

Abstract
Deregulation in different steps of translational control is an emerging mechanism for cancer formation. One example of an oncogene with a direct role in control of translation is the Myc transcription factor. Myc directly increases protein synthesis rates by controlling the expression of multiple components of the protein synthetic machinery, including ribosomal proteins, initiation factors of translation, Pol III and rDNA. However, the contribution of Myc-dependent increases in protein synthesis towards the multi-step process leading to cancer has remained unknown. Recent evidence strongly suggests that Myc oncogenic signaling may monopolize the translational machinery to elicit cooperative effects on cell growth, cell cycle progression, and genome instability as a mechanism for cancer initiation. Moreover, new genetic tools to restore aberrant increases in protein synthesis control are now available, which should enable the dissection of important mechanisms in cancer that rely on the translational machinery.
Background
The Myc proto-oncogene is one of the most frequently activated oncogenes (1). By studying the Myc transcriptional regulatory network, it has become very clear that a fraction of Pol II-dependent genes encode for several components of the translational apparatus (2, 3). EASE (Expression Analysis Systematic Explorer) analysis of Myc-bound promoters shows that out of 430 genes, 13% have a direct role in the control of ribosome biogenesis and/or protein synthesis, which is comparable to the number of cell cycle genes found to be regulated by Myc (12%) (K. Zeller and C. Dang, personal communication). Myc target genes include those encoding translation elongation factors, translation initiation factors, nucleolar assembly components and ribosomal proteins belonging to the small and large subunits (2, 4–8) (Table 1).
Table 1.
Myc regulates the expression of target genes involved in ribosome biogenesis and protein synthesis
| Myc Target Genes | Function |
|---|---|
|
Ribosomal Proteins: (Large and Small subunit). i.e. RPL21; RPL23A; RPL29; RPL34; RPL35; RPL39; RPS14; RPS16; RPS17; RPS19; RPS24; RPS25; RPS3 |
Structural components of the ribosome and protein synthesis control. |
|
Translation factors: Initiation factors, eIF4E, eIF4G, eIF4A, eIF4B, eIF5A. Elongation factors, EEF1B2; EEF1D; EEF1G. |
Proteins that regulate the initiation or elongation steps of protein synthesis. |
| Nucleophosmin (NPM) | Ribosome biogenesis |
| Upstream Binding Factor (UBF) | rRNA transcription |
| DKC1 | rRNA modifications. Component of the H/ACA-box snoRNPs. Control of IRES-dependent translation. |
| Nucleolar antigen Nop52 | pre-rRNA processing |
| Nucleolar protein Nop56 | rRNA modifications. Component of the C/D-box snoRNPs |
| Ribosomal RNA (rRNA) | Structural components of the ribosome |
The ability of Myc to regulate the transcription of several components of the protein synthesis machinery has been validated in several different cell types, and expression changes correlate proportionately with both Myc loss and gain of function (9–15). The effect of Myc in stimulating protein synthesis is also supported by a direct role of Myc in promoting ribosome biogenesis (3, 16–19). Myc overexpression results in a substantial increase in the size of nucleoli (13, 19, 20). Consistent with this phenotype, it has been demonstrated that Myc is a direct activator of Pol I and Pol III, making it a unique transcription factor that is able to regulate all three RNA polymerases (3). Myc also promotes ribosome biogenesis at different levels. For example, Myc can stimulate rRNA modifications and processing by directly controlling the expression of ribonucleases, rRNA-modifying enzymes, and nucleolar proteins involved in ribosome biogenesis such as NPM, Nop52, Nop56, and DKC1 (Table 1) (16). Furthermore, Myc induces UBF expression, which is an essential transcription factor for RNA Pol I-mediated transcription (21). It has also been recently shown that a fraction of the Myc protein is localized in the nucleolus and directly regulates rRNA synthesis by binding to E-box elements located in the rDNA promoter (17–19). In addition, it can activate Pol I transcription by binding and recruiting to the rDNA promoter SL1, which is essential for the assembly of the RNA Pol I pre-initiation complex (18).
While components of the protein synthesis machinery are major targets of Myc family members, to date the immediate cellular effect(s), target mRNAs, and specific steps leading to the biogenesis of cancer that may be directly affected by Myc-dependent increases in protein synthesis have remained unknown. In this respect it is important to point out that the picture becomes more complex as an overwhelming amount of evidence highlights the activity of Myc in coordinating many of the cellular programs necessary for the growth and expansion of cancer cells.
How many of these cellular processes rely on Myc-induced protein synthesis? How can we imagine that alteration of general protein synthesis downstream of Myc perturbs the expression of specific genes relevant to cancer formation? The answers to these questions have been difficult to address genetically due to the ability of Myc to simultaneously activate the expression of multiple components of the translational apparatus, rendering it difficult to restore protein synthesis to normal rates.
Key Findings
In a recent study, we utilized new genetic tools originally employed in Drosophila that were extended to a mammalian system in order to restore aberrant increases in protein synthesis control as a consequence of Myc hyperactivation (22). First described in Drosophila, a class of mutants that are haploinsufficient for translational components are collectively known as Minutes, due to their overall small body size and an associated decrease in protein synthesis rates by approximately 30% (23). In a genetic setting where there is an increase in protein synthesis rates, the expectation would be that in a Minute background this effect would be thwarted because of the overall limit in translation capacity present in Minute cells. In support of this scenario, when drosophila Myc (commonly known as dMyc) is overexpressed in a Minute mutant background, heterozygous for a specific ribosomal protein, dMyc-overexpressing cells lose their growth advantage over normal cells (24). It has until now remained unknown whether, in a mammalian system, haploinsufficiency of specific ribosomal proteins would show an evolutionarily conserved effect on protein synthesis rates and whether such mutations may enable the genetic rescue of increases in protein synthesis, such as would result from Myc hyperactivation, to normal levels.
In 2004, the Glaser lab described the first mouse minute, which resulted from a mutation in the gene encoding for ribosomal protein L24 (Rpl24) (25). Rpl24 heterozygote mice displayed key features of the fly minutes, including smaller overall body size and decreases in overall protein synthesis rates. We took advantage of this first mouse minute to genetically restore protein synthesis rates in Eµ-Myc transgenic mice (in which the Eµ immunoglobulin heavy chain intron transcription enhancer drives the expression of the Myc transgene, which are prone to develop B cell lymphomas, to normal (22). In addition, by employing a second mouse minute, heterozygous for ribosomal protein L38 (Rpl38), we further validated the specific effects of ribosomal protein haploinsufficiency on Myc’s ability to increase protein synthesis. Indeed, by lowering the threshold of protein production in L24+/− and L38+/− mice, the increased protein synthesis rates in Eµ-Myc/+ cells were rescued to normal levels in the Eµ-Myc/+;L24+/− as well as Eµ-Myc/+;L38+/− backgrounds (Fig. 1). Therefore, these data confirmed the validity of utilizing ribosomal protein heterozygous mice to selectively rescue increased protein synthesis rates downstream of oncogenic Myc signaling and facilitated a deeper understanding of the role of deregulations in translation control in cancer formation.
Figure 1. The role of Myc-induced protein synthesis in lymphomagenesis.
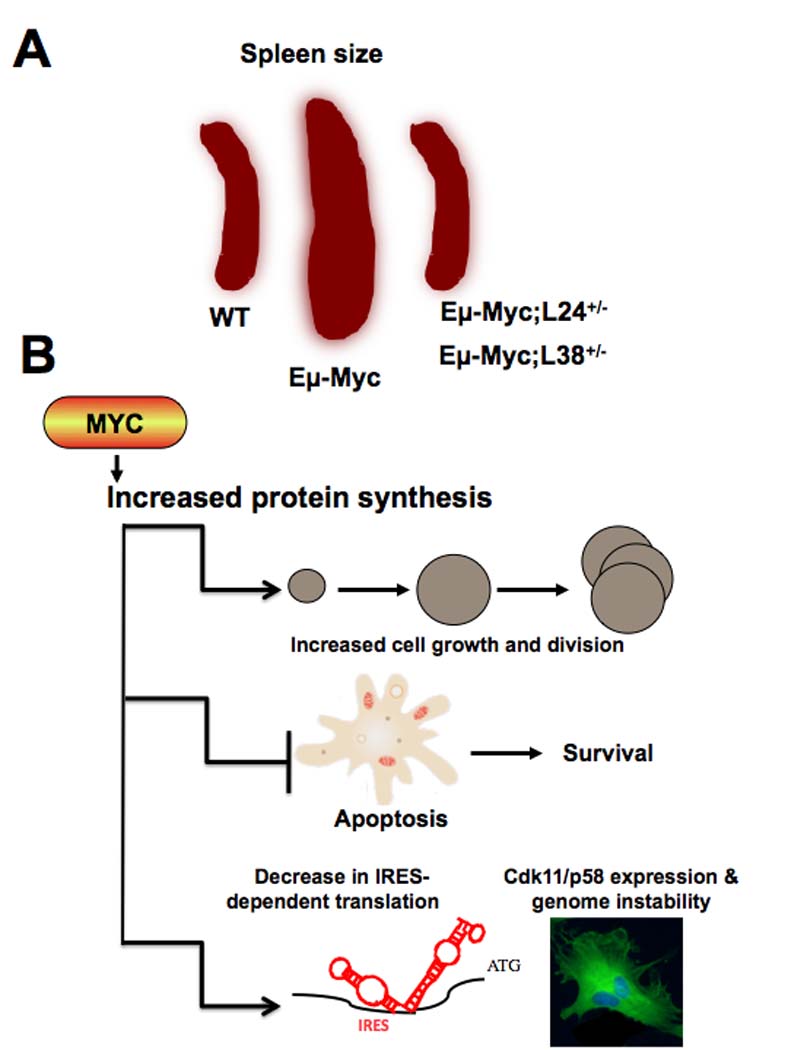
A The restoration of normal protein synthesis in Eµ-Myc/+;L24+/− as well as Eµ-Myc/+;L38+/− mice impairs Myc oncogenic activity towards lymphoma formation manifested by an early rescue in splenomegaly. B Myc-dependent increases in protein synthesis have a pleiotropic effect on distinct cellular and molecular events essential for the expansion of neoplastic clones. The ability of Myc to increase protein synthesis directly augments cell size and is sufficient to accelerate cell cycle progression, enables Myc-overexpressing precancerous cells to have a survival advantage, and increases cap-dependent translation at the expense of IRES-dependent translation of key mRNAs such as Cdk11/p58, which is required for accurate mitotic progression and cytokinesis.
However, a few issues are important to emphasize in terms of employing ribosomal protein mutants as genetic tools for restoring increases in protein synthesis rates. The first is that ribosomal protein loss of function does not necessarily result in reductions in protein synthesis rates in all cases. For example, ribosomal protein L22-deficient mice display normal rates of protein synthesis (26). Moreover, certain ribosomal proteins have been shown to exert extra-ribosomal functions (27). Therefore, not all ribosomal protein mutants can in principle be utilized as genetic tools to lower protein synthesis rates. Caution needs to be taken regarding how such mutations can be utilized, due to the fact that in certain tissue types, deficiency in expression of specific ribosomal proteins can trigger a stress response that results in upregulations in expression of the p53 tumor suppressor gene. For example, S6−/− and L24+/− mice show an increase in p53 activity for small windows of time during embryonic development, such as during gastrulation, and are responsible for certain phenotypes, such as ocular dysplasia and a kinked tail in L24+/− mice (28, 29). These findings may, in principle, raise the question of whether L24+/− mice are tumor resistant due to an increase in p53 activity. Importantly, this cannot be the case because oncogenic lesions are genetically induced after this narrow developmental window of time during which p53 might be stimulated. For example, L24+/− and L38+/− lymphoid cells, such as B or T lymphocytes, do not show any upregulation of either p53 or other stress response genes at steady state or even after activation (29 and data not shown). These findings are consistent with the fact the thymus and hematopoietic development in these mice are perfectly normal (22 and data not shown). Therefore, L24+/− and L38+/− haploinsufficient mice remain a powerful genetic tool to restore protein synthesis to normal levels downstream of oncogenic lesions that impinge directly on the translational apparatus, such as Myc hyperactivation.
In our study, Eµ-Myc;L24+/− and Eµ-Myc;L38+/− mice have been utilized to address whether Myc-induced cell growth is dependent on the ability of Myc to regulate protein synthesis and is coupled to uncontrolled cell cycle progression in cancer. Historically, studies in Drosophila have provided the first compelling evidence for coupling protein synthesis and cell growth downstream of dMyc (30). These studies also show that dMyc regulates cell growth independently of its role in cell division. However, in mammalian cells, it has been more problematic to demonstrate that indeed Myc upregulates cell size by means of increasing global protein synthesis. In this context, an outstanding and unresolved question was whether an increase in cell growth downstream of Myc hyperactivation is coupled to increased cell division, and whether this is relevant for Myc-induced cancer formation. Strikingly, in Eµ-Myc;L24+/− and Eµ-Myc;L38+/− mice, the increase in cell growth in Myc-overexpressing B cells is rescued to normal, and importantly this is directly associated with restoration of cell division rates to near-WT levels (22). These findings suggest that Myc couples cell growth and cell division at least in part through its ability to control protein synthesis (Fig. 1). But how, then, does a deregulation in translational control alter the expression of key mRNAs important for cell cycle progression? And why does this represent a mechanism for cellular transformation? Although there are no formal answers to these questions at present, it can be speculated that increases in cell growth could promote and sustain cell cycle progression downstream of Myc hyperactivation independently from or in addition to Myc’s ability to regulate key cell cycle targets transcriptionally. For example, Cyclin D1 is a translationally regulated gene, and its protein levels, but not its mRNA, increase when cap-dependent translation is augmented (31). Importantly, it has been observed that downstream of Myc activation, Cyclin D1 levels increase by a post-transcriptional mechanism (32). In Myc-overexpressing cells, alterations in translation control could alter accurate timing of cell cycle phases. In particular, as Myc-overexpressing cells are larger, cell growth may no longer represent a limiting factor for Myc-overexpressing cells to enter into cell division, resulting in unscheduled entry into S phase. Interestingly, in addition to the marked suppression in Myc’s ability to increase cell division in the L24+/− and L38+/− genetic backgrounds, the tumor suppressive apoptotic cellular response to Myc’s oncogenic activity (33) is dramatically increased (Fig. 1) (22). It remains to be determined whether deregulation in translation control downstream of Myc activation may affect the expression of apoptotic genes regulated by Myc. These findings suggest that clonal derivatives of precancerous cells may be more efficiently eliminated by programmed cell death in Eµ-Myc;L24+/− and Eµ-Myc;L38+/− mice.
Moreover, the significance for tumorigenesis of how restoring protein synthesis impacts on many cellular processes controlled by Myc is highlighted by the fact that Myc oncogenic capacity is strongly impaired in L24+/− and L38+/− genetic backgrounds (Fig. 1) (22). Indeed, the onset of lymphomas in the Eµ-Myc;L24+/− and Eµ-Myc;L38+/− mice is dramatically delayed compared to Eµ-Myc mice and, most importantly, a significant percentage of these mice do not develop lymphomas even after 1.5 years of age. Notably, the suppression of lymphoma observed in Eµ-Myc;L24+/− and Eµ-Myc;L38+/− mice is specific to a counter-activation of Myc-induced protein synthesis, rather than a general dominant effect that a reduction in protein synthesis might have on any type of cancer model. In fact, ribosomal protein haploinsufficiency in the context of the p53−/− background did not have any effect on tumor formation (22). Unexpectedly, the genome instability index of Myc-derived tumors in the L24+/− or L38+/− backgrounds was drastically lower. While all of the lymphomas analyzed from Eµ-Myc mice had chromosomal abnormalities, Eµ-Myc;L24+/− tumors showed either no chromosomal abnormalities or showed them at a lower frequency (22). The difference in the genome instability index present in mice where Myc can no longer augment protein synthesis revealed a new link between increased protein synthesis and genome instability in cancer.
What is the mechanism by which Myc-dependent increases in protein synthesis lead to genome instability? This may, at least in part, be due to a perturbation in the cytokinetic process associated with impairments in the switch between cap- and IRES-dependent translation which regulates the expression of critical regulators of cytokinesis (22, 34). During mitosis, the most general mechanism for translation initiation, cap-dependent translation, is normally decreased. On the other hand, IRES-dependent translation promotes the expression of specific proteins necessary for mitotic progression and accurate cytokinesis (22, 34, 35).
We found that the elevated rate of protein synthesis during mitosis in Eµ-Myc B lymphocytes was cap-dependent. Therefore, we hypothesized that Myc’s continuous stimulation of cap-dependent translation during mitosis may interfere with the accurate switch to IRES-dependent translation. Indeed, general IRES-dependent translation is not stimulated as a consequence of Myc hyperactivation and affects the expression of target mRNAs that rely on this very precise and orderly switch in translational control. For example, the endogenous IRES-dependent form of Cdk11 (p58-PITSLRE) is specifically expressed in the G2/M phase of the cell cycle (34, 36, 37). In Myc-overexpressing cells, IRES-dependent translation of Cdk11/p58 is impaired (Fig. 1). Several findings have recently revealed a critical function for Cdk11/p58 in mitotic progression, and downregulation of Cdk11/p58 by RNAi results in mitotic abnormalities (38, 39). In agreement with these findings Myc-overexpressing cells display cytokinetic defects, including increased numbers of binucleated cells, and restoring the expression of Cdk11/p58 during mitosis results in a normal mitotic progression (Fig. 1) (22). As a consequence of Myc hyperactivation, the defects in cytokinesis were associated with supernumerary centrosomes, an early hallmark of genome instability, and with chromosomal abnormalities, which are reverted in a background of normal protein synthesis (22). Interestingly, Myc itself possesses an IRES element in its 5’UTR (40). In the context of our studies, it is tempting to speculate that Myc levels may be autoregulated at the translational level through a balance between cap- and IRES-dependent translation. For example, the increase in cap-dependent translation as a consequence of high levels of Myc may ultimately hinder IRES-dependent translation of Myc, thereby creating a negative feedback loop to safeguard cells against increases in Myc protein levels that could cause cellular transformation. Overall, these findings suggest that deregulations in protein synthesis downstream of Myc can have an immediate and profound effect on the acquisition of additional genetic lesions that cooperate with Myc hyperactivation in cancer.
Meaning and Implication
The findings discussed above together with a recent exciting body of research should firmly convince cancer biologists that deregulation of protein synthesis is not just a consequence of overall cellular transformation but indeed can directly contribute to tumorigenesis. This is only the beginning of a novel and exciting field that needs to be filled with answers to many challenging questions, such as: Which specific intracellular or extracellular programs rely on protein synthesis for cancer formation? Which are the specific translational components that have the potential to become oncogenic? Which distinct mRNAs regulated at the translational level are directly relevant for cellular transformation? Which are the oncogenic lesions that directly impinge on the translational apparatus? How can we use our current knowledge to identify new targets for cancer therapy? In the context of the last question we are starting to appreciate the importance of targeting components of the protein synthesis apparatus as a new avenue for therapeutic intervention in cancer. For example, Rapamycin and a new mTOR ATP active site inhibitor, PP242 (41), which targets the mTOR kinase to modulate the activity of critical components of the translational machinery, may also show therapeutic benefits in the context of Myc hyperactivation.
The body of work discussed above offers a glimpse into unexpected and complex molecular and cellular processes caused by deregulations in translational control. For example, these studies have identified a previously unknown and direct relationship between increases in protein synthesis and genome instability (Fig. 1). Moreover, despite an increase in general protein synthesis rates elicited by the Myc oncogene, paradoxically, the downregulation in translation of specific mRNAs such as Cdk11 may play a critical function in cancer initiation. These findings suggest that a “translational code” to cancer remains to be uncovered in which the specific effects of key oncogenic pathways on a repertoire of distinct translation components may lead to unique and/or overlapping functions in the multi-step process of cancer biogenesis. Our ability to tease apart these mechanisms will undoubtedly enable the discovery of new therapeutic interventions for the treatment of specific cancers.
Acknowledgements
I thank Maria Barna for providing critical discussion; Joanna Copley for editing the manuscript; and Chi Dang for sharing unpublished results. This work arose from a direct collaboration with Barna’s lab at UCSF and is supported by NIH R01 HL085572 and NIH R01 CA140456 (D.R.) and the Sandler Foundation (M. Barna).
Literature Cited
- 1.Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 2.Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006;16:253–264. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 3.Gomez-Roman N, Felton-Edkins ZA, Kenneth NS, et al. Activation by c-Myc of transcription by RNA polymerases I, II and III. Biochem Soc Symp. 2006:141–154. doi: 10.1042/bss0730141. [DOI] [PubMed] [Google Scholar]
- 4.Coller HA, Grandori C, Tamayo P, et al. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci U S A. 2000;97:3260–3265. doi: 10.1073/pnas.97.7.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greasley PJ, Bonnard C, Amati B. Myc induces the nucleolin and BN51 genes: possible implications in ribosome biogenesis. Nucleic Acids Res. 2000;28:446–453. doi: 10.1093/nar/28.2.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo QM, Malek RL, Kim S, et al. Identification of c-myc responsive genes using rat cDNA microarray. Cancer Res. 2000;60:5922–5928. [PubMed] [Google Scholar]
- 7.Pajic A, Spitkovsky D, Christoph B, et al. Cell cycle activation by c-myc in a burkitt lymphoma model cell line. Int J Cancer. 2000;87:787–793. doi: 10.1002/1097-0215(20000915)87:6<787::aid-ijc4>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 8.Schuhmacher M, Kohlhuber F, Holzel M, et al. The transcriptional program of a human B cell line in response to Myc. Nucleic Acids Res. 2001;29:397–406. doi: 10.1093/nar/29.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mateyak MK, Obaya AJ, Adachi S, Sedivy JM. Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ. 1997;8:1039–1048. [PubMed] [Google Scholar]
- 10.Iritani BM, Eisenman RN. c-Myc enhances protein synthesis and cell size during B lymphocyte development. Proc Natl Acad Sci U S A. 1999;96:13180–13185. doi: 10.1073/pnas.96.23.13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iritani BM, Delrow J, Grandori C, et al. Modulation of T-lymphocyte development, growth and cell size by the Myc antagonist and transcriptional repressor Mad1. Embo J. 2002;21:4820–4830. doi: 10.1093/emboj/cdf492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuldiner O, Benvenisty N. A DNA microarray screen for genes involved in c-MYC and N-MYC oncogenesis in human tumors. Oncogene. 2001;20:4984–4994. doi: 10.1038/sj.onc.1204459. [DOI] [PubMed] [Google Scholar]
- 13.Kim S, Li Q, Dang CV, Lee LA. Induction of ribosomal genes and hepatocyte hypertrophy by adenovirus-mediated expression of c-Myc in vivo. Proc Natl Acad Sci U S A. 2000;97:11198–11202. doi: 10.1073/pnas.200372597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piedra ME, Delgado MD, Ros MA, Leon J. c-Myc overexpression increases cell size and impairs cartilage differentiation during chick limb development. Cell Growth Differ. 2002;13:185–193. [PubMed] [Google Scholar]
- 15.Schorl C, Sedivy JM. Loss of protooncogene c-Myc function impedes G1 phase progression both before and after the restriction point. Mol Biol Cell. 2003;14:823–835. doi: 10.1091/mbc.E02-10-0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schlosser I, Holzel M, Murnseer M, Burtscher H, Weidle UH, Eick D. A role for c-Myc in the regulation of ribosomal RNA processing. Nucleic Acids Res. 2003;31:6148–6156. doi: 10.1093/nar/gkg794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arabi A, Wu S, Ridderstrale K, et al. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat Cell Biol. 2005;7:303–310. doi: 10.1038/ncb1225. [DOI] [PubMed] [Google Scholar]
- 18.Grandori C, Gomez-Roman N, Felton-Edkins ZA, et al. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat Cell Biol. 2005;7:311–318. doi: 10.1038/ncb1224. [DOI] [PubMed] [Google Scholar]
- 19.Grewal SS, Li L, Orian A, Eisenman RN, Edgar BA. Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat Cell Biol. 2005;7:295–302. doi: 10.1038/ncb1223. [DOI] [PubMed] [Google Scholar]
- 20.Arabi A, Rustum C, Hallberg E, Wright AP. Accumulation of c-Myc and proteasomes at the nucleoli of cells containing elevated c-Myc protein levels. J Cell Sci. 2003;116:1707–1717. doi: 10.1242/jcs.00370. [DOI] [PubMed] [Google Scholar]
- 21.Poortinga G, Hannan KM, Snelling H, et al. MAD1 and c-MYC regulate UBF and rDNA transcription during granulocyte differentiation. Embo J. 2004;23:3325–3335. doi: 10.1038/sj.emboj.7600335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barna M, Pusic A, Zollo O, et al. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature. 2008;456:971–975. doi: 10.1038/nature07449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boring LF, Sinervo B, Schubiger G. Experimental phenocopy of a minute maternal-effect mutation alters blastoderm determination in embryos of Drosophila melanogaster. Dev Biol. 1989;132:343–354. doi: 10.1016/0012-1606(89)90231-5. [DOI] [PubMed] [Google Scholar]
- 24.Moreno E, Basler K. dMyc transforms cells into super-competitors. Cell. 2004;117:117–129. doi: 10.1016/s0092-8674(04)00262-4. [DOI] [PubMed] [Google Scholar]
- 25.Oliver ER, Saunders TL, Tarle SA, Glaser T. Ribosomal protein L24 defect in belly spot and tail (Bst), a mouse Minute. Development. 2004;131:3907–3920. doi: 10.1242/dev.01268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson SJ, Lauritsen JP, Hartman MG, et al. Ablation of ribosomal protein L22 selectively impairs alphabeta T cell development by activation of a p53-dependent checkpoint. Immunity. 2007;26:759–772. doi: 10.1016/j.immuni.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 27.Warner JR, McIntosh KB. How common are extraribosomal functions of ribosomal proteins? Mol Cell. 2009;34:3–11. doi: 10.1016/j.molcel.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panic L, Tamarut S, Sticker-Jantscheff M, et al. Ribosomal protein S6 gene haploinsufficiency is associated with activation of a p53-dependent checkpoint during gastrulation. Mol Cell Biol. 2006;26:8880–8891. doi: 10.1128/MCB.00751-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barkic M, Crnomarkovic S, Grabusic K, et al. The p53 tumor suppressor causes congenital malformations in Rpl24-deficient mice and promotes their survival. Mol Cell Biol. 2009;29:2489–2504. doi: 10.1128/MCB.01588-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P. Drosophila myc regulates cellular growth during development. Cell. 1999;98:779–790. doi: 10.1016/s0092-8674(00)81512-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenwald IB, Lazaris-Karatzas A, Sonenberg N, Schmidt EV. Elevated levels of cyclin D1 protein in response to increased expression of eukaryotic initiation factor 4E. Mol Cell Biol. 1993;13:7358–7363. doi: 10.1128/mcb.13.12.7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perez-Roger I, Kim SH, Griffiths B, Sewing A, Land H. Cyclins D1 and D2 mediate myc-induced proliferation via sequestration of p27(Kip1) and p21(Cip1) Embo J. 1999;18:5310–5320. doi: 10.1093/emboj/18.19.5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 34.Wilker EW, van Vugt MA, Artim SA, et al. 14-3-3sigma controls mitotic translation to facilitate. Nature. 2007;446:329–332. doi: 10.1038/nature05584. [DOI] [PubMed] [Google Scholar]
- 35.Pyronnet S, Sonenberg N. Cell-cycle-dependent translational control. Curr Opin Genet Dev. 2001;11:13–18. doi: 10.1016/s0959-437x(00)00150-7. [DOI] [PubMed] [Google Scholar]
- 36.Cornelis S, Bruynooghe Y, Denecker G, Van Huffel S, Tinton S, Beyaert R. Identification and characterization of a novel cell cycle-regulated internal ribosome entry site. Mol Cell. 2000;5:597–605. doi: 10.1016/s1097-2765(00)80239-7. [DOI] [PubMed] [Google Scholar]
- 37.Tinton SA, Schepens B, Bruynooghe Y, Beyaert R, Cornelis S. Regulation of the cell-cycle-dependent internal ribosome entry site of the PITSLRE protein kinase: roles of Unr (upstream of N-ras) protein and phosphorylated translation initiation factor eIF-2alpha. Biochem J. 2005;385:155–163. doi: 10.1042/BJ20040963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petretti C, Savoian M, Montembault E, Glover DM, Prigent C, Giet R. The PITSLRE/CDK11p58 protein kinase promotes centrosome maturation and bipolar spindle formation. EMBO Rep. 2006;7:418–424. doi: 10.1038/sj.embor.7400639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu D, Valentine M, Kidd VJ, Lahti JM. CDK11(p58) is required for the maintenance of sister chromatid cohesion. J Cell Sci. 2007;120:2424–2434. doi: 10.1242/jcs.007963. [DOI] [PubMed] [Google Scholar]
- 40.Stoneley M, Paulin FE, Le Quesne JP, Chappell SA, Willis AE. C-Myc 5' untranslated region contains an internal ribosome entry segment. Oncogene. 1998;16:423–428. doi: 10.1038/sj.onc.1201763. [DOI] [PubMed] [Google Scholar]
- 41.Feldman ME, Apsel B, Uotila A, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]