lymphocyte at the time it encounters antigen.’ Consequently, their model for thymic sorting must have been that, when intrathymic, the T cells are tolerizable-only, but they leave the thymus as inducible-only. This necessitates a peripheral sorting mechanism that is suppressive and self-specific. In 2004, Miller1 abandoned the Lederberg model substituting circumstances/context for the behavior of peripheral T cells. However, there is a major point to clarify. All models require an initial tolerizable-only stage. Under the Lederberg model, the tolerizable-only state is inherent, whereas under the ARA model, tolerizable-only is the state of a system that lacks eThs. The i cell is born with and always has two pathways open to it, inactivation or activation. The thymus lacks eThs and, therefore, the iT cells, which are born there, behave as tolerizable-only.
SITE OF ENCOUNTER: THYMUS OR EXTRATHYMIC TISSUES
Miller1 views the thymus as the major site responsible for establishing self-tolerance (central) by deletion of ‘high-affinity’ self-reactive T cells. I would prefer the description, by deletion of self-reactive T cells with above a threshold affinity. Be that as it may, although there is no argument against his view, the proportion of the host-derived self that acts as a tolerogenic ligand, (Ps-RI) and (Ps-RII), in thymus is not where the emphasis should be placed because it takes a response to one peripheral self-component to debilitate. If peripheral tolerance does not exist then thymic negative selection would be the whole story. To this end, the role of ectopic expression of peripheral constituents as (Ps-RII) in thymus may be cited. However, it is unlikely that the entire self is ectopically expressed in thymus as (Ps-RII), making it necessary to face peripheral ‘tolerance’ and with it, what would be the selection pressure for the expression of a subset of peripheral constituents ectopically in thymus? If there is a mechanism for peripheral tolerance, what was its limitation that necessitated ectopic thymic expression?
Peripheral components under the control of the transactivating transcription factor, Aire, are ectopically expressed in thymus, presumably as (Ps-RII). We have proposed that the selection pressure for this is the delayed expression of these components as (Ps-RII) in the periphery (see addendum to Langman et al.24 and see discussion in Cohn5,6). In this case, they would be treated as nonself and become a target of attack after the developmental time window closed. The only solution was to ectopically express them as (Ps-RII) while the window was open, thereby deleting the relevant iTh and leaving all other i cells that recognized these components tolerizable-only. While the window is open it is irrelevant whether they are expressed in the thymus and/or the periphery. The ‘system’ is tolerizable-only.
Miller makes a case for the thymic medulla being the primary site of negative selection. However, as he points out, ‘since the medulla... is relatively permeable to soluble proteins, it can be expected that small quantities of tissue specific antigens.....could reach it via the bloodstream,’ resulting in tolerance to them. As this would have to be true for many nonself antigens also, we are left with a smudged picture.
Lastly, only under the ARA model does negative selection in thymus of iTh anti-self make an impact on peripheral tolerance of iB and iTc cells. The eTh delivery of Signal[2] to activate them is obligatory. In its absence, these cells are purged by interaction with ligand (Signal[1]).
TYPES OF CELLS PRESENTING EPITOPES
I read Miller’s1 2004 view, as distinct from his 1996 view2 to be non-Lederbergian in that tolerizable-only cells in the thymus leave to the periphery as tolerizable or inducible, the position taken under the ARA model. Therefore, a decision process distinguishing inactivation from activation must be considered.
Miller1 argues that ‘failsafe mechanisms inducing post-thymic tolerance must exist’ because antigen-presenting cell (APC) processing during housekeeping or waste disposal would convert many peripheral components to T-cell ligands (Ps-RII) and (Ps-RI). This would permit the activation of iT cells that had not been deleted in the thymus.
To face the processing problem raised by Miller,1 we must first discuss its origin. During embryonic development, the cells die by apoptosis. The resultant granules are encapsulated and phagocytized following recognition by the innate system without their contents encountering the i cells of the adaptive immune system. When the developmental time window closes and the neonate encounters the world of nonself, the cells die by necrosis due to several causes, senescence, infection and trauma. Their contents are released and are treated as nonself by the immune system and ridded as a housekeeping or waste disposal mechanism. To respond to these released components of necrosis and denatured or effete proteins, they must be processed to peptide (P) and presented on class II MHC((P-RII)). This is the ligand for eTh required to initiate a response.
If these peptides, presented as (P-RII) and (P-RI) by APC, come from normal host components why does not the animal succumb to autoimmunity. After all, the source of the peptide, whether it originates from the inside or outside of the animal, cannot be determined by the immune system. Miller1 raises a food-for-thought question.
To trigger autoimmunity, the effectors must both be induced and have a target. As all cells of the animal express class I MHC (RI), it is to be expected that the iTc anti-(Ps-RI) would be purged during the sorting process; the animal is tolerant to virtually all intracellular host constituents in the cytotoxic T-cell compartment. The B cells do not see intracellular components and, therefore, as effectors, have no autoimmune target. The helper T cells that are defensive like Th1 and Th17 can trigger autoimmunity but they are not induced normally by the waste requiring a housekeeping humoral response and, in most cases, the cells undergoing necrosis do not, when healthy, express class II MHC (RII) and, therefore, are not targets of eTh1 and eTh17.
As a general comment, under the ARA model, two factors protect against a self-sustaining autoimmunity. First, there is competition between the cell of origin presenting (Ps-RI) as an obligate tolerogen (Signal[1]) for iTc and the APC presenting (Ps-RI) as an immunogen. Second, the pathogen is often ridded before the eTh level anti-(Ps-RII) reaches the threshold required to establish a self-sustaining autoimmunity. These two factors also play a role in preventing the breaking of tolerance by nonself antigens that share epitopes with self. In addition, the target itself plays a role. Pathology may be difficult to observe if the target is rapidly renewable (for example, fibroblasts and epithelial cells) but easily evident and if the target is poorly renewable or non-renewing (for example, endocrine or neuronal cells).
Under the ARA model, the problem of ‘post-thymic tolerance’ was solved by a unitary mechanism. While the developmental time window is open (that is insufficiency of eTh) iT cells encountering their ligands are inactivated whether they be in the thymus or the periphery. When the window closes (that is, a peripheral sufficiency of eTh anti-nonself is present), the state of tolerance is maintained by the persistence of peripheral self ligand, which deletes iTh anti-self as they arise thereby effectively limiting the appearance of eTh anti-self.
‘Peripheral tolerance’ viewed as a failsafe mechanism means that it is a backup for a failed primary mechanism, which, in this case, is central tolerance. To rescue the individual from the failure of a primary mechanism, the failsafe version would have to be superior. This would result either in the evolutionary loss of the less-effective mechanism and its replacement by the superior ‘failsafe’ one, or the immune system could regard the individual, not just the thymus, as the ‘tolerizable-only’ space while the developmental time window is open (the ARA model). As Miller1 has rejected developmental time as a factor in the self–nonself discrimination he is forced to view peripheral tolerance as a special mechanism. Under the Lederberg model, the cells leaving the thymus would be inducible-only, thereby requiring a specific suppressive mechanism anti-peripheral self to regulate them. As any antigen-specific regulatory suppressive mechanism establishing tolerance would have to operate in ARA, the individual would be unresponsive to the self-of-the-species (contrary to the fact).
COSTIMULATORY ACTIVITIES
Miller takes Bretscher and Cohn11 to task by pointing out that they ‘claimed that two signals (antigen-specific) were required for lymphocytes to respond....Lafferty and co-workers25 extended this claim and postulated that the first signal was antigen-specific and the second was a costimulator signal delivered by the APC. The subsequent discovery of costimulator molecules has vindicated this idea.’
It is a constant source of amazement to me that no matter how often I get immunologists to agree that a discussion of the self–nonself discrimination must involve antigen-specific signals, costimulation or its equivalent, which is antigen unspecific, is introduced as Signal[2]. Consider two cells, one anti-self, the other anti-nonself interacting with an APC. How does costimulation decide which one should be activated and which one should be inactivated? Our answer was to place Signal[2] directly or indirectly in the hands of a presorted eTh population.3,19,24,26,27
Miller1 carries the criticism of Bretscher/Cohn one step further. He asks ‘does Signal[1] alone, under physiological conditions and in vivo, exert any effect?’ This question is one of long standing having been raised in the 1970s by Moller and Coutinho for iB cells. Miller argues that peripheralized iT cells never encounter cells that express the ligand-delivering Signal[1] alone (that is, without costimulatory activity). In this case, these cells would ignore what appears to be a potential peripheral self target, a position championed by Zinkernagel and Hengartner.28 This, of course, applies to the activation of helper T cells only. The iTc interacting with their ligands (Ps-RI) would be inactivated, costimulation independent (Signal[1]). The ligand for iTh is (P-RII) expressed mainly on APC and B cells. What the iTh does not encounter as Signal[1], when the developmental time window is open, is nonself to the immune system when the window closes, whether or not it is a host component. When the system is responsive, newly arising self cannot be distinguished from nonself.
The more critical problem that needs to be addressed is that an APC that is believed to process both self and nonself converts every nonself antigen into one that shares epitopes with self. Although solutions have been suggested4,6,8,12 that minimize this pathway to breaking tolerance (that is, a self-sustaining autoimmunity), the problem itself is yet to be faced by the ‘costimulators.’
I do not think that over time one will be able to argue that a mechanism for peripheral T-cell tolerance does not exist. If a potential for Signal[1] alone is functionless in the periphery, then a suppressive model is implied, but it makes little evolutionary or biological sense.29–32 Binding without consequence is unselectable.
THE DANGER CONCEPT
As postulated by Matzinger,33 the immune system distinguishes between harmless and dangerous rather than between self and nonself. Although I agree with Miller that her position is arguable, the two discriminations are not mutually exclusive. The self–nonself discrimination (that is, the sorting of the repertoire) is a first decision step that precedes the second decision step’s coupling of the sorted repertoire to appropriate effector functions. This second decision step is where the harmless–dangerous discrimination can be envisioned to play a role.32,34 Unlike the self–nonself discrimination, which is an antigen-specific somatic learning process, the harmless–dangerous discrimination is an antigen-unspecific germline-encoded mechanism used to determine the class of the response. In this framework, Matzinger33 has made a major contribution.
Anderson et al.35 extended in 2001 the Billingham et al.36 1956 experiment in a uniquely thoughtful study in which they showed that well-healed male grafts on RAG−/− syngeneic females were rejected after being restored with wild-type syngeneic female fetal liver. They point out the difficulties encountered by all models, including their own danger model, to explain the finding and present their explanation. I have pointed to a possible interpretation under the ARA model.29 Here I ask only what might be Miller’s1 explanation based on ‘dependence on the circumstances.’
T-CELL-DEPENDENT SUPPRESSION
Miller1 treats T suppression as do most workers in this field today, to be a peripheral failsafe mechanism keeping autoimmunity in check by limiting the ‘expansion of any autoreactive T cells that may be present.’ My only comment here is that however one views suppression, it cannot be the mechanism used to sort the repertoire.30–32 ‘Natural tolerance’ is not due to suppression.
If T suppression were the sorting mechanism, it would have to function by associative (linked) recognition of antigen to inactivate anti-self cells. Inactivation would operate antigen by antigen not epitope by epitope as dictated by the ARA model. The consequence would be that the individual would be tolerant of the self-of-the-species, enabling transplants between individuals. If anything characterizes the ‘adaptive’ immune system it is the fact that what is self for one individual is nonself for another. Further, were suppression operative on self, the individual would be unresponsive to nonself antigens that share epitopes with self, a lethal situation.
T suppression plays its regulatory role at the level of the determination of the magnitude of the response to nonself, not at the level of the self–nonself discrimination.32
CONCLUSIONS
I have discussed Miller’s1 conclusions except for one that I will address now.
‘Defects in negative selection are factors in the pathogenesis of autoimmune diseases.’
In principle, defects in negative selection leading to autoimmunity fall into two categories.
Defects in the presentation of self antigens
These involve failure to process to peptide, failure to express a restricting element and failure to present while the developmental time window is open (delayed expression). Miller concentrates on this category but, as they were predictable under the ARA model, they pose no paradox.
In this category, it would be well to stress the above-discussed observation of Aire-controlled ectopic expression of peripheral self. If our interpretation is correct that the selective pressure for ectopic expression is a delay in the presentation of (Ps-RII) in the periphery, then strong experimental support would be given to the developmental time model. This would then be added to the Adams/Hanahan experiment, which we usually cite.37
Lastly, as an aside, if defects in antigen presentation that block negative selection have no effect on positive selection then a reasonable argument can be made that specific recognition of peptide is not an element in positive selection.38–42
Defects in the pathway of Signal[1] inactivation
Under the ARA model, both Signal[1] and Signal[2] are required for activation whereas only Signal[1] is required for inactivation. This assures that no cell can be activated that in principle could not have been inactivated. Mutations in anti-self cells that rendered them activatable-only would be lethal. This implies that the defects in negative selection resulting in autoimmunity can only be due to a few precise defects in Signal[1].
Upon binding ligand (L), the TCR/BCR of the i cell delivers Signal[1], which through a series of intracellular steps, terminates in apoptosis of the cell. This can be symbolized as
If the cell responding to Signal[1] receives an eTh-delivered Signal[2] before the pathway to inactivation becomes irreversible, then the i cell would be activated, the first step on the pathway to an effector (e cell). Signal[2] alone has no consequence. This would imply a pathway something like this:
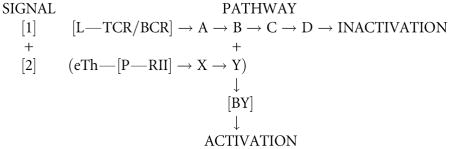
Mutations in the Signal[1] pathway to inactivation at a level before and including stage B result in non-inducibility. Mutations at the level of stage C and beyond create cells that are inducible but not tolerizable, a lethal situation. Although I know of no examples of the latter, a sophisticated look at the literature might reveal such a case. It could well be lethal in utero but detectable in vitro. To this might be added the question, would such non-tolerizable cells be triggerable as effectors. If not triggerable, they pose no problem.
Whatever the origin of the defect in negative selection, induction of autoimmunity is an eTh-dependent process. It can be mediated by defensive cells, eTh1, eTh17, eTc and/or eB, the induction of which is eTh dependent. To understand the genesis of autoimmunity due to failure of negative selection, the origin of the primer eTh anti-self becomes the key question. If iTh anti-self accumulates due to the failure of negative selection then the antigen-independent pathway to eTh becomes prominent, just as it does for eTh anti-nonself.3,24,27 Self antigen processed by APC to (Ps-RII) in the periphery provides the inductive stimulus, which is, in essence, an autocatalytic process with the potential to cascade into autoimmunity.
COMMENTARY ON PRINCIPLE
The problem of the self–nonself discrimination will remain a cross-purpose discussion as long as we fail to agree on the meaning of the terms we use, the framework to adopt for analysis and the necessity to extrapolate from experiment/observation to a maximally general, hopefully default law.
The terms self and nonself are defined by the immune system during a somatic learning process, not by the New Oxford English Dictionary or the immunologist. Self is ‘not-to-be-ridded’; nonself is ‘to-be-ridded.’ Recognition with no consequence is not an evolutionary selection pressure.
The somatically generated large random repertoire necessitates two decision processes, the sorting of the repertoire and the regulation of effector class, both of which depend upon the recognitive elements of the somatic repertoire. The ‘innate’ or germline-selected recognitive elements are blind to a large part of the antigenic universe seen by the ‘adaptive’ or somatically generated repertoire and, therefore, they cannot contribute to the antigen-specific regulation of either decision process. The ‘innate’ system cannot regulate what it cannot see.
The sorting of the repertoire is both necessary and sufficient to make a self–nonself discrimination. The ARA model is a proposed mechanism for the sorting.
Addendum
This essay was reviewed by Jacques Miller and Colin Anderson, both of whom took the position of open reviewing. I find that admirable, being a firm believer in open peer reviews. I will be dealing, however, in this addendum with the below argument of Miller who updated his published papers1,2 and asked me to comment.
Miller questions the developmental time hypothesis with the following argument:
‘...immature or mature DCs under non-activating conditions present self-antigens and induce tolerance whereas MATURE DCs under activating conditions present both self and foreign antigens and induce immunity. Your question, ‘Consider two cells, one anti-self the other anti-nonself interacting with an APC. How does costimulation decide which one should be activated and which one should be inactivated?’ appropriate as it was in 1980, is therefore irrelevant today. Under NORMAL conditions peripheral anti-self T cells are not activated because they are meeting their target self-antigen on mature or immature DCs under non-activating conditions and hence are tolerized. Any anti-self T cell that happens to issue from the thymus and meet its target on a mature DC under ACTIVATING conditions WOULD BE ACTIVATED but would constitute a very minor proportion of the T cell pool, because those T cells with either identical or cross-reacting TCRs, that would have previously met those same targets under non-activating conditions, would no longer be there or functional having been tolerized - hence autoimmunity would not occur. One could even imagine that this lone anti-self T cell would proliferate and then undergo activation-induced cell death, or be the target of T reg cells.
The above does not support the developmental hypothesis.’
We had considered the general case in 199615 by dividing the theories of the self–nonself discrimination into those based on time and those based on space. Miller’s comment is to be placed in a space framework, but as stated, it is not a model of the self–nonself discrimination; so let us complete it as a viable competing model.
There are two assumptions:
The sufficiency or insufficiency of eThs determine the activation or inactivation of all defensive cells, cytotoxic (Tc) or humoral (B cells) because dendritic cells (DCs) do not play a ‘costimulatory’ role in their activation. This is also an assumption of the ARA model. The discussion is limited to the activation/inactivation decision made by T helpers themselves. For all other cells (B and T), a role for developmental time is a default assumption.
The DC has no mechanism to distinguish self from nonself antigens. If it is in a non-activated state, it is unable to take up exogenous components but it can present its own autogenous protein, and presumably, ectopically, all peripheral self proteins. (As an aside, the DC is unlikely to be able to express ectopically all relevant peripheral self, although I will accept that assumption for discussion.) This is the self-only, non-activated state of the DC (that is, tolerigenic-only). Once it is activated to be immunogenic-only, it takes up and presents exogenous antigen (that is, both self and nonself are presented). In sum, activated DCs present all self and nonself; non-activated DCs present all self only. Activated DCs, not eTh, are assumed to be the source of Signal[2] (‘costimulation’).
The space model then is built on the probability that an anti-self Th cell leaving the thymus will be inactivated in the periphery. This probability depends on the competition between tolerigenic-only DC and immunogenic-only DC. As there is a steady-state nonself antigenic load, there is a sizeable steady-state level of activated DC, immunogenic-only, presenting both self and nonself. The steady state of anti-self Th cells leaving the thymus will be sorted stochastically into those anti-self Th cells that will be inactivated and those that will be activated to become eTh anti-self. Over time, this is a recipe for autoimmunity because the response to nonself implies a reasonably high steady-state level of activated DCs that also present self. In fact, under this model, all nonself antigens are processed to immunogenic entities that share determinants with self on a DC.
Miller’s argument that very few anti-self Th cells leaving the thymus will meet an activated DC ‘because they would have previously met the same targets under non-activating conditions’ is to be questioned. In the periphery, this probability depends roughly (anatomy plays a role) on the ratio of the levels of the two states of the DC. If the previous encounter takes place in the thymus but is without consequence (that is, the Th cell is not negatively selected), then that anti-self Th cell is either mislabeled (that is, it is anti-nonself) or it has a non-negligible chance of being activated in the periphery, as pointed out above. If all anti-self Th cells were negatively selected in thymus, then no mechanism for peripheral tolerance need be envisaged. To the extent that this is ruled out, peripheral tolerance must be explained. T helpers’ anti-peripheral self leaves the thymus in a steady state. All explanations must tell us how the T helper learns what is self and what is nonself. Interaction with its ligand (Signal[1]) cannot do that. The DC, which cannot make that discrimination, is a non-teacher. Therefore, it is valid to ask as I did, how does the activated DC signal inactivation to the anti-self Th cell and activation to the anti-nonself Th cell? As Miller points out, it cannot. At best, the DC plays a supportive role; at worst, it is irrelevant. The self–nonself discrimination must be determined by antigen-specific interactions and this requires engagement of the eTh as the source of Signal[2].
It is casuistry to ‘imagine that this lone anti-self T cell would.... undergo activation-induced cell death or be a target of Treg cells’ without telling us why this does not happen to anti-nonself T cells. Are Tregs sorted to be anti-self or anti-nonself or are they unsorted?
How does the ARA model deal with the determination of self–nonself discrimination by the T helper?
The eTh, not the DC, is the source of Signal[2] required for the activation of all i cells including the iTh itself. This raises the question of the source of the primer eTh necessary to initiate responsiveness. While the developmental time window is open (no eTh is present), the interaction of the iTh with self antigen (the only class present) whether in the thymus or periphery results in inactivation. When the window closes (defined by the appearance of eTh anti-nonself), the persistence of self maintains the state of tolerance to self. Given an insufficiency of T helpers’ anti-self, all other anti-self cells (B or T) are inactivated by interaction with ligand. The DC is only a presenter of peptide class II. Whether the DC presents self-only or both self and nonself the interaction of an anti-self T cell with self is inactivating. The source of Signal[2] for Th activation is the eTh anti-nonself, making activation of T helpers an ‘autocatalytic’ process.
All these having been said, the Miller argument does not obviate the primary role of developmental time as postulated under the ARA model.
Acknowledgments
This study was supported by a Grant (RR07716) from the National Center For Research Resources (NCRR), a component of the National Institutes of Health (NIH) and its contents are solely the responsibility of the authors and do not represent the official view of NCRR or NIH.
References
- 1.Miller J. Self–nonself discrimination by T lymphocytes. CR Biologies. 2004;327:399–408. doi: 10.1016/j.crvi.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 2.Miller JFAP, Basten A. Mechanism of tolerance to self. Curr Opin Immunol. 1996;8:815–821. doi: 10.1016/s0952-7915(96)80010-0. [DOI] [PubMed] [Google Scholar]
- 3.Cohn M. The self–nonself discrimination: reconstructing a cabbage from sauerkraut. Res Immunol. 1992;143:323–334. doi: 10.1016/s0923-2494(92)80132-5. [DOI] [PubMed] [Google Scholar]
- 4.Cohn M. A biological context for the self–nonself discrimination and the regulation of effector class by the immune system. Immunol Res. 2005;31:133–150. doi: 10.1385/IR:31:2:133. [DOI] [PubMed] [Google Scholar]
- 5.Cohn M. The common sense of the self–nonself discrimination. Springer Semin Immunopathol. 2005;27:3–17. doi: 10.1007/s00281-005-0199-1. [DOI] [PubMed] [Google Scholar]
- 6.Cohn M. Conceptualizing the self–nonself discrimination by the vertebrate immune system. In: Timmis J, Flower D, editors. Silico Immunology. Springer; New York: 2007. pp. 375–398. [Google Scholar]
- 7.Anderson CC. Time, space and contextual models of the immunity tolerance decision: bridging the geographical divide of Zinkernagel and Hengartner’s ‘Credo 2004’. Scand J Immunol. 2006;63:249–256. doi: 10.1111/j.1365-3083.2006.01742.x. [DOI] [PubMed] [Google Scholar]
- 8.Langman RE, Cohn M. A minimal model for the self–nonself discrimination: a return to the basics. Semin Immunol. 2000;13:189–195. doi: 10.1006/smim.2000.0231. [DOI] [PubMed] [Google Scholar]
- 9.Cohn M. If the ‘adaptive’ immune system can recognize a significant portion of the pathogenic universe to which the ‘innate’ immune system is blind, then... Scand J Immunol. 2004;60:1–2. doi: 10.1111/j.0300-9475.2004.01449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ismail N, Bretscher P. The Th1/Th2 nature of concurrent immune responses to unrelated antigens can be independent. Eur J Immunol. 1999;163:4842–4850. [PubMed] [Google Scholar]
- 11.Bretscher P, Cohn M. A theory of self–nonself discrimination. Science. 1970;169:1042–1049. doi: 10.1126/science.169.3950.1042. [DOI] [PubMed] [Google Scholar]
- 12.Bretscher PA. A two-step, two-signal model for the primary activation of precursor helper T cells. Proc Natl Acad Sci USA. 1999;96:185–190. doi: 10.1073/pnas.96.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langman RE. Was everyone a ‘little-bit-right’ after all? Res Immunol. 1992;143:316–322. doi: 10.1016/s0923-2494(92)80131-4. [DOI] [PubMed] [Google Scholar]
- 14.Langman RE, Cohn M. Two signal models of lymphocyte activation? Immunol Today. 1993;14:235–236. doi: 10.1016/0167-5699(93)90171-G. [DOI] [PubMed] [Google Scholar]
- 15.Langman RE, Cohn M. A short history of time and space in immune discrimination. Scand J Immunol. 1996;44:544–548. doi: 10.1046/j.1365-3083.1996.d01-359.x. [DOI] [PubMed] [Google Scholar]
- 16.Langman RE, Cohn M. If the immune repertoire is large, random, and somatically generated, then. Cell Immunol. 2002;216:15–22. doi: 10.1016/s0008-8749(02)00503-8. [DOI] [PubMed] [Google Scholar]
- 17.Langman RE, Cohn M. The essential self: a commentary on Silverstein and Rose ‘On the mystique of the immunological self’. Immunol Rev. 1997;159:214–217. [Google Scholar]
- 18.Cohn M. The self–nonself discrimination in the context of function. Theor Med Bioeth. 1998;19:475–484. doi: 10.1023/a:1009968722177. [DOI] [PubMed] [Google Scholar]
- 19.Cohn M. Logic of the self–nonself discrimination: principles and history. In: Cambrosio A, Moulin A, editors. Dialogues with Selves. Historical Issues and Contemporary Debates in Immunology. Editions Elsevier; France: 2001. pp. 53–85. [Google Scholar]
- 20.Jerne NK. The natural selection theory of antibody formation. Proc Natl Acad Sci USA. 1955;41:849–856. doi: 10.1073/pnas.41.11.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Talmage DW. Allergy and immunology. Ann Rev Med. 1957;8:239–256. doi: 10.1146/annurev.me.08.020157.001323. [DOI] [PubMed] [Google Scholar]
- 22.Burnet FM. A modification of Jerne’s theory of antibody production using the concept of clonal selection. Aust J Sci. 1957;20:67–69. doi: 10.3322/canjclin.26.2.119. [DOI] [PubMed] [Google Scholar]
- 23.Lederberg J. Genes and antibodies. Science. 1959;129:1649–1653. doi: 10.1126/science.129.3364.1649. [DOI] [PubMed] [Google Scholar]
- 24.Langman RE, Mata JJ, Cohn M. A computerized model for the self–nonself discrimination at the level of the T-helper (Th genesis) II. The behavior of the system upon encounter with nonself antigens. Int’l Immunol. 2003;15:593–609. doi: 10.1093/intimm/dxg059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lafferty KJ, Cunningham AJ. A new analysis of allogeneic interactions. Aust J Exp Biol Med Sci. 1975;53:27–42. doi: 10.1038/icb.1975.3. [DOI] [PubMed] [Google Scholar]
- 26.Cohn M. Antibody diversity 1983: some elementary considerations. In: Yamamura VY, Tada T, editors. Progress in Immunology. Academic Press; Orlando, Florida: 1983. pp. 839–851. [Google Scholar]
- 27.Cohn M, Langman RE, Mata J. A computerized model for the self–nonself discrimination at the level of the T-helper (Th-genesis). I. The origin of ‘primer’ effector T-helpers. Int’l Immunol. 2002;14:1105–1112. doi: 10.1093/intimm/dxf078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zinkernagel RM, Hengartner H. On immunity against infections and vaccines: Credo 2004. Scand J Immunol. 2004;60:9–13. doi: 10.1111/j.0300-9475.2004.01460.x. [DOI] [PubMed] [Google Scholar]
- 29.Cohn M. On ‘Credo 2004’ as viewed under the ‘Development-Context’ model of Colin Anderson. Scan J Immunol. 2006;64:97–103. doi: 10.1111/j.1365-3083.2006.01790.x. [DOI] [PubMed] [Google Scholar]
- 30.Langman RE. The self–nonself discrimination is not regulated by suppression. Cell Immunol. 1987;108:214–219. doi: 10.1016/0008-8749(87)90205-x. [DOI] [PubMed] [Google Scholar]
- 31.Cohn M. Whither T-suppressors: if they didn’t exist would we have to invent them? Cell Immunol. 2004;227:81–92. doi: 10.1016/j.cellimm.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 32.Cohn M. What roles do regulatory T-cells play in the control of the adaptive immune response? Int Immunol. 2008;20:1107–1118. doi: 10.1093/intimm/dxn088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matzinger P. Tolerance, danger and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 34.Cohn M. A rationalized set of default postulates that permit a coherent description of the immune system amenable to computer modeling. Scan J Immunol. 2008;68:371–380. doi: 10.1111/j.1365-3083.2008.02158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson CC, Carroll JM, Gallucci S, Ridge JP, Scheever AW, Matzinger P. Testing time, ignorance-, danger-based models of tolerance. J Immunol. 2001;166:3663–3671. doi: 10.4049/jimmunol.166.6.3663. [DOI] [PubMed] [Google Scholar]
- 36.Billingham RE, Brent L, Medawar PB. Actively acquired tolerance of foreign cells. Nature. 1953;172:603–606. doi: 10.1038/172603a0. [DOI] [PubMed] [Google Scholar]
- 37.Adams TE, Alpert S, Hanahan D. Non-tolerance and autoantibodies to a transgenic self antigen expressed in pancreatic β cells. Nature (London) 1987;325:223–228. doi: 10.1038/325223a0. [DOI] [PubMed] [Google Scholar]
- 38.Langman RE, Cohn M. The standard model of T-cell receptor function: a critical reassessment. Scand J Immunol. 1999;49:570–577. doi: 10.1046/j.1365-3083.1999.00569.x. [DOI] [PubMed] [Google Scholar]
- 39.Cohn M. The Tritope model of restrictive recognition by the TCR. Trends Immunol. 2003;24:127–131. doi: 10.1016/s1471-4906(03)00021-8. [DOI] [PubMed] [Google Scholar]
- 40.Cohn M. An alternative to current thinking about positive selection, negative selection and activation of T-cells. Immunology. 2004;111:1–6. doi: 10.1111/j.1365-2567.2004.01830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cohn M. The Tritope model for restrictive recognition of antigen by T-cells: I. What assumptions about structure are needed to explain function? Mol Immunol. 2005;42:1419–1443. doi: 10.1016/j.molimm.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 42.Cohn M. The Tritope Model for restrictive recognition of antigen by T-cells: II. Implications for ontogeny, evolution and physiology. Mol Immunol. 2008;45:632–652. doi: 10.1016/j.molimm.2006.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]