

Summary
The organization of the eukaryotic genome into nucleosomes dramatically impacts the regulation of gene expression. The delicate balance between transcription and DNA compaction relies heavily on nucleosome dynamics. Surprisingly, little is known about the free energy required to assemble these large macromolecular complexes and maintain them under physiological conditions. Here we describe the thermodynamic parameters that drive nucleosome formation in vitro. To demonstrate the versatility of our approach, we test the effect of DNA sequence and H3K56 acetylation on nucleosome thermodynamics. Furthermore, our studies reveal the mechanism of action of the histone chaperone Nucleosome Assembly Protein 1 (Nap1). We present evidence for a paradigm in which nucleosome assembly requires the elimination of competing, non-nucleosomal histone-DNA interactions by Nap1. This observation is confirmed in vivo, where deletion of the NAP1 gene in yeast results in a significant increase in atypical histone-DNA complexes, as well as in deregulated transcription activation and repression.
Introduction
Nucleosomes are large macromolecular complexes that consist of a histone octamer (formed from one (H3–H4)2 tetramer and two H2A–H2B dimers) that organizes 147 bp of DNA in two tight superhelical turns. Despite the fact that nucleosomes were first described as the basic repeating unit of chromatin over 35 years ago (Kornberg and Lorch, 1999; Olins and Olins, 2003) and their structure is known at great detail (Luger et al., 1997; Richmond and Davey, 2003), very little is known about their thermodynamic properties. Because nucleosomes do not spontaneously self-assemble at physiological salt concentrations due to the preponderance of competing non-nucleosomal histone-DNA complexes (Wilhelm et al., 1978) simple thermodynamic approaches cannot be applied to this system (Thastrom et al., 2004). Technical advances have revealed the rates and equilibrium constants for DNA exposure from the nucleosome (Li et al., 2005; Poirier et al., 2008) but quantitative measurements of histone-histone interactions within the context of the nucleosome have not been published. Thus, it has not been possible to test current hypotheses regarding the effect of DNA sequence, histone variants, or posttranslational modifications on nucleosome stability. For example, acetylation of H3K56 by Rtt109 (in fungi) and by p300 (in humans) has received much attention as a possible signal in transcription regulation, DNA damage repair, cell proliferation, and cancer (Das et al., 2009); and references therein). Due to its location near the DNA at its entry- and exit point from the nucleosome, it has frequently been hypothesized that nucleosomes acetylated at H3K56 might be destabilized (for example, (Ferreira et al., 2007; Tsubota et al., 2007; Williams et al., 2008; Xu et al., 2005). However, this modification has been implicated in aiding nucleosome disassembly as well as nucleosome assembly in vivo (Chen et al., 2008; Williams et al., 2008; Xu et al., 2005). Thus, the effect of H3K56 acetylation on chromatin, and the molecular function of this modification has yet to be resolved.
Nucleosome assembly is a sequential process that begins with the interaction of H3–H4 with DNA to form a (H3–H4)2 tetramer-DNA complex (the ‘tetrasome’). The addition of two H2A–H2B dimers complete a canonical nucleosome that organizes a total of 147 base pairs of DNA (Akey and Luger, 2003). In vitro, nucleosome assembly is accomplished by systematically reducing the ionic strength (Wilhelm et al., 1978). This approach takes advantage of the fact that (H3–H4)2 tetramer binds DNA at higher ionic strength than H2A–H2B dimer. In vivo, nucleosome assembly is orchestrated by an assortment of at least partially redundant assembly factors and histone chaperones (reviewed in (Rocha and Verreault, 2008). It is generally assumed that factor-mediated nucleosome assembly functions by histone deposition via the sequential pathway described above.
Histone chaperones are a heterogeneous class of acidic proteins that bind histones and are implicated in histone trafficking, nucleosome assembly and disassembly (De Koning et al., 2007; Eitoku et al., 2008). The Nap1 family has been particularly well studied in vitro (Park and Luger, 2008; Zlatanova et al., 2007). Nap1 is routinely used for in vitro nucleosome assembly reactions under physiological ionic strength (e.g. (Fujii Nakata et al., 1992; Fyodorov and Kadonaga, 2003); however, the mechanism by which Nap1 assembles chromatin is unknown. Since Nap1 binds H2A–H2B dimer and (H3–H4)2 tetramer with similarly high affinity in vitro (Andrews et al., 2008), sequential deposition of the two histone sub-complexes on DNA seems the obvious (but as yet unproven) mechanism for Nap1-mediated nucleosome assembly. The function of Nap1 in vivo remains enigmatic (e.g. (Altman and Kellogg, 1997; Ohkuni et al., 2003).
Here we present an innovative chaperone-based assay to measure the thermodynamic properties of the nucleosome. We demonstrate the feasibility of our approach by measuring the effect of DNA sequence and histone H3K56 acetylation on nucleosome stability, and we present evidence for a new paradigm of Nap1 function in vitro and in vivo, in which Nap1 promotes nucleosome assembly by the disassembly of nonproductive histone – DNA interactions.
Results
The thermodynamic properties of the nucleosome can be determined in a coupled equilibrium assay
Under physiological conditions, un-chaperoned histones exhibit a strong affinity for DNA, thus precluding the application of simple thermodynamic assays to determine nucleosome stability (Thastrom et al., 2004). To circumvent this obstacle, we decided to study the thermodynamic properties of the nucleosome through monitoring Nap1-mediated nucleosome assembly under physiological conditions. To this end, all of the relevant affinities between histones, DNA and Nap1 (K1–K4, K6; shaded areas in Fig. 1) were determined using a standard fluorescence titration approach. We first quantified the first step in nucleosome assembly, the formation of the tetrasome. We measured the fluorescence change of labeled (H3–H4)2 tetramer as a function of DNA concentration (K3, Fig. 2A; Table 1). Regardless of the DNA sequence, the interaction between H3–H4 and DNA is extremely tight (1 nM). In fact, H3–H4 binds DNA with 10-fold higher affinity than it binds Nap1 (Andrews et al., 2008). An independent competition experiment (Fig. S1A) confirmed that DNA easily outcompeted Nap1 for H3–H4. Thus, formation of the tetrasome is thermodynamically favorable both in the absence and presence of Nap1.
Figure 1. Thermodynamic scheme for Nap1 mediated nucleosome formation.

Nap1 is shown in blue, (H3–H4)2 tetramer in grey, and H2A–H2B dimer in orange. DNA is shown as a black line. The shaded area depicts a mechanism in which simple competition between Nap1 and DNA for histones is sufficient for nucleosome assembly. This scheme is extended by adding the ternary Nap1-H2A–H2B-DNA complex. Equilibrium constants K1–K6 were measured as described in Figures 2–4, and are listed in Table 1. K7 was calculated. See supplementary material for details.
Figure 2. Measurement of the thermodynamic constants for nucleosome formation.

The experimental design for each reaction is shown above each panel, using the symbols described in Fig. 1. Fluorescent labels are indicated by asterisks; FRET is indicated by a red arrow. Closed squares are 601 DNA or (H3–H4)2 tetramer-bound 601 sequence (601-tetrasomes); closed triangles are 5S sequence DNA or (H3–H4)2 tetramer-bound 5S DNA (5S tetrasomes). (A) Normalized fluorescence as a function of DNA binding to H3–H4. (B) The change in FRET between Nap1 and H2B as a function of tetrasome ((H3–H4)2-DNA). (C) The change in FRET between Nap1 and H2B as a function of (H3–H4)2. The Nap1 concentration is kept at the Kd for the Nap1-H2A–H2B dimer complex (K2, Table 1).
Table 1. Apparent equilibrium constants for Nap1 mediated nucleosome assembly.
Apparent equilibrium constants K1, K3, K4, K5, and K6 are from data shown in Figure 2–4, K1 and K2 are from Andrews et al. and K7 was calculated from the other constants. ΔΔG°K5/K6 reflects the free energy change of Nap1 on H2A–H2B dimer-DNA interactions. ΔΔG°H3K56ac reflects the free energy change of the various constants due to H3K56 acetylation (compared to unmodified H3). ΔΔG°601 indicates the effect of the 601 DNA sequence compared to 5S. For information on the Hill coefficients (nh) see supplemental data.
| DNA | Modification | K1 [M] | K2 [M] | K3 [M] | K4 [M] | K5 [M] | K6 [M] | K7 [M] | ΔΔG°K5/K6 (kcal/mol) |
|---|---|---|---|---|---|---|---|---|---|
| 5S | None | 1.0±0.1×10−8 | 7.8±0.4×10−9 | 1.1±0.1×10−9 | 4.0±0.3×10−8 | 2.4±0.2×10−7 | 1.6±0.2×10−8 | ~1.2×10−7 | 1.4 |
| nh=1.4±0.1 | nh=1.2±0.1 | nh=2.0±0.3 | nh=2.0±0.2 | nh=1.6±0.2 | |||||
| 601 | None | 1.0±0.1×10−8 | 7.8±0.4×10−9 | 0.9±0.1×10−9 | 1.3±0.3×10−8 | 4.7±1.9×10−7 | 4.4±0.5×10−8 | ~0.8×10−7 | 1.6 |
| nh=1.4±0.1 | nh=1.1±0.1 | nh=1.6±0.3 | nh=1.6±0.4 | nh=1.9±0.3 | |||||
| 601 | H3K56ac | 1.1±0.1×10−8 | 7.8±0.4×10−9 | 1.9±0.5×10−8 | 1.8±0.2×10−8 | 4.7±1.9×10−7 | 4.4±0.5×10−8 | ~0.8×10−7 | 1.4 |
| nh=2.1±0.5 | nh=1.5±0.2 | nh=1.6±0.4 | nh=1.9±0.3 | ||||||
| ΔΔG°H3K56ac(kcal/mol) | 0 | 0 | 1.8 | 0.2 | 0 | 0 | 0 | ||
| ΔΔG°601(kcal/mol) | 0.1 | 0 | −0.1 | −0.7 | 0.4 | 0.6 | −0.4 | ||
We next monitored the ability of the tetrasome to compete with Nap1 for the H2A–H2B dimer to form a canonical nucleosome. This was done at Nap1 and H2A–H2B concentrations equal to their Kd (K2), so that the amount of free H2A–H2B is equal to the amount of Nap1-bound H2A–H2B. Any disturbance of this equilibrium upon addition of tetrasome is registered through loss of FRET signal between Nap1 (acceptor) and H2B (donor). This results in the binding constant for the H2A–H2B dimer for the tetrasome (K4; Fig. 2B; Table 1). To confirm that the loss of Nap1- H2B signal is equivalent to a gain in nucleosome formation, we moved the fluorescent label from Nap1 to H4. We monitored the appearance of FRET between tetrasome and H2A–H2B dimer, with and without Nap1 (Fig. S1B). In the presence of Nap1 we obtained virtually the same value for K4 as in the experiment with the label on Nap1 (Fig. 2B; 3.5±0.4x10−8 M versus 4.0±0.3x10−8 M, Fig. S1B). In the absence of Nap1, no significant increase in FRET signal was observed, indicating that Nap1 is required for nucleosome assembly, as shown previously (Mazurkiewicz et al., 2006).
We further validated the coupled equilibrium assay by re-measuring the affinity of (H3–H4)2 tetramer to Nap1 by competition with the H2A-H2B-Nap1 complex (K1; Fig. 2C). The value obtained by monitoring FRET between H2B and Nap1 was in excellent agreement with that previously reported (K1 = 0.9±1x10−8 compared to 1±0.6x10−8; (Andrews et al., 2008). Thus, the coupled equilibrium assay effectively measures, for the first time, the affinity of the H2A–H2B dimer for the tetrasome and provides a quantitative measure of nucleosome stability under conditions where the nucleosome is fully intact.
DNA sequence has an effect on nucleosome stability under physiological conditions
It has long been hypothesized that the DNA sequence affects nucleosome stability (Shrader and Crothers, 1989); (Lowary and Widom, 1998). To test this directly in our system, we assayed nucleosome formation on two DNA fragments with equal length but different sequence and nucleosome positioning propensities. The 5S DNA fragment is a naturally occurring sequence (Simpson and Stafford, 1983), while the engineered 601 fragment was originally identified on the basis of its ability to outcompete 5S during salt-dependent deposition (Lowary and Widom, 1998). Our assay reveals that the two DNA sequences form nucleosomes that differ in their apparent stability by a ΔΔG°601 of −0.7 kcal/mol (Table 1, K4; Fig. 2B). This free energy difference supports the observation that 601 is a stronger nucleosome positioning sequence than 5S (Lowary and Widom, 1998) and demonstrates that the coupled equilibrium assay is capable of discerning relatively subtle differences in nucleosome stability.
The acetylation of H3K56 affects the first step of nucleosome formation
To test the hypothesis that the acetylation of H3K56 has a measurable effect on the stability of the nucleosome, we prepared histone H3K56ac in a system established by Neumann and colleagues (Neumann et al., 2008). The presence of the modification was verified by mass spectrometry (Fig. S2), and the modified histone formed nucleosomes that were qualitatively indistinguishable from unmodified nucleosomes (not shown). We determined the affinity of the (H3K56ac-H4)2 tetramer to DNA and Nap1 (Fig. 3A, B), and showed that the acetylation of H3K56 has no effect on the affinity of H3 to Nap1 (K1, Table 1). In contrast, binding to the DNA is about 15-fold weaker for (H3K56ac-H4)2 tetramer compared to unmodified (H3–H4)2 tetramer (K3, Table 1), as evident in a ΔΔGH3K56ac of 1.8 kcal/mol. However, once H3K56ac forms a tetrasome (at DNA concentration at least >5-fold (>100 nM) K3), there is no significant change in nucleosome stability, as measured by the affinity between the H2A–H2B dimer and the H3K56ac-tetrasome; K4, Table 1. Thus, our data indicates that H3K56 acetylation is irrelevant to H2A–H2B dimer interaction with the tetrasome, but once the H2A–H2B dimer is removed, the modified tetrasome is unstable.
Figure 3. H3K56ac destabilizes tetrasome formation.

Fluorescence studies of H3K56ac-H4 in tetrasome and nucleosome formation. (A) tetrasome formation as monitored by the normalized fluorescence change from Alexa-488 labeled K3K56ac-H4 as a function of DNA. (B) nucleosome assembly, as monitored by the FRET ratio between Nap1 and H2A–H2B as a function of H3K56ac-H4. Thermodynamic constants are shown in Table 1, experimental details are found in the supplemental section.
Nap1 disfavors the interaction of H2A/H2B with DNA in vitro
Due to their highly basic character, all histones are likely to have a high affinity for DNA. Unlike the (H3–H4)2 tetramer DNA complexes (see above), the interaction of H2A–H2B dimer with DNA is unlikely to lead to nucleosome formation. H2A–H2B dimers bind DNA with high affinities (between 10 and 50 nM; depending on the DNA sequence, Fig. 4A, K6, Table 1) and thus these complexes may be counter-productive for canonical nucleosome formation if not prevented. To examine these values in the presence of Nap1, we used the coupled equilibrium assay where we allowed DNA to compete with Nap1 for H2A–H2B dimer. Unexpectedly, the data obtained from this assay is inconsistent with a simple model in which Nap1 and DNA are both competing for H2A–H2B, since we observed no loss of FRET between H2B and Nap1 at increasing concentrations of DNA (Fig. 4B; Fig. S3A). The Nap1-H2A–H2B interaction remains unperturbed in the presence of DNA. To measure the extent by which Nap1 alters the ability of H2A–H2B to interact with DNA, we repeated the experiment with all H2A–H2B dimer in complex with Nap1 (i.e. Nap1 concentration >5-fold K2). Under these conditions, the observed change in signal is due to DNA interacting with a Nap1-histone complex (Fig. 4C; Table 1). The binding constant for the interaction between chaperone-bound H2A–H2B and DNA is approximately ten fold reduced compared to that of free H2A–H2B with DNA (ΔΔG°(K5/K6) of ~1.5 kcal/mol). Together, the data summarized in Table 1 reveal an unexpected mechanism for Nap1 mediated nucleosome assembly through preventing the strong non-productive interactions between H2A–H2B and DNA.
Figure 4. Nap1 disfavors the interaction between H2A–H2B dimer and DNA.
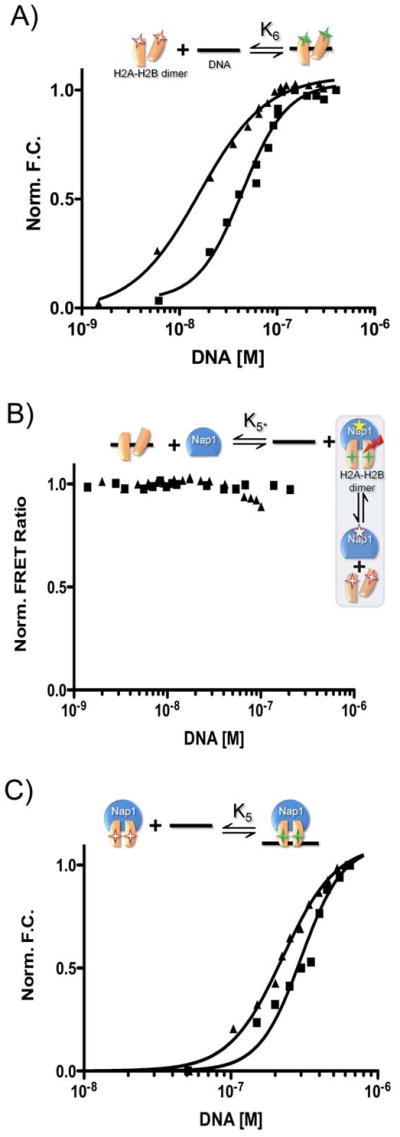
The experimental design for each reaction is shown above each panel, using the symbols described in Fig. 1. Fluorescent labels are indicated by asterisks; FRET is indicated by a red arrow. Closed squares are 601-tetrasomes; closed triangles are 5S-tetrasomes. (A) Normalized fluorescence as a function of DNA binding to H2A–H2B. (B) The change in FRET between Nap1 and H2B as a function of DNA. The experiment was originally designed to confirm K6; however, the data indicate the Nap1-H2A–H2B complex remains intact in the presence of DNA and thus the small amount of signal change is due to DNA binding to the Nap1-H2A–H2B complex. Therefore, the binding event really monitored is K5, and is thus termed K5*. (C) Fluorescence change as a function of DNA binding to a Nap1-H2A–H2B complex (Nap1 is 10-fold > K2). Binding constants derived from the shown experiments are summarized in Table 1. See supplemental information for experimental details.
To confirm this finding by an independent experimental approach, we performed gel-shift analysis of histone-DNA complexes in the presence and absence of Nap1 (Fig. S3B, C). A constant amount of DNA was incubated with increasing amounts of purified H2A–H2B dimer, resulting in the appearance of multiple bands with slower electrophoretic mobility. These represent histone-DNA complexes, as reported previously (Aragay et al., 1988). The addition of Nap1 to these samples resulted in the complete disappearance of the shifted bands, consistent with the removal of H2A–H2B dimers from the DNA (compare lanes 1–3 with 4–6, upper panel). This was correlated with the occurrence of a supershifted fluorescent Nap1 band, most likely due to its interaction with H2A–H2B dimers (compare lanes 6 and 8, lower panel).
The deletion of NAP1 results in excessive H2A–H2B binding to DNA and aberrant transcription in yeast
To investigate whether Nap1 regulates chromatin structure by this mechanism in vivo, we examined histone occupancy in a wild type yeast strain and compared it to a strain deleted for NAP1 (nap1Δ). We focused on the GAL locus because its chromatin architecture, the destabilization of positioned nucleosomes during transcriptional induction, and the reassembly of nucleosomes upon return to repression, are well-characterized (Bryant and Ptashne, 2003; Cavalli and Thoma, 1993; Lee et al., 2004; Lohr and Hopper, 1985; Schwabish and Struhl, 2004). We tested a variety of locations, including coding sequences and promoters, using chromatin immunoprecipitation (ChIP) assays and antibodies specific for H2A, H2B and H3 (Fig. 5A). Strikingly, H2A and H2B levels are significantly increased in a nap1Δ strain compared to wild type levels (Fig. 5B, C), while H3 levels are unchanged or even depleted (depending on the location probed) in the nap1Δ strain (Fig. 5D). Thus, in the absence of NAP1, there is a marked increase in H2A–H2B occupancy without a corresponding increase in H3. This is consistent with a role of Nap1 in disfavoring the interaction of H2A–H2B with the DNA.
Figure 5. Deletion of NAP1 alters histone occupancy in vivo.

(A) schematic of the GAL gene locus, showing the relative position of the coding sequence for GAL1, GAL7 and GAL10. The location of the amplicons used for ChIP assays are indicated (A–K). (B) ChIP analyses of histone H2A were performed on wild type (wt; grey bars) or nap1Δ cells (black bars). Each column corresponds to the location of a real-time PCR amplicon as shown in (A). Error bars indicate standard deviations from three independent biological replicates. Histone H2B (C) and H3 (D) occupancies were determined as for H2A.
We next tested whether this altered histone occupancy affected transcription. We performed S1 nuclease protection assays on GAL1, GAL7 (Fig. 6), and GAL10 (not shown) before and after gene activation. Intriguingly, the nap1Δ strain has enhanced activation kinetics for the GAL genes compared to a wild type strain, suggesting that dimer-enriched chromatin is more permissive to transcription than canonical chromatin. We also examined transcriptional repression. As expected in wild type cells, GAL expression was repressed by adding glucose (Fig. 6). In contrast, the nap1Δ strain has a severe defect in repressing transcription of the GAL genes. This gene locus is well characterized for the eviction of canonical nucleosomes after activation (Bryant and Ptashne, 2003; Cavalli and Thoma, 1993; Lee et al., 2004; Lohr and Hopper, 1985; Schwabish and Struhl, 2004). In wild type cells, histone occupancy via ChIP assay is high when the genes are inactive, and a rapid decrease of histone H2A, H2B and H3 occupancy is observed upon transcriptional activation (Fig. S4). Similarly, there is a decrease in occupancy after switching the nap1Δ strain to galactose. Thus, these atypical histone-DNA complexes are efficiently evicted upon transcription activation.
Figure 6. Deletion of NAP1 changes the kinetics of GAL gene expression.

(A) S1 nuclease protection assays were used to analyze the expression levels of GAL1 and GAL7 transcripts in a wild type or nap1Δ strain. To examine activation of transcription, galactose was added for the time (in minutes) indicated. To examine repression of transcription, glucose was added after 60 minutes of growth in galactose. A representative gel is shown. (B, C) The amount of transcript for GAL1 and GAL7 at each time point is plotted for the wild type and nap1Δ strain. mRNA levels were normalized using the signal from the intron of tRNAW. Error bars indicate standard deviations from three independent biological replicates.
Discussion
Together, our data suggest that Nap1 promotes nucleosome assembly not by delivering histones, but through disfavoring non-nucleosomal interactions between H2A–H2B dimers and DNA, both in vitro and in vivo. Several early studies have characterized the interactions of H2A–H2B dimer with DNA (Aragay et al., 1988; Oohara and Wada, 1987; Samso and Daban, 1993). Our in vitro experiments demonstrate that these histone-DNA interactions must be prevented for nucleosome assembly to occur. Moreover, predictions from our in vitro experiments are in perfect accord with results obtained in vivo, where we observe significantly enriched H2A and H2B levels (but not H3 levels) at endogenous genes in a nap1Δ strain. This atypical H2A–H2B enriched chromatin appears to present a lower barrier for the transcription machinery, resulting in deregulated gene activation and repression. Accumulation of H2A–H2B is observed in the single knockout strain; thus, this particular function of Nap1 is non-redundant with the other histone binding proteins in yeast. It is also interesting to speculate that localized inhibition of Nap1 (perhaps through one of its many interaction partners) may be capable of locally generating this atypical chromatin architecture in a wild type cell. The demonstration of non-nucleosomal chromatin structures profoundly changes the view of histones and histone chaperones in regulating DNA accessibility. Many studies have used the occupancy of H3 in a chromatin immunoprecipitation assay to infer that no histones are bound to the DNA. The existence of the atypical chromatin complexes described here must be taken into account when interpreting these published results. Our thermodynamic data also explain why Nap1 is found predominantly in complex with H2A–H2B in vivo (Ito et al., 1996; Mosammaparast et al., 2001), despite its high affinity for both H2A–H2B and H3–H4 in vitro (Andrews et al., 2008).
The thermodynamic assay developed and employed here is applicable to other histone chaperones and provides a means to clarify the nomenclature in the chromatin field. There are a plethora of proteins currently classified as histone chaperones, many of which may simply bind histones and/or aid in direct assembly. In keeping with our results, as well as the original definition of a histone chaperone by Laskey et al. (Laskey et al., 1978), we feel this term should be reserved for those proteins that prevent incorrect histone-DNA interactions. This terminology is also consistent with the functions of general protein chaperones, which prevent both newly synthesized polypeptide chains and assembled subunits from aggregating into nonfunctional structures. As such, and somewhat ironically because of its name, our thermodynamic analysis and in vivo experiments assign Nucleosome Assembly Protein 1 (Nap1) as the first true histone chaperone.
The comparison of the thermodynamics of nucleosome assembly on two different DNA sequences confirms the notion that DNA sequence has a measurable effect on nucleosome stability under physiological conditions. Previous studies have used competitive salt gradients to determine that the 601 DNA sequence is a more favorable sequence for nucleosome formation than 5S by a ΔΔG° of −2.9 kcal/mol (Lowary and Widom, 1998). However, these observations are the result of a convolution of K3, K4, and K6 and their respective salt dependence, while our experiments are independent of ionic strength and address each constant independently. Thus, while the two experimental approaches are expected to give the same trend, the ΔΔG° values cannot be compared directly.
In light of recent efforts to understand the relationship between DNA sequence and in vivo nucleosome positions, our assay will undoubtedly be a useful tool to test key hypotheses stemming from whole genome analyses (e.g. Kaplan et al., 2008; Zhang et al., 2009). Our thermodynamic analysis also clarifies the role of H3K56 acetylation in promoting chromatin dynamics during transcription, DNA repair and replication. With the known turnover of H2A–H2B in vivo (reviewed in Kimura, 2005; Thiriet and Hayes, 2006), our results with acetylated H3K56 indicate that this modification increases the fluidity of chromatin by disfavoring the association of the tetramer with DNA. This alone could explain the observed increased interaction of (H3K56ac-H4)2 tetramer with CAF-1 (Li et al., 2008). The future use of our multidisciplinary approach will fill important gaps in the field of chromatin biology, as it will allow us to systematically test additional key hypotheses regarding the effect of DNA sequence, histone posttranslational modifications and histone variants on nucleosome stability under physiologically relevant conditions.
Experimental Procedures
Reagents
Wild type and derivatives of Xenopus laevis histones were prepared as described (Dyer et al., 2004). For histone labeling see (Andrews et al., 2008). For fluorescence titrations and data analysis see supplemental information. H3K56ac was expressed from the system developed and kindly given to us by Neumann and colleagues (Neumann et al., 2008) and purified from inclusion bodies using Ni-NTA resin. All histones were refolded into tetramers and dimers as described (Dyer et al., 2004).
Fluorescence assays
Assays and data treatment are described under supplementary information. Briefly, interaction between the various components was measured either by a change in fluorescence signal upon complex formation (K3, K5 and K6), or by changes in fluorescence resonance energy transfer (FRET) efficiency (K1, K4, and K5). These latter constants were measured in a ‘coupled equilibrium assay’ monitoring the displacement of H2A–H2B from Nap1. Changes in FRET signal between Nap1 and H2A–H2B are measured as a function of another protein or protein-DNA complex. For these experiments, Nap1 is kept close to the KdNap-H2A–H2B (8 nM, K1) and H2A–H2B is ~0.5 nM or less. Assays are schematically depicted in Figures 2–4.
Yeast transcriptional assays
The wild type (BY4741 from ATCC) and nap1Δ cells were grown in YP raffinose (2%), then washed and transferred to YP galactose (2%). During the one hour galactose induction, cells were collected every 10 minutes. For glucose repression, glucose was added to the media to a final concentration of 0.2%, and cells were collected every 10 minutes. S1 nuclease protection assays to quantify the expression levels of GAL7, GAL1 and tRNAw were performed as described before (Zhang et al., 2008). Transcript levels were quantitated by phosphorimaging and normalized to the level of the tRNAW intron. All studies were performed in triplicate, starting with independent cultures of cells.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assays were performed as described (Strahl Bolsinger et al., 1997; Zhang et al., 2008). Wild type and nap1Δ cells (50 ml) were grown at 30°C in glucose media for the uninduced condition, and in raffinose to an OD600 of 0.8 to 1.0, then induced with galactose for 30 minutes and one hour. Commercially available antibodies were utilized (anti-H2A and anti-H2B: Active Motif 39235 and 39237; Anti-H3: Abcam 1791). Occupancy for H2A, H2B and H3 at the GAL10 TATA box amplicon (oligo F, Fig. 5A) in the wild type strain was set to 1.0. All studies were performed in triplicate, starting with independent cultures of cells. Oligo sequences are available upon request.
Supplementary Material
Acknowledgments
We are grateful to Jason Chin and Heinz Neumann for the kind gift of their system to genetically encode N(epsilon)-acetyllysine in recombinant proteins. We thank Sheena D’Arcy and Kitty Brown for comments, Alison White and Pamela Dyer for help with DNA and protein preparation, and Teri McLain from the W.M.Keck Protein expression and purification facility for histones. Supported by the Ruth L. Kirschstein service award GM083532 to AJA, GM061909 to KL, and GM067777 to LAS and KL. Also supported by the Howard Hughes Medical Institute (KL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akey CW, Luger K. Histone chaperones and nucleosome assembly. Curr Opin Struct Biol. 2003;13:6–14. doi: 10.1016/s0959-440x(03)00002-2. [DOI] [PubMed] [Google Scholar]
- Altman R, Kellogg D. Control of mitotic events by Nap1 and the Gin4 kinase. J Cell Biol. 1997;138:119–130. doi: 10.1083/jcb.138.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews AJ, Downing G, Brown K, Park YJ, Luger K. A thermodynamic model for Nap1-histone interactions. J Biol Chem. 2008;283:32412–32418. doi: 10.1074/jbc.M805918200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragay AM, Diaz P, Daban JR. Association of nucleosome core particle DNA with different histone oligomers. Transfer of histones between DNA-(H2A, H2B) and DNA-(H3, H4) complexes. J Mol Biol. 1988;204:141–154. doi: 10.1016/0022-2836(88)90605-5. [DOI] [PubMed] [Google Scholar]
- Bryant GO, Ptashne M. Independent recruitment in vivo by Gal4 of two complexes required for transcription. Mol Cell. 2003;11:1301–1309. doi: 10.1016/s1097-2765(03)00144-8. [DOI] [PubMed] [Google Scholar]
- Cavalli G, Thoma F. Chromatin transitions during activation and repression of galactose-regulated genes in yeast. Embo J. 1993;12:4603–4613. doi: 10.1002/j.1460-2075.1993.tb06149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, Tyler JK. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell. 2008;134:231–243. doi: 10.1016/j.cell.2008.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009 doi: 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Koning L, Corpet A, Haber JE, Almouzni G. Histone chaperones: an escort network regulating histone traffic. Nat Struct Mol Biol. 2007;14:997–1007. doi: 10.1038/nsmb1318. [DOI] [PubMed] [Google Scholar]
- Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, Luger K. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 2004;375:23–44. doi: 10.1016/s0076-6879(03)75002-2. [DOI] [PubMed] [Google Scholar]
- Eitoku M, Sato L, Senda T, Horikoshi M. Histone chaperones: 30 years from isolation to elucidation of the mechanisms of nucleosome assembly and disassembly. Cell Mol Life Sci. 2008;65:414–444. doi: 10.1007/s00018-007-7305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira H, Somers J, Webster R, Flaus A, Owen-Hughes T. Histone tails and the H3 alphaN helix regulate nucleosome mobility and stability. Mol Cell Biol. 2007;27:4037–4048. doi: 10.1128/MCB.02229-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii Nakata T, Ishimi Y, Okuda A, Kikuchi A. Functional analysis of nucleo-some assembly protein, NAP-1. The negatively charged COOH-terminal region is not necessary for the intrinsic assembly activity. J Biol Chem. 1992;267:20980–20986. [PubMed] [Google Scholar]
- Fyodorov DV, Kadonaga JT. Chromatin assembly in vitro with purified recombinant ACF and NAP-1. Methods Enzymol. 2003;371:499–515. doi: 10.1016/S0076-6879(03)71037-4. [DOI] [PubMed] [Google Scholar]
- Ito T, Bulger M, Kobayashi R, Kadonaga JT. Drosophila NAP-1 is a core his-tone chaperone that functions in ATP-facilitated assembly of regularly spaced nucleosomal arrays. Mol Cell Biol. 1996;16:3112–3124. doi: 10.1128/mcb.16.6.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, Leproust EM, Hughes TR, Lieb JD, Widom J, et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2008 doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H. Histone dynamics in living cells revealed by photobleaching. DNA Repair (Amst) 2005 doi: 10.1016/j.dnarep.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- Laskey RA, Honda BM, Mills AD, Finch JT. Nucleosomes are assembled by an acidic protein which binds histones and transfers them to DNA. Nature. 1978;275:416–420. doi: 10.1038/275416a0. [DOI] [PubMed] [Google Scholar]
- Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat Genet. 2004;36:900–905. doi: 10.1038/ng1400. [DOI] [PubMed] [Google Scholar]
- Li G, Levitus M, Bustamante C, Widom J. Rapid spontaneous accessibility of nucleosomal DNA. Nat Struct Mol Biol. 2005;12:46–53. doi: 10.1038/nsmb869. [DOI] [PubMed] [Google Scholar]
- Li Q, Zhou H, Wurtele H, Davies B, Horazdovsky B, Verreault A, Zhang Z. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell. 2008;134:244–255. doi: 10.1016/j.cell.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr D, Hopper JE. The relationship of regulatory proteins and DNase I hypersensitive sites in the yeast GAL1-10 genes. Nucleic Acids Res. 1985;13:8409–8423. doi: 10.1093/nar/13.23.8409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary PT, Widom J. New DNA sequence rules for high affinity binding to his-tone octamer and sequence-directed nucleosome positioning. J Mol Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- Luger K, Maeder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–259. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Mazurkiewicz J, Kepert JF, Rippe K. On the mechanism of nucleosome assembly by histone chaperone NAP1. J Biol Chem. 2006;281:16462–16472. doi: 10.1074/jbc.M511619200. [DOI] [PubMed] [Google Scholar]
- Mosammaparast N, Jackson KR, Guo Y, Brame CJ, Shabanowitz J, Hunt DF, Pemberton LF. Nuclear import of histone H2A and H2B is mediated by a network of karyopherins. J Cell Biol. 2001;153:251–262. doi: 10.1083/jcb.153.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H, Peak-Chew SY, Chin JW. Genetically encoding N(epsilon)-acetyllysine in recombinant proteins. Nat Chem Biol. 2008;4:232–234. doi: 10.1038/nchembio.73. [DOI] [PubMed] [Google Scholar]
- Ohkuni K, Shirahige K, Kikuchi A. Genome-wide expression analysis of NAP1 in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2003;306:5–9. doi: 10.1016/s0006-291x(03)00907-0. [DOI] [PubMed] [Google Scholar]
- Olins DE, Olins AL. Chromatin history: our view from the bridge. Nat Rev Mol Cell Biol. 2003;4:809–814. doi: 10.1038/nrm1225. [DOI] [PubMed] [Google Scholar]
- Oohara I, Wada A. Spectroscopic studies on histone-DNA interactions. I. The interaction of histone (H2A, H2B) dimer with DNA: DNA sequence dependence. J Mol Biol. 1987;196:389–397. doi: 10.1016/0022-2836(87)90699-1. [DOI] [PubMed] [Google Scholar]
- Park YJ, Luger K. Histone chaperones in nucleosome eviction and histone exchange. Curr Opin Struct Biol. 2008 doi: 10.1016/j.sbi.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier MG, Bussiek M, Langowski J, Widom J. Spontaneous access to DNA target sites in folded chromatin fibers. J Mol Biol. 2008;379:772–786. doi: 10.1016/j.jmb.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature. 2003;423:145–150. doi: 10.1038/nature01595. [DOI] [PubMed] [Google Scholar]
- Rocha W, Verreault A. Clothing up DNA for all seasons: Histone chaperones and nucleosome assembly pathways. FEBS Lett. 2008;582:1938–1949. doi: 10.1016/j.febslet.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Samso M, Daban JR. Unfolded structure and reactivity of nucleosome core DNA-histone H2A, H2B complexes in solution as studied by synchrotron radiation X-ray scattering. Biochemistry. 1993;32:4609–4614. doi: 10.1021/bi00068a018. [DOI] [PubMed] [Google Scholar]
- Schwabish MA, Struhl K. Evidence for eviction and rapid deposition of histones upon transcriptional elongation by RNA polymerase II. Mol Cell Biol. 2004;24:10111–10117. doi: 10.1128/MCB.24.23.10111-10117.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrader TE, Crothers DM. Artificial nucleosome positioning sequences. PNAS. 1989;86:7418–7422. doi: 10.1073/pnas.86.19.7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson RT, Stafford DW. Structural features of a phased nucleosome core particle. PNAS. 1983;80:51–55. doi: 10.1073/pnas.80.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl Bolsinger S, Hecht A, Luo K, Grunstein M. SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev. 1997;11:83–93. doi: 10.1101/gad.11.1.83. [DOI] [PubMed] [Google Scholar]
- Thastrom A, Gottesfeld JM, Luger K, Widom J. Histone-DNA Binding Free Energy Cannot Be Measured in Dilution-Driven Dissociation Experiments. Biochemistry. 2004;43:736–741. doi: 10.1021/bi0302043. [DOI] [PubMed] [Google Scholar]
- Thiriet C, Hayes JJ. Histone dynamics during transcription: exchange of H2A/H2B dimers and H3/H4 tetramers during pol II elongation. Results Probl Cell Differ. 2006;41:77–90. doi: 10.1007/400_009. [DOI] [PubMed] [Google Scholar]
- Tsubota T, Berndsen CE, Erkmann JA, Smith CL, Yang L, Freitas MA, Denu JM, Kaufman PD. Histone H3-K56 acetylation is catalyzed by histone chaperone-dependent complexes. Mol Cell. 2007;25:703–712. doi: 10.1016/j.molcel.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm FX, Wilhelm ML, Erard M, Duane MP. Reconstitution of chromatin: assembly of the nucleosome. Nucleic Acids Res. 1978;5:505–521. doi: 10.1093/nar/5.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SK, Truong D, Tyler JK. Acetylation in the globular core of histone H3 on lysine-56 promotes chromatin disassembly during transcriptional activation. PNAS. 2008;105:9000–9005. doi: 10.1073/pnas.0800057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Zhang K, Grunstein M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell. 2005;121:375–385. doi: 10.1016/j.cell.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Zhang L, Fletcher AG, Cheung V, Winston F, Stargell LA. Spn1 regulates the recruitment of Spt6 and the Swi/Snf complex during transcriptional activation by RNA polymerase II. Mol Cell Biol. 2008;28:1393–1403. doi: 10.1128/MCB.01733-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Moqtaderi Z, Rattner BP, Euskirchen G, Snyder M, Kadonaga JT, Liu XS, Struhl K. Intrinsic histone-DNA interactions are not the major determinant of nucleosome positions in vivo. Nat Struct Mol Biol. 2009;16:847–852. doi: 10.1038/nsmb.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatanova J, Seebart C, Tomschik M. Nap1: taking a closer look at a juggler protein of extraordinary skills. Faseb J. 2007;21:1294–1310. doi: 10.1096/fj.06-7199rev. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.