

Abstract
P2X receptor activation protects in heart failure models. MRS2339 3, a 2-chloro-AMP derivative containing a (N)-methanocarba (bicyclo[3.1.0]hexane) system, activates this cardioprotective channel. Michaelis–Arbuzov and Wittig reactions provided phosphonate analogues of 3, expected to be stable in vivo due to the C-P bond. After chronic administration via a mini-osmotic pump (Alzet), some analogues significantly increased intact heart contractile function in calsequestrin-overexpressing mice (genetic model of heart failure) compared to vehicle-infused mice (all inactive at the vasodilatory P2Y1 receptor). Two phosphonates, (1’S,2’R,3’S,4’R,5’S)-4’-(6-amino-2-chloropurin-9-yl)-2’,3’-(dihydroxy)-1’-(phosphonomethylene)-bicyclo[3.1.0]hexane 4 and its homologue 9, both 5’-saturated, containing a 2-Cl substitution, improved echocardiography-derived fractional shortening (20.25% and 19.26%, respectively, versus 13.78% in controls), while unsaturated 5’-extended phosphonates, all 2-H analogues, and a CH3-phosphonate were inactive. Thus, chronic administration of nucleotidase-resistant phosphonates conferred a beneficial effect, likely via cardiac P2X receptor activation. Thus, we have greatly expanded the range of carbocyclic nucleotide analogues that represent potential candidates for the treatment of heart failure.
Introduction
The actions of extracellular nucleotides in cell signaling are mediated by two classes of cell surface purinergic receptors: P2X receptors are ligand-gated ion channels activated by extracellular ATP, and P2Y receptors are G protein-coupled receptors activated by both adenine and uracil nucleotides.1,2 In the heart, a variety of P2 receptors are expressed.3
Cardiac P2X receptors represent a novel and potentially important therapeutic target for the treatment of heart failure. A P2X receptor on the cardiomyocyte mediates cardioprotection and is activated by ATP or its potent analogue 2-MeSATP 1 (Chart 1), as demonstrated using the calsequestrin (CSQ) model of cardiomyopathy. Extracellular ATP can cause an ionic current in murine, rat, and guinea pig cardiac ventricular myocytes.4–6 The P2X4 receptor is an important subunit of the native cardiac P2X receptor, which mediates ionic current induced by extracellular ATP.4 This P2X current was up-regulated in cardiac ventricular myocytes of the CSQ hearts. Furthermore, cardiac myocyte-specific overexpression of the P2X4 receptor can mimic the beneficial effects following chronic infusion of P2X agonist analogues. This analysis suggested that regulation of this cardiac P2X receptor is protective in cardiac hypertrophy or failure.
Chart 1.

Representative adenine nucleotide derivatives that display activity at various P2 receptors. Compound 3 served as the lead compound for the present set of phosphonate analogues.
(1’S,2’R,3’S,4’R,5’S)-4-(6-amino-2-chloro-9H-purin-9-yl)-1-[phosphoryloxymethyl] bicyclo[3.1.0]hexane-2,3-diol, (MRS2339, 3) is an (N)-methanocarba monophosphate derivative of 2-chloro-AMP 2 that contains a rigid bicyclic ring system (bicyclo[3.1.0]hexane) in place of ribose that we reported to to be effective in an in vivo model of cardiomyopathy.8 This ring system impedes hydrolysis of the 5’-phosphate in a model compound by its nucleotidase.9 Compound 3 induced a current in the CSQ myocyte similar to that by compound 1, characteristic of the action of the P2X4 receptor.8 Chronically administered compound 3 rescued the hypertrophic and heart failure phenotype in the CSQ-overexpressing mouse.7 When administered via an Alzet mini-osmotic pump, it significantly increased longevity as compared to vehicle-injected mice. The improvement in survival was associated with decreases in heart weight/body weight ratio and in cross-section area of the cardiac myocytes. Compound 3 was devoid of any vasodilator action in aorta ring preparations indicating that its salutary effect in heart failure was not due to any vascular unloading.
Activation of this myocyte P2X receptor leads to the opening of a nonselective cation channel permeable to Na+, K+, and Ca2+. The current is inward at negative membrane potentials, reverses near 0 mV, and becomes outward at positive potentials.4 The continuous activation of this receptor channel under the resting or negative membrane potentials would produce an inward current while its activation during depolarized portions of the action potential should lead to an outward current. These ionic currents represent a possible ionic mechanism by which the cardiomyocyte P2X channel achieves its protective effect.
We have explored the structure activity relationship (SAR) of phosphonate analogues of 3 in a model of cardioprotection. Although a (N)-methanocarba nucleoside 5’-monophosphate was shown to be a poor substrate of 5’-nucleotidase (CD73),9 replacement of the phosphoester group of 3 with a phosphonate would be expected to further increase the in vivo half-life because of the stability of the C-P bond. Phosphonate analogues of nucleotides and other known ligands, in some cases, have been shown to display activity at P2 receptors.14–17
Results
Chemical Synthesis
The phosphonate derivatives on varied carbon skeletons (Table 1) were synthesized by the methods shown in Scheme 1 – Scheme 6. In some cases, the phosphorous atom was bonded directly to the 5’ carbon atom (Scheme 1, Scheme 2), while in other cases, a carbon atom was added at that position to form either a saturated or unsaturated nucleotide analogue (Scheme 3–Scheme 5). Alternately, a methylphosphonate group was included in compounds 11 and 12, which were otherwise equivalent to 4 and 5, respectively (Scheme 6). The 2 position contained either hydrogen (in compounds 5, 8, 10, 12) or Cl (as in the known active compound 3 and the novel analogues 4, 7, 9, 11). The reference compound 3 was synthesized by a modification of the reported method,13 which lead to improved yields (Supporting Information).
Table 1.
Phosphonate analogues: structure and effects on in vivo heart function as determined by echocardiography-derived FS in CSQ heart failure mice.
| No | Structure | FS in % in CSQ Micea |
n = |
|---|---|---|---|
| 3 | 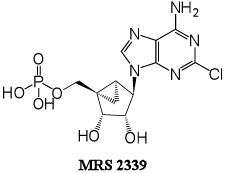 |
15.47 ± 1.15 | 10 |
| 4 | 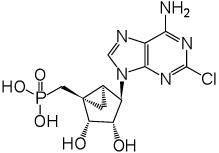 |
20.25 ±1.19 | 8 |
| 5 | 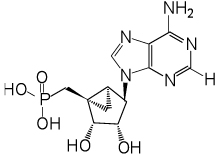 |
16.23 ± 0.93 | 13 |
| 7 | 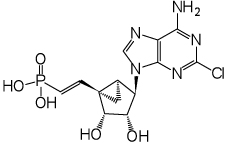 |
12.12 ±1.2 | 11 |
| 8 | 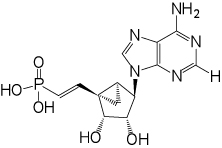 |
13.88 ± 2.12 | 8 |
| 9 | 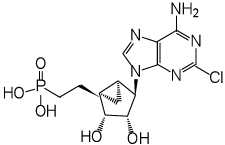 |
19.26 ± 1.23 | 16 |
| 10 | 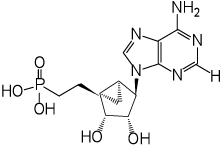 |
11.15 ±1.44 | 12 |
| 11 | 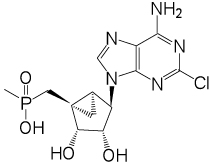 |
15.0 ± 1.2 | 10 |
| 12 | 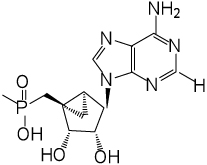 |
ND |
at 3.3 µM. The vehicle control mice displayed a %FS of 13.78 ± 1.19% (n=16).
ND – not determined.
Scheme 1.

Reagents and Conditions. a) tert-Butylchlorodiphenylsilane, imidazole, DMAP, an. CH2Cl2, 93%; b) DIBAL-H, an. THF, 82 %; c) Methanesulfonyl chloride, triethylamine, an. CH2Cl2, 96%; d) NaI, 65 °C, an. 1,4-dioxane, 95%; e) Triethylphosphite, 94%; f) TBAF, THF, 88%.
Scheme 6.

Reagents and Conditions. a) CBr4, triphenylphosphine, triethylamine, 81%; b) Diethylmethylphosphite, 110 °C, 95%; c) TBAF, THF, 91%, d) Triphenylphosphine, 2,6-dichloropurine, diisopropyl azodicarboxylate, an. THF, 75%; e) 2 M NH3 in i-PrOH, 70 °C, 60%; f) Iodotrimethylsilane, an. CH2Cl2, 20% combined yield.
Scheme 2.

Reagents and Conditions. a) Triphenylphosphine, 2,6-dichloropurine, diisopropyl azodicarboxylate, an. THF, 75%; b) 2 M NH3 in i-PrOH, 70 °C, 80% for 9, 79% for 11; c) Iodotrimethylsilane, an. CH2Cl2, 23% for 4 and 27% for 5; d) Triphenylphosphine, 6-chloropurine, diisopropyl azodicarboxylate, an. THF, 87%.
Scheme 3.

Reagents and Conditions. a) Dess-Martin periodinane, an. CH2Cl2, 80%; b) Tetraisopropyl methylenediphosphonate, NaH, an. THF, 83%; c) 2 M NH3 in i-PrOH, 70 °C, 80%; d) 10% Pd/C, H2 (3 bar), MeOH:2M aq. NaOH (1:1, v/v), 79%; e) Iodotrimethylsilane, an. CH2Cl2, 47% for 10 and 28% for 7.
Scheme 5.

Reagents and Conditions. a) 10% Pd/C, H2 (3 bar), MeOH, 72%; b) Triphenylphosphine, 2,6-dichloropurine, diisopropyl azodicarboxylate, an. THF, 40%; c) 2 M NH3 in i-PrOH, 70 °C, 71%; d) Iodotrimethylsilane, an. CH2Cl2, 45% combined yield.
Known alcohol 1313 was protected as a O-tert-butyldimethylsilyl ether using TBDPS-Cl, imidazole and DMAP to get compound 14, followed by the reduction of the ethyl ester using DIBAL-H in anhydrous THF resulting in compound 15 in very good yield (Scheme 1). In order to introduce an iodo group at the 5’ position, a classical two step procedure was implemented. This involved the initial activation of the 5’-alcohol as a mesylate followed by an SN2 nucleophilic attack of iodide on the activated 5’-position resulting in the 5’-iodo compound 17 in 95% yield. The iodo compound 17 was subjected to classical Michaelis–Arbuzov reaction conditions with excess triethylphosphite and heating up to 110 °C for 17 h to provide a phosphonate diester 18 in excellent 94% yield (Scheme 1). Desilylation using TBAF resulted in alcohol 19, which was a suitable substrate for Mitsunobu base coupling reactions.
The alcohol 19 was used as a common key intermediate to synthesize phosphonates 4 and 5 (Scheme 2). Synthetic procedures for phosphonates 4 and 5 involved initial Mitsunobu base coupling reaction using triphenylphosphine, diisopropyl azodicarboxylate, and the corresponding purine base followed by amination at the 6 position of the purine ring using 2M NH3 in isopropanol (Scheme 2). Finally, the simultaneous deprotection of both the phosphonate diester and the acetonide of 21 and 23 was achieved upon treatment with freshly opened iodotrimethylsilane to get target phosphonates 4 and 5, respectively (Scheme 2). Our efforts to use alternative relatively milder reagents, bromotrimethylsilane and Dowex-50 ion exchange resin, resulted in partial deprotection (results not shown).
The synthetic routes to the elongated saturated and unsaturated phosphonate derivatives 7–10 are shown in Scheme 3–Scheme 5. The saturated phosphonate 10 was synthesized by oxidation of known alcohol 2413 to the 5’-aldehyde 25 in 80% yield. It is noteworthy that not even a small amount of decomposition of the aldehyde was observed after storage at room temperature (rt) for several days. The α,β-unsaturated alkyl phosphonate ester 26 was prepared from aldehyde 25 in a Wittig-type reaction using tetraisopropyl methylenediphosphonate and sodium hydride in anhydrous THF.22,23 The E-configuration of the resulting alkene could be inferred from the large coupling constant (3J = 17.1 Hz). Amination followed by hydrolysis of phophonate diester and acetonide resulted in α,β-unsaturated alkyl phosphonate 7 (Scheme 3).
Catalytic hydrogenation of 27 in the presence of H2 (3 bar), palladium on carbon and MeOH:2M aq. NaOH (1:1, v/v)24,25 resulted in the expected olefin reduction and dechlorination to give the corresponding saturated phosphonate diester 28. Compound 28 was converted to the long chain saturated alkyl phosphonate 10 using the previously described iodotrimethylsilane deprotection reaction conditions. To our surprise, our various efforts to synthesize 36 (Scheme 5) from 27 by olefin reduction, running the reaction at atmospheric pressure and using less weight percent of catalyst, either resulted in an incomplete reaction or generated an inseparable mixture of dehalogenated and halogenated products (results not shown).
Hence, in order to synthesize phosphonates 8 and 9, we decided to install the phosphonate diester before the Mitsunobu base coupling reaction, as described in Scheme 4 and Scheme 5. The 5’-alcohol of compound 15 was oxidized using Dess-Martin periodinane reaction to get aldehyde 29. Similar to the aldehyde 25, aldehyde 29 also displayed considerable stability at rt. The α,β-unsaturated alkyl phosphonate diester 30 was obtained using the previously described Wittig-type reaction conditions. Desilylation under standard conditions resulted in the formation of alcohol 31, which served as the key intermediate for the synthesis of long chain unsaturated and saturated alkyl phosphonates 8 and 9, respectively.
Scheme 4.

Reagents and Conditions. a) Dess-Martin periodinane, an. CH2Cl2, 72%; b) Tetraisopropyl methylenediphosphonate, NaH, an. THF, 48%; c) TBAF, THF, 88%; d) Triphenylphosphine, 6-chloropurine, diisopropyl azodicarboxylate, an. THF, 85%; e) 2 M NH3 in i-PrOH, 70 °C, 85%; f) Iodotrimethylsilane, an. CH2Cl2, 78%.
A Mitsunobu base coupling reaction on compound 31 using triphenylphosphine, 6-chloropurine, and diisopropyl azodicarboxylate followed by amination and hydrolysis of the phophonate diester and acetonide resulted in formation of long chain α,β-unsaturated alkyl phosphonate 8. Sequential catalytic hydrogenation of the resulting vinyl phosphonate diester 31 in the presence of palladium on carbon, a Mitsunobu base coupling reaction, and amination provided the long chain saturated phosphonate diester 36. Simultaneous deprotection of the phosphonate diester and the acetonide using iodotrimethylsilane resulted in the desired phosphonate 9 along with the formation of corresponding dehalogenated product phosphonate 10.
The synthetic approach to methylphosphonates 11 and 12 is shown in Scheme 6. It involved initial 5’-bromination using CBr4 and treatment with triphenylphosphine and triethylamine to result in 5’-bromosugar 37 in 81% yield. A subsequent Michaelis–Arbuzov reaction using diethyl methylphosphite, followed by desilylation with TBAF gave the 5’-methylphosphonate monoester 38 with 1-alcohol 39 as an inseparable mixture of diastereomers. A further Mitsunobu base coupling reaction with 2,6-dichloropurine, followed by amination and final deprotection gave the desired methylphosphonate 11 along with corresponding dehalogenated methylphosphonate 12.
Biological Evaluation
Various phosphonate derivatives were infused subcutaneously individually via an Alzet minipump in CSQ mice. After 28 days of infusion, the in vivo heart function was assessed using echocardiography-derived fractional shortening (FS), which is the ratio of the change in the diameter of the left ventricle between the contracted and relaxed states. Thus, a lower percentage represents a decrease in function. Two of the phosphonates, 4 and 9, were able to cause an improved FS as compared to vehicle (Figure 1, Table 1) and in comparison to the reference nucleotide 3. Other analogues tested in this model (2-H analogues 5, 8, and 10, and 2-Cl analogues 7 and 11) had lower FS values. Compounds 7, 8, and 10 were not protective at this dose, i.e. FS in vehicle control-treated CSQ mice was similar to that from mice treated chronically with these nucleotides. Thus, in the saturated phosphonate series, the orientation of the phosphorous relative to the methanocarba ring was somewhat structurally permissive, although inclusion of an olefin in the spacer prevented the cardioprotective action. In 11, one OH group of the phosphonate has been replaced with CH3. This reduces the overall charge on the molecule and is intended to make it more bioavailable. Evidently, the binding site of the receptor requires both oxygens for the most favorable improvement in FS. Therefore, as summarized in Figure 1, the most significant improvement in FS was associated with the saturated homologues containing a 2-Cl substitution and an unmodified phosphonate group, 4 and 9.
Figure 1.

Beneficial effects of 2-Cl substituted phosphonate derivatives of (N)-methanocarba AMP in heart failure mice. Various derivatives of phosphonates were dissolved in sterile NS at 3.3 µM and were infused subcutaneously individually via an Alzet minipump in CSQ mice as described in Methods. After 28 days of infusion, the in vivo heart function was assessed using echocardiography-derived FS. (A) The 2-Cl substituted 5’-phosphonate derivative 4 was able to improve in vivo cardiac contractile performance in heart failure mice as compared to normal saline-treated heart failure animals (one-way ANOVA with posttest comparison, P<0.05) while the unsubstituted 5 was ineffective (P>0.05). (B) Similarly, 2-Cl substituted higher homologue 9 was able to enhance cardiac contractile function (P<0.05), while the parent unsubstituted 10 did not improve the contractile function (P>0.05). Data were mean ± SE.
An echocardiographic image of compound 4- versus normal saline (NS)-infused CSQ hearts is shown in Figure 2. An increased shortening of both the septum and left ventricular (LV) free wall was evident in the heart from mice treated with 4 in comparison to that from vehicle (NS)-infused mice.
Figure 2.

Chronic infusion of compound 4 resulted in improved echocardiographically derived FS in CSQ heart failure mice. Following chronic subcutaneous infusion of NS or compound 4, two-dimensional directed M-mode echocardiography was carried out as described in Methods. The heart rate (HR) is indicated on each figure. Representative M-mode echocardiography was shown for a CSQ animal infused with NS (A) and for a CSQ mouse infused with compound 4 (B). A heart from the NS-infused mice showed less shortening of both septum and LV free wall than did compound 4-infused mice.
We also evaluated the ability of the nucleotide analogues to activate the human P2Y1 receptor (Table 1). This receptor is vasodilatory, and agonist action at this subtype would be expected to be relevant to the observed cardiac effects. Compound 3 was previously reported to activate phospholipase C (PLC) mediated by the human P2Y1 receptor.9 The phosphonate derivatives were tested in a FLIPR assay of calcium flux induced in 1321N1 astrocytoma cells stably expressing the human P2Y1 receptor. The known P2Y1 receptor agonist 2-MeSADP induced a Ca2+ flux with an EC50 of 10.3±0.4 nM (n = 3) in the transfected cells (Figure 3), but in control 1321N1 astrocytoma cells there was no change in intracellular Ca2+ in response to 10 µM 2-MeSADP. At concentrations up to 10 µM, the phosphonate analogues 4 – 11 produced no effect in the same assay. However, compound 3 was active in this assay as an agonist, with an EC50 of 722±55 nM (n = 3). The maximal effect of 3 was ~80–90% of that of the full agonist 2-MeSADP.
Figure 3.

Changes in intracellular calcium in 1321N1 human astrocytoma cells stably expressing the hP2Y1 receptor. Fluorescence in response to a known hP2Y1 receptor agonist 2-MeSADP (EC50 10.3±0.4 nM), compound 3 (EC50 722±55 nM), or the phosphonate analogues (compounds 4 – 11, all inactive at 10 µM) was quantified using a FLIPR-Tetra.
Discussion
Retrosynthetic analysis
We set out to synthesize 5’-phosphonate and 5’-methyl phosphonate derivatives of (N)-methanocarba-adenosine 5, 12 or the corresponding 2-Cl derivatives 4, 11 using the previously reported intermediate 13.13 At least two synthetic pathways leading to these target molecules can be envisioned viz. routes A and B (Scheme 7A) based on the installation of phosphonate group. Route A involves phosphonate installation at the nucleoside level to generate key intermediate 42, which could be used as common intermediate for the generation of both phosphonates and methyl phosphonates 43 using Michaelis–Arbuzov reaction conditions. Route B involves phosphonate installation at the sugar level on halogenated intermediates 17 and 37 to get intermediates 18 and 38, respectively. Route B does not have a common intermediate, like route A, and, as a result, it involves a longer and more laborious synthetic sequence. Generally, the Michaelis–Arbuzov reaction conditions to generate phosphonate derivatives require long reaction times (24 to 48 h) at elevated temperatures (120 to 180 °C). Moreover, removal of trialkyl phosphite reagent needs high temperatures and high vacuum. Because of these harsh conditions, a Michaelis–Arbuzov reaction at the nucleoside level generally results in very poor yields (less than 25% yield) of the desired phosphonates29 along with the formation of a dark-colored thermal degraded products of the nucleosides. On the other hand, Michaelis–Arbuzov reactions at the sugar level generally result in very good yields.29 Hence, although route B is time consuming and contains more synthetic steps than route A, we have decided to obtain these phosphonate derivatives via route B, believing that it would be reliable with good yields.
Scheme 7.


A) Retrosynthetic analysis of 5’-phosphonate and 5’-methyl phosphonates of (N)-methanocarba adenine or 2-Cl adenine derivatives. Use of route B, in which the phosphonate was introduced prior to the nucleobase, proved successful. B) Retrosynthetic analysis of saturated and unsaturated long chain 5’-phosphonates of (N)-methanocarba adenine or 2-Cl adenine derivatives. Use of the shorter synthetic route C, in which the nucleobase was introduced prior to the phosphonate, proved successful.
Long chain saturated and unsaturated phosphonates of (N)-methanocarba adenine or 2-Cl adenine derivatives 7–10 could be achieved from the same starting compound 13 as for the phosphonates 4, 5, 11 and 12. Similar to the synthesis of phosphonates 4, 5, 11 and 12, these phosphonates could possibly be obtained via two synthetic routes viz, routes C and D (Scheme 7B), based on the installation of the phosphonate group. The long chain unsaturated phosphonate could be introduced by oxidation of the 5’-alcohol of either nucleoside 24 or compound 15 followed by the Wittig–type reaction using tetraisopropyl methylenediphosphonate and NaH to provide phosphonate diester 26 or 30, respectively.22,23 One could expect this reaction to proceed smoothly at both nucleoside and sugar stages. Since route C has a common intermediate 26 for the synthesis of long chain saturated and unsaturated 5’-phosphonates 7–10, we have decided to explore the synthesis of these phosphonates by the shorter synthetic route C.
Biological analysis
There is increasing evidence that chronic activation of the native cardiac P2X receptor by nucleotide analogues protects against the progression of heart failure. The P2X4 receptor is an essential subunit of this native receptor, but we do not know what other P2X subtypes are present. The cardiac myoctye receptor is not identical to the vascular P2X4 receptor, which has a key role in the response of endothelial cells to changes in blood flow.27
The synthetic nucleotide analogue 3 activates the native cardiac P2X receptor, as indicated in electrophysiological experiments with normal cardiac myocytes and those that overexpress CSQ and based on its in vivo ability to improve the heart failure phenotype of these animals. The rigid carbocyclic ring system contained in this derivative stabilizes nucleotides toward the action of nucleotidases. Therefore, compound 3 is expected to be more stable than the corresponding riboside. In the present study, we have synthesized fully hydrolysis-resistant adenosine monophosphate derivatives based on phosphonate linkages. The C-O-P bond of 3 was found to be stable over 24 h in aqueous medium at pH 1.5 to simulate the acidity of the stomach, however incubation at 37°C in the presence of mammalian cell membranes (1321N1 astrocytoma cells) resulted in considerable hydrolysis of the 5’-phosphate of 3 (data not shown). Therefore, a more stable structural alternate to the 5’-phosphate linkage was sought.
An in vivo screen of cardiac function was used to test the novel analogues. Thus, the results of this chronic study likely reflect both pharmacodynamic and pharmacokinetic factors. Several of the novel phosphonate analogues displayed the same agonist activity as 3 in protecting the heart muscle when chronically administered in the CSQ model. The SAR analysis showed that considerable cadioprotection was associated with specific structural features of the phosphonate derivatives. The variation in the chain length and saturation at the 5’ carbon provided consistent results in the in vivo screen. Two of the phosphonates, 4 and 9, both saturated homologues containing a 2-Cl substitution, improved FS, while the unsaturated phosphonates and 2-H analogues were inactive. The most favorable FS (20.25%, compared to 13.78% in controls) was observed for (1’S,2’R,3’S,4’R,5’S)-4’-(6-amino-2-chloropurin-9-yl)-2’,3’-(dihydroxy)-1’-(phosphonomethylene)-bicyclo[3.1.0]hexane 4, which is the equivalent of 3 in which the 5’-O has been excised. The higher homologue 9 displayed a FS of 19.26%. Thus, it is possible to extend the SAR around compound 3, for chronic activation of the cardiac P2X receptor leading to a beneficial effect in heart failure.
The activity measured here does not reflect direct measurements at a P2X receptor, and the likely association with this mechanism is due to the close resemblance to compound 3, which clearly activates a P2X4 receptor-dependent ion flux that has been shown to protect in this model of cardiomyopathy. It is not feasible to study the analogues at a recombinant homotrimeric P2X4 receptor system,12 because the endogenous cardiac P2X receptor is thought to be composed of P2X4 receptor subunits in heteromeric association with a yet unidentified P2X subtype. The P2X4 receptor is known to associate with other P2X receptor subtypes, and these heterotrimers, for example heteromers with P2X1 subunits, are pharmacologically distinct from P2X4 homotrimers.26 More recently, the existence of P2X4/P2X7 heteromers has also been described.31
The P2X4 receptor also is distributed widely through the central and peripheral nervous systems, the epithelial layers of ducted glands and airways, bladder smooth muscle, gastrointestinal tract, uterus, and fat cells.32 Its activation in spinal microglial cells participates in the pathogenesis of chronic pain.33 Might the effects of overactivation of P2X4 receptors oustide the myocardium complicate the use of such a phosphonate derivative for treatment of cardiac failure and cardiomyopathy? We have no evidence that these monophosphate analogues activate a P2X4 receptor channel at any other location. This is a topic for further investigation.
Another site of action of adenine nucleotides in cardiac tissue is the metabotropic P2Y1 receptor, which causes a nitric oxide-dependent relaxation of the vascular smooth muscle. Therefore, we tested the nucleotides as P2Y1 receptor agonists to account for the possibility that the observed cadiovascular effects of the phosphonate derivatives were a result of activation of an endothelial P2Y1 receptor. Compound 3 was initially characterized in assays of PLC as a weak hP2Y1 receptor agonist (EC50 1.89 µM),9 and that conclusion is consistent with the potency obseved here in inducing calcium transients in the same cell line. All of the phosphonate derivatives tested were inactive at the P2Y1 receptor. This suggests the use of these compounds as more selective pharmacological probes of the endogenous cardiac P2X receptor than compound 3. However, it is worth noting that the cardioprotection provided by 3 was shown to be independent of the P2Y1 receptor by its inability to dilate aortic rings and by use of a P2Y1-selective antagonist MRS2500. This antagonist could not block the membrane current evoked by 3 under voltage clamp in mouse cardiac myocytes.8
In conclusion, we have greatly expanded the range of carbocyclic nucleotide analogues that represent potential candidates for the treatment of heart failure. A more chemically and biologically stable linkage than the phosphate group in compound 3 has been introduced in the form of the phosphonate groups,30 which in several cases preserve heart contractile function in a genetic model of heart failure. Facile routes for the synthesis of phosphonate analogues of 3 in the conformationally constrained (N)-methanocarba series were developed using Michaelis–Arbuzov and Wittig reactions. A further advantage of the phosphonate linkage is that the undesired activity as agonist of the P2Y1 receptor has been eliminated. The beneficial effects of these nucleotidase-resistant agonists can now be explored in additional models of cardiac failure and cardiomyopathy.
Experimental Procedures
General methods
Compound 13 was either synthesized as reported13 or obtained as a custom synthesis from Natland International Corporation (Research Triangle Park, NC). All other reagents and solvents (regular and anhydrous) were of analytical grade and obtained from commercial suppliers and used without further purification. Reactions were conducted under an atmosphere of argon whenever anhydrous solvents were used. All reactions were monitored by thin-layer chromatography (TLC) using silica gel coated plates with a fluorescence indicator which were visualized: a) under UV light, b) by dipping in 5% conc. H2SO4 in absolute ethanol (v/v) followed by heating, or c) by dipping in a solution of anisaldehyde:H2SO4 (1:2, v/v) in MeOH followed by heating. Silica gel column chromatography was performed with silica gel (SiO2, 200–400 mesh, 60Å) using moderate air pressure. Evaporation of solvents was carried out under reduced pressure at a temperature below 50 °C. After column chromatography, appropriate fractions were pooled, evaporated and dried at high vacuum for at least 12 h to give the obtained products in high purity. 1H NMR and 31P NMR ascertained sample purity. No corrections in yields were made for solvent of crystallization. 1H NMR and 31P NMR spectra were recorded at 300 MHz and 121.5 MHz, respectively. Chemical shifts are reported in parts per million (ppm) relative to tetramethylsilane or deuterated solvent as the internal standard (dH: CDCl3 7.26 ppm). For compounds 38 – 41, the integral of the H3’-signal of the least predominant isomer was set to 1.0. Systematic compound names for bicyclic nucleosides are given according to the von Baeyer nomenclature.21 High resolution mass spectroscopic (HRMS) measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) using external calibration with polyalanine. Observed mass accuracies are those expected on the basis of known performance of the instrument as well as the trends in masses of standard compounds observed at intervals during the series of measurements. Reported masses are observed masses uncorrected for this time-dependent drift in mass accuracy.
Purification of the nucelotide derivatives for biological testing was performed by HPLC with a Luna 5µ RP-C18(2) semipreparative column (250 × 10.0 mm; Phenomenex, Torrance, CA) under the following conditions: flow rate of 2 mL/min; 10 mM triethylammonium acetate (TEAA)-CH3CN from 100:0 (v/v) to 70:30 (v/v) in 30 min and isolated in the triethylammonium salt form. Analytical purity of compounds was checked using a Hewlett–Packard 1100 HPLC equipped with Zorbax SB-Aq 5 µm analytical column (50×4.6 mm; Agilent Technologies Inc, Palo Alto, CA). Mobile phase: linear gradient solvent system: 5 mM TBAP (tetrabutylammonium dihydrogenphosphate)-CH3CN from 80:20 to 40:60 in 13 min; the flow rate was 0.5 mL/min. Peaks were detected by UV absorption with a diode array detector at 254, 275, and 280 nm. All derivatives tested for biological activity showed >99% purity by HPLC analysis (detection at 254 nm).
(1’S,2’R,3’S,4’R,5’S)-4’-(6-Amino-2-chloropurin-9-yl)-2’,3’-(dihydroxy)-1’-(phosphonomethylene)-bicyclo[3.1.0]hexane (4)
Nucleoside 21 (30 mg, 0.064 mmol) was coevaporated with anhydrous toluene (3 × 3 mL) and dissolved in anhydrous CH2Cl2 (3 mL). To this soution was added iodotrimethylsilane (91 µl, 0.64 mmol). After stirring for 17 h, the reaction mixture was cooled to 0 °C followed by the addition of ice-cold H2O (25 mL) and CH2Cl2 (25 mL). The phases were separated, and the aqueous phase washed with CH2Cl2 (1 × 35 mL) and diethyl ether (3 × 35 mL). The resulting aqueous phase evaporated to dryness and purified by HPLC (retention time: 19.1 min) to afford 4 (8.5 mg, 23%) as a white solid material. ESI-HRMS m/z 374.0397 [M - H]−, C12H14ClN5O5P–: Calcd. 374.0421); 1H NMR (D2O) δ 8.21 (s, 1H), 4.71 (s, 1H), 4.57 (d, 1H, J = 6.6 Hz), 4.01 (d, 1H, J = 6.6 Hz), 3.19 (q, 24H, J = 7.2 Hz), 2.23 (t, 1H, 15.5 Hz), 1.63–1.77 (m, 2H), 1.42–1.49 (m, 1H), 1.26 (t, 36H), 0.96–1.04 (m, 1H). 31P NMR (D2O) δ 23.68. Purity >99% by HPLC (retention time: 4.51 min).
(1’S,2’R,3’S,4’R,5’S)-4’-(6-Aminopurin-9-yl)-2’,3’-(dihydroxy)-1’-(phosphonomethylene)-bicyclo[3.1.0]hexane (5)
Nucleoside 23 (25 mg, 0.057 mmol) was coevaporated with anhydrous toluene (3 × 3 mL) and dissolved in anhydrous CH2Cl2 (3 mL). Iodotrimethylsilane (83 µl, 0.57 mmol) was added. After stirring for 15 h, the reaction mixture was cooled to 0 °C followed by the addition of ice-cold H2O (25 mL) and CH2Cl2 (25 mL). The phases were separated, and the aqueous phase was washed with CH2Cl2 (1 × 35 mL) and diethyl ether (3 × 35 mL). The resulting aqueous phase was evaporated to dryness and purified by HPLC (retention time: 17.5 min) to afford 5 (6.8 mg, 27%) as a white solid material. ESI-HRMS m/z 340.0817 [M - H]−, C12H15N5O5P−: Calcd. 340.0811); 1H NMR (D2O) δ 8.36 (s, 1H), 8.20 (s, 1H) 4.75 (s, 1H), 4.63 (d, 1H, J = 6.1 Hz), 4.09 (d, 1H, J = 6.1 Hz), 3.19 (q, 6H, J = 7.2 Hz), 1.95–2.18 (m,2H), 1.75–1.84 (m, 1H), 1.40–1.46 (m, 1H),1.26 (t, 9H, J = 7.2 Hz ), 0.92–1.02 (m, 1H). 31P NMR (D2O) δ 25.36. Purity >99% by HPLC (retention time: 2.9 min)
(1’S,2’R,3’S,4’R,5’S)-4’-(6-Amino-2-chloropurin-9-yl)-2’,3’-(dihydroxy)-1’-[(E)-phosphonoethenyl]-bicyclo[3.1.0]-hexane (7)
Nucleoside 27 (12 mg, 0.023 mmol) was coevaporated with anhydrous toluene (3 × 2 mL) and dissolved in anhydrous CH2Cl2 (2 mL). Iodotrimethylsilane (35 µl, 0.24 mmol) was added. After stirring for 18 h, the reaction mixture was cooled to 0 °C, followed by the addition of ice-cold H2O (15 mL) and CH2Cl2 (15 mL). The phases were separated, and the aqueous phase was washed with CH2Cl2 (1 × 25 mL) and diethyl ether (3 × 35 mL). The resulting aqueous phase was evaporated to dryness and purified by HPLC (retention time: 22.8 min) to afford 7 (2.5 mg, 28%) as a white solid material. ESI-HRMS m/z 386.0403 [M - H]−, C13H14N5ClO5P−: Calcd. 386.0421); 1H NMR (D2O) δ 7.99 (s, 1H), 6.21–6.36 (m, 1H), 6.06 (t, 1H, J = 17.5 Hz), 4.84–4.89 (m,1H), 4.06 (d, 1H, J = 6.6 Hz), 3.22 (q, 3H, J = 7.2 Hz), 1.99–2.06 (m,1H), 1.78–1.87 (m, 1H), 1.29 (t, 6H, J = 7.2 Hz ), 1.21–1.26 (m, 1H). 31P NMR (D2O) δ 14.68. Purity >99% by HPLC (retention time: 4.3 min)
(1’S,2’R,3’S,4’R,5’S)-4’-(6-Aminopurin-9-yl)-2’,3’-(dihydroxy)-1’-[(E)-phosphonoethenyl]-bicyclo[3.1.0]hexane (8)
Nucleoside 33 (20 mg, 0.042 mmol) was coevaporated with anhydrous toluene (3 × 5 mL) and dissolved in anhydrous CH2Cl2 (5 mL). Iodotrimethylsilane (60 µl, 0.42 mmol) was added. After stirring for 17 h, the reaction mixture was cooled to 0 °C, followed by the addition of ice-cold H2O (25 mL) and CH2Cl2 (25 mL). The phases were separated and the aqueous phase was washed with CH2Cl2 (1 × 35 mL) and diethyl ether (3 × 35 mL). The resulting aqueous phase was evaporated to dryness and purified by HPLC (retention time: 16.5 min) to afford 8 (11.8 mg, 78%) as a white solid material. ESI-HRMS m/z 352.0821 [M - H]−, C13H15N5O5P +: Calcd. 352.0811); 1H NMR (D2O) δ 8.30 (s, 1H), 8.06 (s, 1H), 6.30–6.44 (m, 1H), 6.07 (t, 1H, J =17.5), 4.97 (s, 1H ), 4.89 (d, 1H, J = 7.2 Hz), 4.09 (d, 1H, J = 7.2 Hz), 3.21 (q, 3H, J = 7.2 Hz), 2.03–2.10 (m, 2H), 1.84–1.89 (m, 1H), 1.29 (t, 7H, J = 7.2 Hz ). 31P NMR (D2O) δ 15.71. Purity >99% by HPLC (retention time: 3.5 min)
(1’S,2’R,3’S,4’R,5’S)-4’-(6-Aminopurin-9-yl)-2’,3’-(dihydroxy)-1’-(phosphonoethenyl)-bicyclo[3.1.0]hexane (10)
Nucleoside 28 (15 mg, 0.032 mmol) was coevaporated with anhydrous toluene (3 × 2 mL) and dissolved in anhydrous CH2Cl2 (2 mL). Iodotrimethylsilane (45 µl, 0.32 mmol) was added. After stirring for 15 h, the reaction mixture was cooled to 0 °C followed by the addition of ice-cold H2O (15 mL) and CH2Cl2 (15 mL). The phases were separated, and the aqueous phase was washed with CH2Cl2 (1 × 25 mL) and diethyl ether (3 × 35 mL). The resulting aqueous phase was evaporated to dryness and purified by HPLC (retention time: 16.6 min) to afford 10 (6.7 mg, 47%) as a white solid material. ESI-HRMS m/z 354.0970 [M - H]−, C13H17N5O5P−: Calcd. 354.0967); 1H NMR (CDCl3) δ 8.20 (s, 1H), 8.14 (s, 1H), 4.76 (s, 1H), 4.58 (d, 1H, J = 6.1 Hz), 4.09 (d, 1H, J = 6.1 Hz), 3.21 (q, 3H, J = 7.2 Hz), 1.69–2.14 (m, 4H), 1.59–1.69 (m, 1H), 1.39–1.349 (m, 1H), 1.29 (t, 6H, J = 7.2 Hz ), 0.81–0.92 (m, 1H). 31P NMR (D2O) δ 27.95. Purity >99% by HPLC (retention time: 2.91 min)
(1’S,2’R,3’S,4’R,5’S)-4’-(6-Amino-2-chloropurin-9-yl)-2’,3’-dihydroxy-1’-(methylphosphonicacid)-bicyclo[3.1.0]hexane (11) and (1’S,2’R,3’S,4’R,5’S)-4’-(6-Aminopurin-9-yl)-2’,3’-dihydroxy-1’-(methylphosphonicacid)-bicyclo[3.1.0]hexane (12)
Nucleoside 29 (15 mg, 0.034 mmol) was coevaporated with anhydrous toluene (3 × 3 mL) and dissolved in anhydrous CH2Cl2 (4 mL). Iodotrimethylsilane (91 µl, 0.33 mmol) was added. After stirring for 19 h, the reaction mixture was cooled to 0 °C, followed by the addition of ice-cold H2O (25 mL) and CH2Cl2 (25 mL). The phases were separated and the aqueous phase was washed with CH2Cl2 (1 × 35 mL) and diethyl ether (3 × 35 mL). The resulting aqueous phase was evaporated to dryness and purified by HPLC (retention time: 16.8 min) to afford 11 (1.3 mg, 11%) and 12 (0.8 mg, 20%, combined yield) as a white solid material.
Analytical data of compound 11
ESI-HRMS m/z 372.0625 [M-H]−, C13H16ClN5O4P−: Calcd. 372.0628); 1H NMR (D2O) δ 8.23 (s, 1H), 4.76–4.79 (m, 1H), 4.63 (d, 1H, J = 6.2 Hz), 4.12 (d, 1H, J = 6.2 Hz), 3.21 (q, 2H, J = 7.2 Hz), 2.34 (t, 1H, J = 15.3 Hz), 1.75–1.86 (m, 1H), 1.68–1.75 (m, 1H), 1.51–1.56 (m, 1H) 1.35 (d, 3H, J = 13.2 Hz), 1.26 (t, 1H, J = 7.2 Hz ) 0.96–1.04 (m, 1H). 31P NMR (D2O) δ 46.01. Purity >99% by HPLC (retention time: 4.19 min).
Analytical data of compound 12
ESI-HRMS m/z 338.1016 [M-H]−, C13H17N5O4P−: Calcd. 338.1018); 1H NMR (D2O) δ 8.26 (s, 1H), 8.25 (s, 1H), 4.76–4.79 (m, 1H), 4.63 (d, 1H, J = 6.2 Hz), 4.12 (d, 1H, J = 6.2 Hz), 3.21 (q, 2H, J = 7.2 Hz), 2.34 (t, 1H, J = 15.3 Hz), 1.75–1.86 (m, 1H), 1.68–1.75 (m, 1H), 1.51–1.56 (m, 1H) 1.35 (d, 3H, J = 13.2 Hz), 1.26 (t, 1H, J = 7.2 Hz ) 0.96–1.04 (m, 1H). 31P NMR (D2O) δ 46.0. Purity >99% by HPLC (retention time: 5.91 min).
Ethyl-(1S,2R,3S,4S,5S)-2,3-O-(isopropylidene)-4-O-(tert-butyldimethylsilyl)-bicyclo[3.1.0]hexanecarboxylate (14)
Known alcohol 13 (0.83 g, 3.40 mmol) was coevaporated with anhydrous toluene (2 × 10 mL) and dissolved in anhydrous CH2Cl2 (25 mL). Imidazole (0.69 g, 10.20 mmol), DMAP (0.04 g, 0.34 mmol) and tert-butylchlorodiphenylsilane (1.74 mL, 6.81 mmol) were added. After stirring at rt for 16 h, the reaction mixture was diluted with CH2Cl2 (50 mL) and washed with sat. aq. NaHCO3 (1 × 30 mL). The aqueous phase was back-extracted with CH2Cl2 (2 × 50 mL). The combined organic phase was evaporated to dryness, and the resulting crude residue was purified by silica gel column chromatography (0–8% EtOAc in petroleum ether, v/v) to afford compound 14 (1.52 mg, 93%) as a colorless oil. Rf = 0.3 (10% EtOAc in CH2Cl2, v/v); ESI-HRMS m/z 519.1981 ([M + K]+, C28H36O5Si·K+: Calcd. 519.1969); 1H NMR (CDCl3) δ 7.69–7.80 (m, 4H, Ph), 7.32–7.45 (m, 6H, Ph), 5.11 (d, 1H, J = 6.6 Hz), 4.42 (t, 1H, J = 6.1 Hz), 3.99–4.18 (m, 3H), 2.17–2.25 (m, 1H), 1.91 (t, 1H, J = 5.5 Hz), 1.57 (s, 3H), 1.45–1.53 (m, 1H), 1.18–1.24 (m, 6H), 1.07 (s, 9H).
(1S,2R,3S,4S,5S)-1-Hydroxymethyl-2,3-O-(isopropylidene)-4-O-(tert-butyldimethylsilyl)-bicyclo[3.1.0]hexane (15)
Compound 14 (0.98 g, 2.04 mmol) was coevaporated with anhydrous toluene (2 × 20 mL), dissolved in anhydrous THF (30 mL) and cooled to −70 °C. DIBAL-H (1.5 M in toluene 10.8 mL, 16.32 mmol) was added slowly to this solution over 20 min. After stirring at −70 °C for 3 h, the reaction was quenched with the very careful addition of ice-cold MeOH (20 mL), followed by warming the reaction mixture to rt. 1 M cold H2SO4 (20 mL) was added to the mixture and it was stirred for 1 h, followed by addition of CH2Cl2 (100 mL). The phases were separated, and the aqueous phase was extracted with CH2Cl2 (3 × 35 mL). The combined organic phase was evaporated to dryness, and the resulting residue was purified by silica gel column chromatography (0–45% EtOAc in petroleum ether, v/v) to afford compound 15 (0.74 g, 82%) as a colorless oil. Rf = 0.4 (50% EtOAc in petroleum ether, v/v); ESI-HRMS m/z 461.2112 ([M + Na]+, C26H34O4Si·Na+: Calcd. 461.2124); 1H NMR (CDCl3) δ 7.71–7.78 (m, 4H, Ph), 7.31–7.44 (m, 6H, Ph), 4.73 (d, 1H, J = 6.5 Hz), 4.44 (t, 1H, J = 6.5 Hz), 4.09 (t, 1H, J = 6.5 Hz), 3.59–3.67 (m, 1H), 3.41–3.49 (m, 1H), 1.57–1.59 (m, 4H), 1.33 (t,1H, J = 5.5 Hz), 1.20 (s, 3H), 1.07 (s, 9H), 0.56–0.68 (m, 1H).
(1S,2R,3S,4S,5S)-2,3-O-(Isopropylidene)-1-methanesulfonyloxymethyl-4-O-(tert-butyldimethylsilyl)-bicyclo[3.1.0]hexane (16)
Compound 15 (0.59 g, 1.36 mmol) was coevaporated with anhydrous toluene (2 × 20 mL), dissolved in anhydrous CH2Cl2 (30 mL) and cooled to 0 °C. Triethylamine (0.95 mL, 6.79 mmol) and methanesulfonyl chloride (0.22 mL, 2.72 mol) were added at 0 °C over 10 min. After warming the reaction mixture to rt, it was stirred for 17 h. Then, ice-cold H2O (25 mL) was added and the mixture was extracted with EtOAc (2 × 45 mL). The combined organic phase was washed with sat. aq. NaHCO3 (2 × 35 mL) and evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–50% EtOAc in petroleum ether, v/v) to afford compound 16 (0.68 g, 96%) as a colorless oil. Rf = 0.5 (50% EtOAc in petroleum ether, v/v); ESI-HRMS m/z 555.1623 ([M + K]+, C27H36O6SSi·K+: Calcd. 555.1639); 1H NMR (CDCl3) δ 7.69–7.77 (m, 4H, Ph), 7.31–7.45 (m, 6H, Ph), 4.69 (d, 1H, J = 6.5 Hz), 4.47 (t, 1H, J = 6.5 Hz), 4.37–4.43 (dd, 1H, J = 10.9 Hz), 4.07 (t, 1H, J = 6.5 Hz), 3.90–3.95 (dd, 1H, J = 10.9 Hz), 2.99 (s, 3H), 1.67–1.72 (m, 2H), 1.55 (s, 3H), 1.20 (s, 3H), 1.08 (s, 9H), 0.72–0.79 (m, 1H).
(1S,2R,3S,4S,5S)-1-Iodomethyl-2,3-O-(isopropylidene)-4-O-(tert-butyldimethylsilyl)-bicyclo[3.1.0]hexane (17)
Compound 16 (0.68 g, 1.32 mmol) was coevaporated with anhydrous toluene (2 × 20 mL), and the residue dissolved in anhydrous 1,4-dioxane (25 mL). NaI (0.59 g, 3.94 mol) was added to the mixture, and it was heated to 65 °C. After stirring for 17 h, the reaction mixture was cooled to rt and diluted with H2O (25 mL) and CH2Cl2 (75 mL). The phases were separated, and the aqueous phase was extracted with CH2Cl2 (3 × 35 mL). The combined organic phase was evaporated to dryness, and the resulting residue was purified by silica gel column chromatography (0–20% EtOAc in petroleum ether, v/v) to afford compound 17 (0.69 g, 95%) as a colorless oil. Rf = 0.5 (20% EtOAc in petroleum ether, v/v); ESI-HRMS m/z 549.1322 ([M + H]+, C26H33IO3Si·H+: Calcd. 549.1322); 1H NMR (CDCl3) δ 7.68–7.77 (m, 4H, Ph), 7.32–7.46 (m, 6H, Ph), 4.69 (d, 1H, J = 6.5 Hz), 4.43 (t, 1H, J = 6.5 Hz), 4.09 (t, 1H, J = 6.5 Hz), 3.55–3.60 (dd, 1H, J = 10.5 Hz), 3.97–4.02 (dd, 1H, J = 10.5 Hz), 2.02 (t, 1H, J = 4.9 Hz), 1.54–1.57 (s, 4H), 1.20 (s, 3H), 1.07 (s, 9H), 0.83–0.90 (m, 1H).
Diethyl-(1S,2R,3S,4S,5S)-2,3-O-(isopropylidene)-4-O-(tert-butyldimethylsilyl)-bicyclo[3.1.0]hexane phosphonate (18)
Compound 17 (0.68 g, 1.23 mmol) was dissolved in triethylphosphite (17 mL), and the mixture was heated to 110 °C. After stirring for 17 h, the reaction mixture was cooled to rt and evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–90% EtOAc in petroleum ether, v/v) to afford compound 18 (0.65 g, 94%) as a colorless oil. Rf = 0.3 (EtOAc); ESI-HRMS m/z 559.2665 ([M + H]+, C30H43O6PSi·H+: Calcd. 559.2645); 1H NMR (CDCl3) δ 7.71–7.77 (m, 4H, Ph), 7.30–7.43 (m, 6H, Ph), 4.80 (d, 1H, J = 6.5 Hz), 4.47 (t, 1H, J = 6.5 Hz), 4.10 (t, 1H, J = 6.5 Hz), 3.91–4.05 (m, 4H), 2.22 (t, 1H, J = 16.5 Hz), 1.63–1.71 (m,1H), 1.57–1.61 (m, 2H), 1.55 (s, 3H), 1.22 (t, 3H, J = 7.2 Hz), 1.20 (t, 3H, J = 7.2 Hz), 1.19 (s, 3H), 1.07 (s, 9H), 0.53–0.60 (m, 1H). 31P NMR (CDCl3) δ 29.93.
Diethyl-(1S,2R,3S,4S,5S)-4-hydroxy-2,3-O-(isopropylidene)-bicyclo[3.1.0]hexane phosphonate (19)
Compound 18 (0.65 g, 1.16 mmol) was dissolved in a mixture of THF (20 mL) and tetrabutylammonium fluoride (1 M in THF, 2.91 mL, 2.91 mmol). After stirring for 17 h, the reaction mixture was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–7% MeOH in EtOAc, v/v) to afford compound 19 (0.33 g, 88%) as a colorless oil. Rf = 0.3 (5% MeOH in EtOAc, v/v); ESI-HRMS m/z 321.1466 [M + H]+, C14H25O6P·H+: Calcd. 321.1467); 1H NMR (CDCl3) δ 5.02 (d, 1H, J = 6.1 Hz), 4.50–4.58 (m, 2H), 4.02–4.17 (m, 4H), 2.32–2.37 (m, 1H), 2.26 (t, 1H, J = 16.5 Hz), 1.88–1.96 (m,1H), 1.61–1.74 (m, 1H), 1.54 (s, 3H), 1.32 (t, 6H, J = 7.2 Hz), 1.28 (s, 3H),1.21–1.27 (m, 1H), 0.60–0.67 (m, 1H). 31P NMR (CDCl3) δ 29.01.
Diethyl-(1’S,2’R,3’S,4’R,5’S)-4’-(2,6-dichloropurin-9-yl)-2’,3’-O-(isopropylidene)-bicyclo[3.1.0]hexane phosphonate (20)
Diisopropyl azodicarboxylate (97 µL, 0.49 mmol) was added at rt to a mixture of triphenylphosphine (128 mg, 0.49 mmol) and 2,6-dichloropurine (92 mg, 0.49 mmol) in anhydrous THF (3 mL). After stirring for 30 min, a solution of compound 19 (78 mg, 0.25 mmol) in THF (3 mL) was added to the mixture. After stirring for 51 h, the reaction mixture was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–4% MeOH in EtOAc, v/v) to afford nucleoside 20 (90 mg, 75%) as a white solid material. Rf = 0.5 (5% MeOH in EtOAc, v/v); ESI-HRMS m/z 491.1013 [M + H]+, C19H25Cl2N4O5P·H+: Calcd. 491.1018); 1H NMR (CDCl3) δ 8.82 (s, 1H), 5.39 (d, 1H, J = 6.5 Hz), 5.10 (s, 1H), 4.61 (d, 1H, J = 6.5 Hz), 4.02–4.21 (m, 4H), 2.46 (t, 1H, J = 16.5 Hz), 1.91–2.06 (m,1H), 1.74–1.82 (m, 1H), 1.54 (s, 3H), 1.32 (t, 3H, J = 7.2 Hz), 1.26 (t, 3H, J = 7.2 Hz), 1.24 (s, 3H),1.08–1.21 (m, 1H), 0.97–1.06 (m, 1H).
Diethyl-(1’S,2’R,3’S,4’R,5’S)-4’-(6-amino-2-chloropurin-9-yl)-2’,3’-O-(isopropylidene)-bicyclo[3.1.0]hexane phosphonate (21)
Nucleoside 20 (90 mg, 0.19 mmol) was treated with 2 M NH3 in i-PrOH (5 mL), and the mixture was heated to 70 °C and stirred for 17 h. The reaction mixture was evaporated to dryness, and the resulting residue was purified by silica gel column chromatography (0–5% MeOH in CH2Cl2, v/v) to afford nucleoside 21 (70 mg, 80%) as a white solid material. Rf = 0.5 (5% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 472.1519 [M + H]+, C19H27ClN5O5P·H+: Calcd. 472.1517); 1H NMR (CDCl3) δ 8.31 (s, 1H), 5.98 (s, 2H), 5.36 (d, 1H, J = 7.1 Hz), 4.97 (s, 1H), 4.61 (d, 1H, J = 6.5 Hz), 4.03–4.19 (m, 4H), 2.39 (t, 1H, J = 16.5 Hz), 2.03–2.17 (m, 1H), 1.70–1.77 (m, 1H), 1.52 (s, 3H), 1.32 (t, 3H, J = 7.2 Hz), 1.25 (t, 3H, J = 7.2 Hz), 1.23 (s, 3H), 1.18–1.21 (m, 1H), 0.96–1.04 (m, 1H).
Diethyl-(1’S,2’R,3’S,4’R,5’S)-4’-(6-chloropurin-9-yl)-2’,3’-O-(isopropylidene)-bicyclo[3.1.0]hexane phosphonate (22)
Diisopropyl azodicarboxylate (100 µL, 0.50 mmol) was added at rt to a mixture of triphenylphosphine (133 mg, 0.50 mmol) and 6-chloropurine (96 mg, 0.50 mmol) in anhydrous THF (3 mL). After stirring the mixture for 30 min, a solution of compound 19 (81 mg, 0.26 mmol) in THF (3 mL) was added. After stirring for 17 h, the reaction mixture was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–4% MeOH in EtOAc, v/v) to afford nucleoside 22 (100 mg, 87%) as a white solid material. Rf = 0.5 (5% MeOH in EtOAc, v/v); ESI-HRMS m/z 457.1417 [M + H]+, C19H26ClN4O5P·H+: Calcd. 457.1408); 1H NMR (CDCl3) δ 8.84 (s, 1H), 8.78 (s, 1H), 5.39 (d, 1H, J = 6.5 Hz), 5.15 (s, 1H), 4.62 (d, 1H, J = 6.5 Hz), 4.07–4.21 (m, 4H), 2.44 (t, 1H, J = 16.5 Hz), 1.94–2.18 (m,1H), 1.83–1.90 (m, 1H), 1.58 (s, 3H), 1.33 (t, 3H, J = 7.2 Hz), 1.24–1.30 (m, 4H), 0.97–1.06 (m, 1H).
Diethyl-(1’S,2’R,3’S,4’R,5’S)-4’-(6-aminopurin-9-yl)-2’,3’-O-(isopropylidene)-bicyclo[3.1.0]hexane phosphonate (23)
Nucleoside 22 (100 mg, 0.22 mmol) was treated with 2 M NH3 in i-PrOH (5 mL) and heated up to 70 °C. After stirring for 19 h, the reaction mixture was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–6% MeOH in CH2Cl2, v/v) to afford nucleoside 23 (75 mg, 79%) as a white solid material. Rf = 0.4 (5% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 438.1912 [M + H]+, C19H28N5O5P·H+: Calcd. 438.1906); 1H NMR (CDCl3) δ 8.38 (s, 1H), 8.36 (s, 1H), 5.54 (s, 2H), 5.36 (d, 1H, J = 7.2 Hz), 5.03 (s, 1H), 4.63 (d, 1H, J = 7.2 Hz), 4.06–4.20 (m, 4H), 2.38 (t, 1H, J = 16.5 Hz), 1.97–2.11 (m, 1H), 1.78–1.85 (m,1H), 1.68 (s, 3H), 1.32 (t, 3H, J = 7.2 Hz), 1.27 (t, 3H, J = 7.2 Hz), 1.23 (s, 3H),1.18–1.21 (m, 1H), 0.95–1.02 (m, 1H).
(1’S,2’R,3’S,4’R,5’S)-4’-(2,6-Dichloropurin-9-yl)-1’-formyl-2,3-O-(isopropylidine)-bicyclo[3.1.0]hexane (25)
Known nucleoside13 24 (150 mg, 0.41 mmol) was coevaporated with anhydrous toluene (2 × 8 mL) and dissolved in anhydrous CH2Cl2 (8 mL). Dess-Martin periodinane (257 mg, 0.61 mmol) was added. After stirring for 1 h, the reaction mixture was diluted with EtOAc (50 mL) and washed with an aqueous mixture of Na2S2O3 and NaHCO3 (3 × 35 mL). The aqueous phase was then extracted with EtOAc (2 × 35 mL). The combined organic phase was evaporated to dryness, and the resulting residue was purified by silica gel column chromatography (0–100% EtOAc in petroleum ether, v/v) to afford compound 25 (120 mg, 80%) as a white solid material. Rf = 0.6 (EtOAc); ESI-HRMS m/z 369.0527 ([M + H]+, C15H14Cl2N4O3·H+: Calcd. 369.0521); 1H NMR (CDCl3) δ 9.62 (s, 1H), 8.05 (s, 1H), 5.94 (d, 1H, J = 7.2 Hz), 4.97 (s, 1H), 4.83 (d, 1H, J = 7.2 Hz), 2.22–2.29 (m, 1H), 1.73 (t, 1H, J = 6.1 Hz), 1.57 (s, 3H), 1.30 (s, 3H).
(1’S,2’R,3’S,4’R,5’S)-4’-(2,6-Dichloropurin-9-yl)-1’-[diisopropyl-(E)-ethenylphosphonate]-2’,3’-O-(isopropylidine)-bicyclo[3.1.0]hexane (26)
Tetraisopropyl methylenediphosphonate (165 µL, 0.51 mmol) was added to a suspension of NaH (60% dispersion in mineral oil, 25 mg, 1.02 mmol) in anhydrous THF (2 mL) at 0 °C. After H2 evolution ceased, a solution of aldehyde 25 (125 mg, 0.34 mmol) in anhydrous THF (3 mL) was added dropwise carefully at 0 °C. After stirring at 0 °C for 1 h, the mixture was warmed to rt. After stirring at rt for 1 h, the reaction mixture was cooled to 0 °C, and ice-cold H2O (20 mL) was added. The phases were separated, and the aqueous phase was extracted with EtOAc (3 × 35 mL). The combined organic phase was evaporated to dryness, and the resulting residue was purified by silica gel column chromatography (0–4% MeOH in EtOAc, v/v) to afford nucleoside 26 (150 mg, 83%) as a white solid material. Rf = 0.3 (EtOAc); ESI-HRMS m/z 531.1313 ([M + H]+, C22H29Cl2N4O5P·H+: Calcd. 531.1331); 1H NMR (CDCl3) δ 8.04 (s, 1H), 6.50–6.65 (m, 1H), 5.97 (t, 1H, J = 17.1 Hz), 5.53 (d, 1H, J = 7.2 Hz), 4.98 (s, 1H), 4.77 (d, 1H, J = 7.2 Hz), 4.60–4.74 (m, 2H), 1.82–1.90 (m, 1H), 1.59 (s, 3H), 1.22–1.38 (m, 16H), 0.83–0.90 (m, 1H). 31P NMR (CDCl3) δ 16.64.
(1’S,2’R,3’S,4’R,5’S)-4’-(6-Amino-2-chloropurin-9-yl)-1’-[diisopropyl-(E)-ethenylphosphonate]-2,3-O-(isopropylidine)-bicyclo-[3.1.0]-hexane (27)
Nucleoside 26 (100 mg, 0.19 mmol) was treated with 2 M NH3 in i-PrOH (5 mL) and heated to 70 °C. After stirring for 16 h, the reaction mixture was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–8% MeOH in CH2Cl2, v/v) to afford nucleoside 27 (85 mg, 88%) as a white solid material. Rf = 0.3 (5% MeoH in EtOAc, v/v); ESI-HRMS m/z 512.1821 ([M + H]+, C22H31ClN5O5P·H+: Calcd. 512.1830); 1H NMR (CDCl3) δ 7.69 (s, 1H), 6.52–6.68 (m, 1H), 5.94 (t, 1H, J = 17.5 Hz), 5.75 (s, 2H), 5.51 (d, 1H, J = 7.2 Hz), 4.91 (s, 1H), 4.76 (d, 1H, J = 7.2 Hz), 4.59–4.73 (m, 2H), 1.80–1.89 (m, 1H), 1.61 (s, 3H), 1.21–1.37 (m, 15H), 1.08–1.17 (m, 1H), 0.77–0.95 (m, 1H).
(1’S,2’R,3’S,4’R,5’S)-4’-(6-Aminopurin-9-yl)-1’-(diisopropyl-phosphonoethenyl)-2’,3’-O-(isopropylidine)-bicyclo[3.1.0]hexane (28)
Nucleoside 27 (20 mg, 0.04 mmol) was dissolved in a mixture of MeOH and aqueous 2 M NaOH (3 mL, 2:1, v/v). 10% Pd/C (20 mg) and H2 (3 bar) were added to this solution. After stirring the mixture for 19 h, the catalyst was removed by filtration through a Celite pad, which was washed with MeOH (40 mL), and the filtrate was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–10% MeOH in EtOAc, v/v) to afford nucleoside 28 (15 mg, 79%) as white solid material. Rf = 0.5 (15% MeOH in EtOAc, v/v); ESI-HRMS m/z 480.2385 ([M + H]+, C22H34N5O5P·H+: Calcd. 480.2376); 1H NMR (CDCl3) δ 8.32 (s, 1H), 7.79 (s, 1H), 5.80 (s, 2H), 5.19 (d, 1H, J = 7.2 Hz), 4.83 (s, 1H), 4.74 (d, 1H, J = 7.2 Hz), 4.63–4.73 (m, 2H), 1.60–2.35 (m, 4H), 1.52 (s, 3H), 1.44–1.51 (m, 1H), 1.33 (s, 6H), 1.31 (s, 6H), 1.23 (s, 3H), 1.04–1.09 (m, 1H), 0.76–0.83 (m, 1H). 31P NMR (CDCl3) 30.04.
(1S,2R,3S,4S,5S)-1-Formyl-2,3-O-(isopropylidene)-4-O-(tert-butyldimethylsilyl)-bicyclo[3.1.0]hexane (29)
Compound 15 (0.63 g, 1.43 mmol) was coevaporated with anhydrous toluene (2 × 25 mL) and dissolved in anhydrous CH2Cl2 (25 mL). Dess-Martin periodinane (0.91 g, 2.13 mmol) was added to this solution. After stirring for 4 h, the reaction mixture was diluted with EtOAc (50 mL) and washed with an aqueous mixture of Na2S2O3 and NaHCO3 (3 × 50 mL). The aqueous phase was extracted with EtOAc (2 × 50 mL). The combined organic phase was evaporated to dryness and the resulting residue was purified by silica gel column chromatography (0–25% EtOAc in petroleum ether, v/v) to afford aldehyde 29 (452 mg, 73%) as a colorless oil. Rf = 0.6 (50% EtOAc in petroleum ether, v/v); ESI-HRMS m/z 459.1986 ([M + Na]+, C26H32O4Si·Na+: Calcd. 459.1968); 1H NMR (CDCl3) δ 8.92 (s, 1H), 7.68–7.78 (m, 4H, Ph), 7.31–7.48 (m, 6H, Ph), 5.13 (d, 1H, J = 6.5 Hz), 4.41 (t, 1H, J = 6.5 Hz), 4.16 (t, 1H, J = 6.5 Hz), 2.19–2.28 (m, 1H), 2.10–2.18 (m, 1H), 1.55 (s, 3H), 1.43–1.51 (m, 1H), 1.23 (s, 3H), 1.09 (s, 9H).
(1S,2R,3S,4S,5S)-1-[Diisopropyl-(E)-phosphonoethenyl]-2,3-O-(isopropylidene)-4-O-(tert-butyldimethylsilyl)-bicyclo[3.1.0]hexane (30)
Tetraisopropyl methylenediphosphonate (475 µL, 1.47 mmol) was added to a suspension of sodium hydride (71 mg, 2.95 mmol, 60 % dispersion in mineral oil) in anhydrous THF (6 mL) at 0 °C. After H2 evolution ceased, a solution of aldehyde 29 (0.43 g, 0.98 mmol) in anhydrous THF (4 mL) was added dropwise carefully at 0 °C. After stirring at 0 °C for 1 h, the mixture was warmed to rt. After stirring at rt for 1h, the mixture was cooled to 0 °C, and ice-cold H2O (20 mL) was added. The phases were separated, and the aqueous phase was extracted with EtOAc (3 × 35 mL). The combined organic phase was evaporated to dryness and the resulting residue was purified by silica gel column chromatography (0–70% EtOAc in petroleum ether, v/v) to afford nucleoside 30 (0.24 mg, 48%) as a white solid material. Rf = 0.4 (70% EtOAc in petroleum ether, v/v); ESI-HRMS m/z 599.2938 ([M + H]+, C33H47O6PSi·H+: Calcd. 599.2958); 1H NMR (CDCl3) δ 7.69–7.77 (m, 4H, Ph), 7.31–7.45 (m, 6H, Ph), 6.24–6.40 (m, 1H), 5.66 (t, 1H, J = 17.5 Hz), 4.75 (d, 1H, J = 6.5 Hz), 4.52–4.64 (m, 2H) 4.42 (t, 1H, J = 6.5 Hz), 4.11 (t, 1H, J = 6.5 Hz), 1.93–1.99 (m, 1H), 1.78–1.85 (m, 1H), 1.57 (s, 3H), 1.19–1.32 (m, 15H), 1.07 (s, 9H), 0.93–1.01 (m, 1H).
(1S,2R,3S,4S,5S)-1-[Diisopropyl-(E)-phosphonoethenyl]-4-(hydroxy)-2,3-O-(isopropylidene)-bicyclo[3.1.0]hexane (31)
Compound 30 (0.45 g, 0.76 mmol) was dissolved in THF (10 mL) and tetrabutylammonium fluoride (1.0 M in THF, 2.3 mL, 2.3 mmol) was added. After stirring for 13 h, the reaction mixture was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–7% MeOH in EtOAc, v/v) to afford compound 31 (0.26 g, 98%) as a colorless oil. Rf = 0.3 (EtOAc); ESI-HRMS m/z 361.1790 ([M + H]+, C17H29O6P·H+: Calcd. 361.1780); 1H NMR (CDCl3) δ 6.34–6.49 (m, 1H), 5.74 (t, 1H, J = 17.5 Hz), 4.99 (d, 1H, J = 6.5 Hz), 4.47–4.69 (m, 4H), 2.40 (d, 1H, J = 9.5 Hz), 2.07–2.15 (M, 1H), 1.59–1.63 (m, 1H), 1.58 (s, 3H), 1.22–1.34 (m, 15H), 0.93–1.07 (m, 1H).
(1’S,2’R,3’S,4’R,5’S)-4’-(6-Chloropurin-9-yl)-1’-[diisopropyl-(E)-phosphonoethenyl] -2’,3’-O-(isopropylidene)-bicyclo[3.1.0]hexane (32)
Diisopropyl azodicarboxylate (90 µL, 0.45 mmol) was added at rt to a mixture of triphenylphosphine (117 mg, 0.45 mmol) and 6-chloropurine (70 mg, 0.45 mmol) in anhydrous THF (5 mL). After stirring for 30 min, a solution of the compound 31 (80 mg, 0.23 mmol) in THF (5 mL) was added. After stirring for 60 h, the reaction mixture was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–55% acetone in petroleum ether, v/v) to afford nucleoside 32 (92 mg, 85%) as a white solid material. Rf = 0.4 (60% acetone in petroleum ether, v/v); ESI-HRMS m/z 519.1532 ([M + Na]+, C22H30ClN4O5P·Na+: Calcd. 519.1540); 1H NMR (CDCl3) δ 8.71 (s, 1H), 8.07 (s, 1H), 6.49–6.63 (m, 1H), 5.96 (t, 1H, J =17.5), 5.53 (d, 1H, J = 6.5 Hz), 5.02 (s, 1H ), 4.79 (d, 1H, J = 6.5 Hz), 4.61–4.73 (m, 2H), 1.86–1.92 (m, 1H), 1.58–1.63 (m, 1H), 1.54 (s, 3H), 1.22–1.38 (m, 16H).
(1’S,2’R,3’S,4’R,5’S)-4’-(6-Aminopurin-9-yl)-1’-[diisopropyl-(E)-phosphonoethenyl]-2’,3’-O-(isopropylidene)-bicyclo[3.1.0]hexane (33)
Nucleoside 32 (90 mg, 0.19 mmol) was treated with 2 M NH3 in i-PrOH (7 mL) and heated to 70 °C. After stirring for 17 h, the reaction mixture was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–12% MeOH in CH2Cl2, v/v) to afford nucleoside 33 (74 mg, 85%) as a white solid material. Rf = 0.2 (10% MeOH in EtOAc, v/v); ESI-HRMS m/z 478.2198 ([M + H]+, C22H32N5O5P·H+: Calcd. 478.2219); 1H NMR (CDCl3) δ 8.30 (s, 1H), 7.73 (s, 1H), 6.51–6.66 (m, 1H), 5.96 (t, 1H, J =17.5), 5.47–5.54 (m, 3H), 4.95 (s, 1H ), 4.78 (d, 1H, J = 6.5 Hz), 4.60–4.72 (m, 2H), 1.86–1.94 (m, 1H), 1.53–1.57 (m, 4H), 1.24–1.37 (m, 16H).
(1S,2R,3S,4S,5S)-1-(Diisopropyl-phosphonoethenyl)-4-(hydroxy)-2,3-O-(isopropylidene)-bicyclo[3.1.0]hexane (34)
Compound 31 (30 mg, 0.083 mmol) was dissolved in MeOH (3 mL). 10% Pd/C (25 mg) and H2 (3 bar) was added. After stirring the mixture for 17 h, the catalyst was removed by filtration through a Celite pad, which was washed with MeOH (40 mL), and the filtrate was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–90% acetone in petroleum ether, v/v) to afford nucleoside 34 (22 mg, 72%) as white solid material. Rf = 0.3 (5% MeOH in EtOAc, v/v); ESI-HRMS m/z 363.1933 ([M + H]+, C17H31O6P·H+: Calcd. 363.1937); 1H NMR (CDCl3) δ 4.61–4.76 (m, 2H), 4.43–4.54 (m, 2H), 4.17–4.35 (m, 1H), 2.32 (d, J = 9.8 Hz, 1H), 1.43–1.93 (m, 8H), 1.22–1.37 (m, 15H), 1.07–1.14 (m,1H), 0.47–0.56 (m, 1H).
(1’S,2’R,3’S,4’R,5’S)-4’-(2,6-Dichloropurin-9-yl)-1’-(diisopropyl-phosphonoethenyl)-2’,3’-O-(isopropylidene)-bicyclo[3.1.0]hexane (35)
Diisopropyl azodicarboxylate (93 µL, 0.47 mmol) was added at rt to a mixture of triphenylphosphine (123 mg, 0.47 mmol) and 2,6-dichloropurine (89 mg, 0.47 mmol) in anhydrous THF (4 mL). After stirring for 30 min, a solution of the compound 34 (85 mg, 0.24 mmol) in THF (4 mL) was added. After stirring for 65 h, the reaction mixture was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–5% MeOH in EtOAc, v/v) to afford nucleoside 35 (50 mg, 40%) as a white solid material. Rf = 0.4 (5% MeOH in EtOAc, v/v); ESI-HRMS m/z 533.1497 ([M + H]+, C22H31Cl2N4O5P·H+: Calcd. 533.1487); 1H NMR (CDCl3) δ 8.09 (s, 1H), 5.20 (d, 1H, J = 7.2 Hz), 4.86 (s, 1H), 4.73 (d, 1H, J = 7.2 Hz), 4.63–4.73 (m, 2H), 2.25–2.44 (m, 1H), 1.79–2.09 (m, 2H), 1.58–1.70 (m, 1H), 1.52 (s, 3H), 1.43–1.52 (m, 1H), 1.28–1.35 (m, 12H), 1.24 (s, 3H), 1.04–1.11 (m,1H), 0.78–0.87 (m, 1H).
(1’S,2’R,3’S,4’R,5’S)-4’-(6-Amino-2-chloropurin-9-yl)-1’-(diisopropyl-phosphonoethenyl)-2’,3’-O-(isopropylidene)-bicyclo[3.1.0]hexane (36)
Nucleoside 35 (50 mg, 0.094 mmol) was treated with 2 M NH3 in i-PrOH (5 mL) and heated to 70 °C. After stirring for 19 h, the reaction mixture was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–10% MeOH in CH2Cl2, v/v) to afford nucleoside 36 (34 mg, 71%) as a white solid material. Rf = 0.4 (8% MeOH in EtOAc, v/v); ESI-HRMS m/z 514.1978 ([M + H]+, C22H33ClN5O5P·H+: Calcd. 514.1986); 1H NMR (CDCl3) δ 7.73 (s, 1H), 5.84 (s, 2H), 5.20 (d, 1H, J = 6.5 Hz), 4.76 (s, 1H), 4.72 (d, 1H, J = 6.5 Hz), 4.62–4.71 (m, 2H), 2.24–2.40 (m, 1H), 1.75–2.08 (m, 1H), 1.56–1.74 (m, 5H), 1.40–1.47 (m, 1H), 1.28–1.35 (m, 12H), 1.24 (s, 3H), 1.01–1.06 (m,1H), 0.74–0.82 (m, 1H).
(1’S,2’R,3’S,4’R,5’S)-4-(6-Amino-2-chloropurin-9-yl)-2’,3’-(dihydroxy)-1’-(phosphonoethenyl)-bicyclo[3.1.0]hexane (9)
Nucleoside 23 (25 mg, 0.049 mmol) was coevaporated with anhydrous toluene (3 × 4 mL) and dissolved in anhydrous CH2Cl2 (4 mL). Iodotrimethylsilane (70 µl, 0.49 mmol) was added. After stirring for 19 h, the reaction mixture was cooled to 0 °C followed by the addition of ice-cold H2O (25 mL) and CH2Cl2 (25 mL). The phases were separated, and the aqueous phase was washed with CH2Cl2 (1 × 35 mL) and diethyl ether (3 × 35 mL). The resulting aqueous phase was evaporated to dryness and purified by HPLC (retention time: 21.5 min) to afford 9 (8.5 mg) and 10 (1.3 mg, 53%, combined yield) as a white solid materials. ESI-HRMS m/z 388.0574 [M - H]−, C13H16ClN5O5P−: Calcd. 388.0578); 1H NMR (D2O) δ 8.15 (s, 1H), 4.74 (s, 1H), 4.61 (d, 1H, J = 7.2 Hz), 4.11 (d, 1H, J = 7.2 Hz), 3.21 (q, 3H, J = 7.2 Hz), 1.70–2.11 (m, 4H), 1.63–1.71 (m, 1H), 1.33–1.38 (m, 1H), 1.29 (t, 3H, J = 7.2 Hz ), 0.81–0.91 (m, 1H). 31P NMR (D2O) δ 28.26. Purity >99% by HPLC (retention time: 4.6 min)
(1S,2R,3S,4S,5S)-1-Bromomethyl-2,3-O-(isopropylidene)-4-O-(tert-butyldimethylsilyl)-bicyclo[3.1.0]hexane (37)
Compound 15 (0.30 g, 0.69 mmol) was coevaporated with anhydrous toluene (3 × 10 mL) and dissolved in anhydrous CH2Cl2 (8 mL). CBr4 (0.46 g, 1.36 mmol) triphenylphosphine (0.36 g, 1.36 mmol), and triethylamine (0.3 mL, 2.07 mmol) were added. After stirring for 17 h, the reaction mixture was diluted with CH2Cl2 (50 mL) and sat. aqueous NaCl (25 mL). The phases were separated and aqueous phase was extracted with CH2Cl2 (3 × 25 mL). The combined organic phase was evaporated to dryness and the resulting residue was purified by silica gel column chromatography (0–20% EtOAc in petroleum ether, v/v) to afford compound 37 (0.28 g, 81%) as a colorless oil. Rf = 0.8 (50% EtOAc in petroleum ether, v/v); ESI-HRMS m/z 523.1296 ([M + Na]+, C26H33BrO3Si·Na+: Calcd. 523.1280); 1H NMR (CDCl3) δ 7.67.77 (m, 4H, Ph), 7.30–7.45 (m, 6H, Ph), 4.77 (d, 1H, J = 6.5 Hz), 4.44 (t, 1H, J = 6.5 Hz), 4.08 (t, 1H, J = 6.5 Hz), 3.76 (d, 1H, J = 10.5 Hz), 3.13 (d, 1H, J = 10.5 Hz), 1.80–1.87 (m, 1H), 1.60–1.70 (m, 1H), 1.55 (s, 3H), 1.21 (s, 3H), 1.05 (s, 9H), 0.71–0.86 (m, 1H).
(1S,2R,3S,4S,5S)-1-C-(Ethoxymethylphosphinyl)-2,3-O-(isopropylidene)-4-O-(tert-butyldimethylsilyl)-bicyclo[3.1.0]hexane (38)
Compound 37 (0.28 g, 0.56 mmol) was dissolved in diethylmethylphosphite (4 mL) and heated up to 110 °C. After stirring for 17 h, the reaction mixture was cooled to rt and evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–90% EtOAc in petroleum ether, v/v) to afford inseparable diastereomeric mixture of compound 38 (0.28 g, 95%) as a colorless oil. Rf = 0.6 (5% MeOH in EtOAc, v/v); ESI-HRMS m/z 529.2532 ([M + H]+, C29H41O5PSi·H+: Calcd. 529.2539); 1H NMR (CDCl3) δ 7.68–7.77 (m, 6.8H, Ph), 7.29–7.46 (m, 10.2H, Ph), 4.75 (d, 0.7H, J = 6.5 Hz), 4.68 (d, 1H, J = 6.5 Hz), 4.43–4.49 (m, 1.7H), 3.87–4.14 (m, 5.1 H), 1.73–1.92 (m, 3.4H), 1.62 (s, 5.1H), 1.57–1.60 (m,1.7H), 1.48 (d, 3H, J = 3.4 Hz), 1.44 (d, 2.1H, J = 3.4 Hz), 1.26 (t, 3H, J = 7.2 Hz), 1.22 (t, 2.1H, J = 7.2 Hz), 1.19 (s, 2.1H), 1.18 (s, 3H), 1.09–1.13 (m, 1.7H), 1.07 (s, 15.3H), 0.52–0.69 (m, 1H).
(1S,2R,3S,4S,5S)-1-C-(Ethoxymethylphosphinyl)-4-hydroxy-2,3-O-(isopropylidene)-bicyclo[3.1.0]hexane (39)
Compound 38 (0.30 g, 0.57 mmol) was dissolved in THF (10 mL) and tetrabutylammonium fluoride (1.0 M in THF, 1.70 mL, 1.70 mmol) was added. After stirring for 21 h, the reaction mixture was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–15% MeOH in EtOAc, v/v) to afford an inseparable diastereomeric mixture of compound 39 (0.15 g, 91%) as a colorless oil. Rf = 0.2 (15% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 291.1366 ([M + H]+, C13H23O5P·H+: Calcd. 291.1361); 1H NMR (CDCl3) δ 4.89–5.03 (m, 2H), 4.50–4.60 (m, 4H), 3.97–4.14 (m, 4H), 2.34–2.40 (m, 2H), 1.95 (t, 2H, J = 7.7 Hz), 1.90 (t, 2H, J = 7.7 Hz),1.78–1.87 (m, 2H), 1.66 (s, 6H), 1.55 (d, 3H, J = 3.9 Hz), 1.50 (d, 3H, J = 3.9 Hz), 1.29–1.35 (m, 6H), 1.28 (s, 6H), 1.23–1.26 (m, 2H), 0.60–0.71 (m, 2H).
(1’S,2’R,3’S,4’R,5’S)-1’-C-(Ethoxymethylphosphinyl)-4’-(2,6-dichloropurin-9-yl)-2’,3’-O-(isopropylidene)-bicyclo[3.1.0]hexane (40)
Diisopropyl azodicarboxylate (360 µL, 1.82 mmol) was added at rt to a mixture of triphenylphosphine (0.48 g, 1.82 mmol) and 2,6-dichloropurine (0.35 g, 1.82 mmol) in anhydrous THF (5 mL). After stirring for 30 min, a solution of the compound 39 (0.27 g, 0.91 mmol) in THF (5 mL) was added. After stirring for 60 h, the reaction mixture was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–10% MeOH in EtOAc, v/v) to afford inseparable diastereomeric mixture of nucleoside 40 (0.25 mg, 60%) as a white solid material. Rf = 0.2 (10% MeOH in EtOAc, v/v); ESI-HRMS m/z 461.0899 ([M + H]+, C18H23Cl2N4O4P·H+: Calcd. 461.0912); 1H NMR (CDCl3) δ 8.79 (s, 0.5H), 8.51 (s, 1H), 5.45 (d, 0.5H, J =7.2 Hz), 5.32 (d, 1H, J = 7.2 Hz), 5.06 (s, 0.5H), 4.96 (s, 1H), 4.66 (d, 1.5H, J = 7.2 Hz), 3.98–4.21 (m, 3H), 2.40–2.51 (m, 0.5H), 2.15–2.24 (m, 1H), 1.93–2.11 (m, 1.5H), 1.63–1.75 (m, 0.5H), 1.61 (s, 5.5H), 1.59 (d, 3H, J = 3.9 Hz), 1.55 (d, 1.5H, J = 3.9 Hz), 1.35 (t, 3H, J = 7.2 Hz), 1.25 (t, 1.5H, J = 7.2Hz), 1.24 (s, 4.5H), 1.18–1.23 (m, 1.5H), 0.95–1.13 (m, 1.5H).
(1’S,2’R,3’S,4’R,5’S)-4’-(6-Amino-2-chloropurin-9-yl)-1’-C-(ethoxymethylphosphinyl)-2’,3’-O-(isopropylidene)-bicyclo[3.1.0]hexane (41)
Nucleoside 40 (0.20 g, 0.44 mmol) was treated with 2M NH3 in i-PrOH (8 mL) and heated to 70 °C. After stirring for 15 h, the reaction mixture was evaporated to dryness. The resulting residue was purified by silica gel column chromatography (0–7% MeOH in CH2Cl2, v/v) to afford nucleoside 41 (150 mg, 79%) as a white solid material. Rf = 0.4 (10% MeOH in CH2Cl2, v/v); ESI-HRMS m/z 442.1416 ([M + H]+, C18H26ClN5O4P·H+: Calcd. 442.1411); 1H NMR (CDCl3) δ 8.21 (s, 0.5H), 8.00 (s, 1H), 5.96 (s, 3H), 5.40 (d, 0.5H, J = 6.5 Hz), 5.30 (d, 1H, J = 6.5 Hz), 4.91 (s, 0.5H), 4.81 (s, 1H), 4.62–4.71 (d, 1.5H, J = 7.2 Hz), 4.10–4.22 (m, 2H), 3.98–4.09 (m, 1H), 3.60–3.80 (m, 1H), 2.64 (t, 1H, J = 15.3 Hz), 2.35 (t, 0.5H, J = 15.3 Hz), 2.01–2.17 (m, 0.5H), 1.87 (t, 1.5H, J = 15.8 Hz), 1.75 (s, 4.5H), 1.55–1.69 (m, 4.5H), 1.36 (t, 3H, J = 7.2 Hz), 1.25 (t, 1.5H, J = 7.2Hz), 1.24 (s, 4.5H), 1.17–1.22 (m, 1.5H), 0.96–1.07 (m, 1.5H).
Biological evaluation
CSQ mice and compound administration
Mice displaying the CSQ model of severe cardiomyopathy and heart failure were bred and maintained according to a previously described method (7). The CSQ transgenic (TG) mice were originally provided by Dr. Larry Jones10,11 and developed hypertrophy followed by a lethal heart failure phenotype with death near the age of 3 months.
Compound 3 and its analogues were dissolved in phosphate-buffered saline, pH=7.4 at 3.3 µM (200 µL total volume), filtered for sterility for in vivo administration at 6 µL per day for 28 days via a mini-osmotic pump (Alzet) in the CSQ mice. Intact heart function in vivo was assessed by echocardiography following infusion of nucleotide or vehicle.
Mouse echocardiography
Transthoracic echocardiography was performed using a linear 30-MHz transducer according to manufacturer’s instructions (Vevo 660 High Resolution Imaging System from VisualSonics, Toronto, Canada) similar to previously described methods.18,19 Two dimensional-targeted M-mode echocardiographic measurements were carried out at mid-papillary muscle level. Mice were anesthetized with 1% isoflurane using a vaporizer as previously described.20 Left ventricular end-diastolic (LVEDD) and end-systolic (LVESD) diameters, and FS (defined as LVEDD-LVESD/LVEDD) were measured. Parameters were measured digitally on the M-mode tracings and were averaged from more than 3 cardiac cycles.
Activation of human P2Y1 receptors
Activity at the hP2Y1 receptor was quantified in 1321N1 human astrocytoma cells stably expressing this receptor, obtained from Prof. T. K. Harden, University of North Carolina School of Medicine, Chapel Hill, NC. The procedure for measuring intracellular calcium using a FLIPR in response to nucleotide derivatives is similar to that described by Mamedova et al.28 Cells were grown overnight in 100 ml of medium in 96-well flatbottom plates at 37 °C at 5%CO2 or until they reached ~80% confluency. The calcium-4 assay kit (Molecular Devices, Sunnyvale, CA) was used as directed with no washing of cells. Cells were loaded with 40 mL dye with probenecid in each well and incubated for 1 h at rt. The compound plate was prepared with dilutions of various compounds in Hank’s Buffer at pH 7.2. Samples were run in duplicate with a FLIPR-Tetra (Molecular Devices) at rt. Cell fluorescence (excitation = 485 nm; emission = 525 nm) was monitored following exposure to a compound. Increases in intracellular calcium are reported as the maximum fluorescence value after exposure minus the basal fluorescence value before exposure.
Data analysis
Unless otherwise indicated, data were provided as mean ± standard error of the mean. For analysis of multiple groups, one-way ANOVA and posttest comparison were used. Student’s t-test for paired or unpaired samples was used to evaluate the effects of experimental interventions; P<0.05 was taken as statistically significant.
Supplementary Material
Acknowledgments
Mass spectral measurements were carried out by Dr. John Lloyd and Dr. Noel Whittaker (NIDDK). We thank Dr. T. Kendall Harden (Univ. of North Carolina, Chapel Hill, NC) for the gift of tranfected 1321N1 astrocyoma cells and Dr. William Trenkle (NIDDK) for helpful discussion. This research was supported in part by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases. This work was supported in part by RO1-HL48225 and Ray Neag Distinguished Professorship to Bruce T. Liang.
Abbreviations
- 5’-AMP
adenosine 5’-monophosphate
- CSQ
calsequestrin
- DIBAL-H
diisobutylaluminium hydride
- DMEM
Dulbecco’s modified Eagle medium
- FS
fractional shortening
- HEPES
N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- HPLC
high performance liquid chromatography
- HRMS
high resolution mass spectroscopy
- LV
left ventricular
- LVEDD
left ventricular end-diastolic diameter
- LVESD
left ventricular end-systolic diameter
- NS
normal saline
- PLC
phospholipase C
- SAR
structure activity relationship
- TBAF
tetrabutylammonium fluoride
- TBAP
tetrabutylmmonium dihydrogen phosphate
- TBDPS-Cl
tert-butyl(chloro)diphenylsilane
- THF
tetrahydrofuran
- TEAA
triethylammonium acetate
Footnotes
Supporting Information Available: Updated synthetic methods for compound 3 and NMR spectral data and HPLC traces of selected nucleotide derivatives are included. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.North RA. Molecular physiology of P2X receptors. Physiol. Rev. 2002;82:1013–1067. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- 2.Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- 3.Vassort G. Adenosine 5’-triphosphate: A P2-purinergic agonist in the myocardium. Physiol. Rev. 2001;81:767–806. doi: 10.1152/physrev.2001.81.2.767. [DOI] [PubMed] [Google Scholar]
- 4.Shen J-B, Pappano A, Liang BT. Extracellular ATP-stimulated current in wild type and P2X4 receptor transgenic mouse ventricular myocytes. implication for a cardiac physiologic role of P2X4 receptors. FASEB J. 2006;20:277–284. doi: 10.1096/fj.05-4749com. [DOI] [PubMed] [Google Scholar]
- 5.Scamps F, Vassort G. Pharmacological profile of the ATP-mediated increase in L-type calcium current amplitude and activation of a non-specific cation current in rat ventricular cells. Br. J. Pharmacol. 1994;113:982–986. doi: 10.1111/j.1476-5381.1994.tb17089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parker KE, Scarpa A. An ATP-activated nonselective cation channel in guinea pig ventricular myocytes. Amer. J. Physiol. 1995;269:H789–H797. doi: 10.1152/ajpheart.1995.269.3.H789. [DOI] [PubMed] [Google Scholar]
- 7.Yang A, Sonin D, Jones L, Liang BT. A beneficial role of cardiac P2X4 receptors in heart failure: Rescuing the calsequestrin-overexpression model of cardiomyopathy. Am. J. Physiol. 2004;287:H1096–H1103. doi: 10.1152/ajpheart.00079.2004. [DOI] [PubMed] [Google Scholar]
- 8.Shen JB, Cronin C, Sonin D, Joshi BV, Carolina M, Nieto G, Harrison D, Jacobson KA, Liang BT. P2X purinergic receptor-mediated ionic current in cardiac myocytes of calsequestrin model of cardiomyopathy. Implications for the treatment of heart failure. Am. J. Physiol. Heart Circ. Physiol. 2007;292:H1077–H1084. doi: 10.1152/ajpheart.00515.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ravi RG, Kim HS, Servos J, Zimmermann H, Lee K, Maddileti S, Boyer JL, Harden TK, Jacobson KA. Adenine nucleotides analogues locked in a Northern methanocarba conformation: Enhanced stability and potency as P2Y1 receptor agonists. J. Med. Chem. 2002;45:2090–2100. doi: 10.1021/jm010538v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones LR, Suzuki YJ, Wang W, Kobayashi YM, Ramesh V, Franzini-Armstrong C, Cleemann L, Morad M. Regulation of Ca2+ signaling in transgenic mouse cardiac myocytes overexpressing calsequestrin. J. Clin. Invest. 1998;101:1385–1393. doi: 10.1172/JCI1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knollman BC, Knollmann-Ritschel BEC, Weismann NJ, Jones LR, Morad M. Remodeling of ionic currents in hypertrophied and failing hearts of transgenic mice overexpressing calsequestrin. J. Physiol. 2000;525:483–498. doi: 10.1111/j.1469-7793.2000.t01-1-00483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soto F, Garcia-Guzman M, Gomez-Hernandez JM, Hollmann M, Karschin C, Stühmer W. P2X4: an ATP-activated ionotropic receptor cloned from rat brain. Proc. Natl. Acad. Sci. USA. 1996;93:3684–3688. doi: 10.1073/pnas.93.8.3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joshi BV, Melman A, Mackman RL, Jacobson KA. Synthesis of ethyl (1S,2R,3S,4S,5S)-2,3-O-(isopropylidene)-4-hydroxy-bicyclo[3.1.0]hexane-carboxylate from L-ribose: A versatile chiral synthon for preparation of adenosine and P2 receptor ligands. Nucleos. Nucleot. Nucleic Acids. 2008;27:279–291. doi: 10.1080/15257770701845253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sauer R, El-Tayeb A, Kaulich M, Müller CE. Synthesis of uracil nucleotide analogs with a modified, acyclic ribose moiety as P2Y2 receptor antagonists. Bioorg. Med. Chem. 2009;17:5071–5079. doi: 10.1016/j.bmc.2009.05.062. [DOI] [PubMed] [Google Scholar]
- 15.Cosyn L, Van Calenbergh S, Joshi BV, Ko H, Carter RL, Harden TK. Jacobson K.A. Synthesis and P2Y receptor activity of nucleotide 5′-phosphonate derivatives. Bioorg. Med. Chem. Lett. 2009;19:3002–3005. doi: 10.1016/j.bmcl.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu B, Stephens A, Kirschenheuter G, Greslin AF, Cheng X, Sennelo J, Cattaneo M, Zighetti ML, Chen A, Kim SA, Kim HS, Bischofberger N, Cook G, Jacobson KA. Acyclic analogues of adenosine bisphosphates as P2Y receptor antagonists: Phosphate substitution leads to multiple pathways of inhibition of platelet aggregation. J. Med. Chem. 2002;45:5694–5709. doi: 10.1021/jm020173u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim YC, Brown SG, Harden TK, Boyer JL, Dubyak G, King BF, Burnstock G, Jacobson KA. Structure activity relationships of pyridoxal phosphate derivatives as potent and selective antagonists of P2X1 receptors. J. Med. Chem. 2001;44:340–349. doi: 10.1021/jm9904203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu B, Mei Q, Smith E, Barry WH, Liang BT. A novel cardiac inotropic phenotype with cardiac transgenic expression of human P2X4 Receptor transgenic mouse. FASEB J. 2001;15:2739–2741. doi: 10.1096/fj.01-0445fje. [DOI] [PubMed] [Google Scholar]
- 19.Curcio A, Noma T, Prasad SVN, Wolf MJ, Lemaire A, Perrino C, Mao L, Rockman HA. Competitive displacement of phosphoinoside 3-kinase from β-adrenergic receptor kinase-1 improves postinfarction adverse myocardial remodeling. Am. J. Phsyiol. Heart Circ. Physiol. 2006;291:H1754–H1760. doi: 10.1152/ajpheart.01199.2005. [DOI] [PubMed] [Google Scholar]
- 20.Tsoporis JN, Marks A, Haddad A, Dawood F, Liu PP, Parker TG. S100B expression modulates left ventricular remodeling after myocardial infarction in mice. Circ. 2005;111:598–606. doi: 10.1161/01.CIR.0000154554.65287.F5. [DOI] [PubMed] [Google Scholar]
- 21.Moss GP. Extension and revision of the von Baeyer system for naming polycyclic compounds (including bicyclic compounds) Pure Appl. Chem. 1999;71:513–529. [Google Scholar]
- 22.Bichlmaier I, Kurkela T, Joshi M, Siiskonen A, Rϋffer T, Lang H, Suchanova B, Vahermo M, Finel M, Yli-Kauhaluoma J. Isoform-Selective Inhibition of the Human UDP-glucuronosyltransferase 2B7 by Isolongifolol Derivatives. J. Med. Chem. 2007;50:2655–2664. doi: 10.1021/jm061204e. [DOI] [PubMed] [Google Scholar]
- 23.Samadder P, Bittman R, Byun H-S, Arthur G. Synthesis and use of novel ether phospholipid enantiomers to probe the molecular basis of the antitumor effects of alkyllysophospholipids: Correlation of differential activation of c-Jun NH2-terminal protein kinase with antiproliferative effects in neuronal tumor cells. J. Med. Chem. 2004;47:2710–2713. doi: 10.1021/jm0302748. [DOI] [PubMed] [Google Scholar]
- 24.Salvatori D, Volpini R, Vincenzetti S, Vita A, Costanzi S, Lambertucci C, Cristalli G, Vittori S. Adenine and deazaadenine nucleoside and deoxynucleoside analogues: Inhibition of viral replication of sheep MVV (In vitro model for HIV) and bovine BHV-1. Bioorg. Med. Chem. 2002;10:2973–2980. doi: 10.1016/s0968-0896(02)00131-1. [DOI] [PubMed] [Google Scholar]
- 25.Hoffmann MFH, Brückner AM, Hupp T, Engels B, Diederichsen U. Specific purine-purine base pairing in linear alanyl-peptide nucleic acids. Helv. Chim. Acta. 2000;83:2580–2593. [Google Scholar]
- 26.Nicke A, Kerschensteiner D, Soto F. Biochemical and functional evidence for heteromeric assembly of P2X1 and P2X4 subunits. J. Neurochem. 2005;92:925–933. doi: 10.1111/j.1471-4159.2004.02939.x. [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto K, Sokabe T, Matsumoto T, Yoshimura K, Shibata M, Ohura N, Fukuda T, Sato T, Sekine K, Kato S, Isshiki M, Fujita T, Kobayashi M, Kawamura K, Masuda H, Kamiya A, Ando J. Impaired flow-dependent control of vascular tone and remodeling in P2X4-deficient mice. Nat. Med. 2006;12:133–137. doi: 10.1038/nm1338. [DOI] [PubMed] [Google Scholar]
- 28.Mamedova LK, Gao ZG, Jacobson KA. Regulation of death and survival in astrocytes by ADP acting at P2Y1 and P2Y12 receptors. Biochem. Pharmacol. 2006;72:1031–1041. doi: 10.1016/j.bcp.2006.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peyrottesm S, Gallier F, Béjaud J, Périgaud C. ‘Use of microwave irradiation for sugar and nucleoside phosphonates synthesis’. Tett. Lett. 2006;47:7719–7721. [Google Scholar]
- 30.De Clercq E. The acyclic nucleoside phosphonates from inception to clinical use: historical perspective. Antiviral Res. 2007;75:1–13. doi: 10.1016/j.antiviral.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 31.Guo C, Masin M, Qureshi OS, Murrell-Lagnado RD. Evidence for Functional P2X4/P2X7 Heteromeric Receptors. Mol. Pharmacol. 2007;72:1402–1405. doi: 10.1124/mol.107.035980. [DOI] [PubMed] [Google Scholar]
- 32.Bo X, Kim M, Nori SL, Schoepfer R, Burnstock G, North RA. Tissue distribution of P2X4 receptors studied with an ectodomain antibody. Cell Tissue Res. 2003;313:159–165. doi: 10.1007/s00441-003-0758-5. [DOI] [PubMed] [Google Scholar]
- 33.Tsuda M, Tozaki-Saitoh H, Inoue K. Pain and purinergic signaling. Brain Res. Rev. 2010 doi: 10.1016/j.brainresrev.2009.11.003. doi:10.1016/j.brainresrev.2009.11.003 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.