

Abstract
Connexin43 (Cx43) is the most abundantly expressed gap junction protein. The C-terminal tail of Cx43 is important for regulation of gap junctions via phosphorylation of specific tyrosine and serine residues and through interactions with cellular proteins. The C-terminus of Cx43 has been shown to interact with the PDZ2 domain of the tight and adherens junction associated zona occludens 1 (ZO-1) protein. Analysis of the PDZ2 binding domain of Cx43 indicated that positions −3 and −2, and the final hydrophobic amino acid at the C-terminus, are critical for ZO-1 binding. In addition, the C-termini of connexins 40 and 45, but not Cx32, interacted with ZO-1. To evaluate the functional significance of the Cx43-ZO-1 interaction, Cx43 wild type (Cx43wt) and mutants lacking either the C-terminal hydrophobic isoleucine (Cx43ΔI382) or the last five amino acids (Cx43Δ378–382), required for ZO-1 binding in vitro, were introduced into a Cx43-deficient MDCK cell line. In vitro binding studies and coimmunoprecipitation assays indicated that these Cx43 mutants failed to interact with ZO-1. Confocal and deconvolution microscopy revealed that a fraction of Cx43wt colocalized with ZO-1 at the plasma membrane. A similar colocalization pattern was observed for the Cx43ΔI382 and Cx43Δ378–382 mutants, which were translocated to the plasma membrane and formed functional gap junction channels. The wt and mutant Cx43 appeared to have similar turnover rates. However, the P2 and P3 phosphoisoforms of the Cx43 mutants were significantly reduced compared to Cx43wt. These studies indicated that the interaction of Cx43 with ZO-1 may contribute to the regulation of Cx43 phosphorylation.
Keywords: Gap junctions, connexin, ZO-1, PDZ domain, phosphorylation
Introduction
Connexin43 (Cx43) is a member of the family of gap junction proteins which form membrane channels that permit the intercellular exchange of ions and small molecular weight molecules, including the second messengers 1,4,5-inositol triphosphate, Ca2+, and cyclic nucleotides (1, 2). Each connexin family member forms channels with unique functional characteristics, and heteromeric channels comprised of more than one connexin present an additional level of functional complexity (3, 4). Gap junctional communication has been implicated in many diverse biological processes including synchronization of contraction of the heart and uterus (5), generation and propagation of action potentials in excitable cells (6), exocrine and endocrine gland secretion (7), organ morphogenesis (8), embryonic pattern formation and differentiation (9), as well as growth regulation and tumor suppression (10, 11).
Cx43 function is regulated through a number of mechanisms including protein degradation (12), intracellular Ca2+ levels (13), pH (14), applied voltage (15), and posttranslational modification by phosphorylation (16). Several studies have reported that phosphorylation is not only important for the regulation of channel gating (reviewed in 16, 17), but also essential for the assembly of connexins into connexons, the subsequent formation of functional channels (18), and connexin processing during mitosis (19–21). The phosphorylation of Cx43 on Ser255, Ser279 and Ser282 by activated mitogen-activated protein (MAP) kinase (22), on Ser368 by activated PKC (23), and on Tyr247 and Tyr265 by the v-Src tyrosine kinase (24) has established that specific phosphorylation in the C-terminus is a primary means of regulating Cx43 function.
We performed a search for additional Cx43 interacting proteins to further extend our knowledge of the function and regulation of gap junctions. Using a yeast two-hybrid screen and the C-terminus of Cx43 as bait we identified the PDZ2 domain and flanking sequences of zona occludin 1 (ZO-1) as a novel connexin interacting partner (unpublished results reported at the International Gap Junction Meeting, Key Largo, FL. 1997). Similar results from other investigators have clearly documented the interaction of Cx43 with ZO-1 (25–27); however, the functional significance of this interaction remains elusive.
Here, we further define the domain in the Cx43 C-terminus (CT) that interacts with the PDZ2 domain of ZO-1, and demonstrate that Cx40 and Cx45, but not Cx32, also bind to ZO-1 through the PDZ2 domain. In addition, minimally mutated Cx43 deletion mutants lacking the ZO-1 PDZ2 binding domain localize to the plasma membrane and form functional gap junction channels in Madin-Darby canine kidney (MDCK) cells. Interestingly, phosphorylation of the Cx43 deletion mutants was diminished, suggesting that the Cx43-ZO-1 interaction plays a key role in mediating Cx43 phosphorylation. The data also suggest that the extent of phosphorylation observed in wildtype Cx43 (Cx43wt) is not essential for Cx43 retention in the plasma membrane. These studies shed new light on the role of the Cx43-ZO-1 interaction in the regulation of Cx43 and suggest a potential role for ZO-1 in the regulation of other connexins that have also been shown to interact with the ZO-1 protein.
Materials and Methods
Cell Culture
MDCK cells lacking endogenous Cx43 were obtained from Dale Laird (University of Western Ontario) and cultured in DMEM (GIBCO, high glucose) supplemented with 2mM glutamine and 10% fetal bovine serum. Sf9 insect cells were cultured in optimized serum-free medium (Sf-900 II SFM, Life Technology, Inc.) at 27°C.
Construction of Plasmids Expressing LexA Fusions Proteins
The cDNA fragments encoding the carboxyl-terminal tail of Cx43wt (Cx43CT, amino acids 222–382), Cx43CTΔI382 deletion mutant containing amino acids 222–381 (deletion of I382), Cx43CTΔ378–382 comprising amino acids 222–377 (deletion of the last five amino acids), and point substitution mutants D379A, L380A, and E381A of Cx43CT were PCR amplified from clone G2B (28), containing the full length rat Cx43 cDNA, using the common forward primer, 5′-GT ATCAGAATTCGCTTTGAACATCATTGAG-3′ and the following specific reverse primers:
5′-TATTCAGGATCCTTAAATCTCCAGGTCATC-3 (wt),
5′-TATTCAGGATCCTTACTCCAGGTCATCAGGCCG-3′ (ΔI382),
5′-TATTCAGGATCCTTAAGGCCGAGGCCTGCTGC-3′ (Δ378–382),
5′-TATTCAGGATCCTTAAATCTCCAGGGCATCA GGCCG-3′ (D379A),
5′-TATTCAGGATCCTTAAATCTCGGCGTCATCAGGCCGA GGC-3′ (L380A), and
5′-TATTCAGGATCCTTAAATGGCCAGGTCATCAGGCCGAG-3′ (E381A).
The PCR products were gel purified and cloned into the EcoRI and BamHI sites of the LexA fusion vector pBTM116 (29). The fidelity and reading frame of all LexA-Cx43CT fusion constructs were confirmed by DNA sequencing.
The cDNA fragment encoding the carboxyl-terminal tails of Cx32 (amino acids 202–283), Cx40 (amino acids 197–356), and Cx45 (amino acids 236–396) were PCR amplified from the mouse Cx32, rat Cx40, and mouse Cx45 cDNAs, respectively (30–32). The cDNA fragments were gel purified, cloned in frame into the EcoRI and BamHI sites of the LexA fusion vector pBTM116, and sequenced.
Yeast Two-Hybrid and His3 Reporter Gene Assay
The genotype of the S. cerevisiae yeast reporter strain L40 is MATa trp1 leu2 his3 LYS2:lexA-His3 URA3::lexA-lacZ (provided by S. Hollenberg, Vollum Institute, Oregon Health Sciences University). Yeast strains were grown at 30°C in YPDA medium (1% yeast extract, 2% Bacto-Peptone, 2% glucose, and 0.1 mg/ml adenine) or in synthetic minimal medium supplemented with appropriate amino acids (33, 34).
The cDNA fragment encoding the carboxyl-terminal tail of Cx43 (amino acids 222–382) was cloned into pBTM116 and used in the two-hybrid assay. The plasmids expressing LexA fusions of the C-terminal tails of different connexins or mutants of Cx43 described above were cotransformed with pVP16-ZO-1aa163–310 (the PDZ2 domain and flanking sequences identified in the two-hybrid screen) into the yeast reporter strain L40 as described (35). The yeast transformants were first plated on synthetic medium lacking leucine and tryptophan followed by incubation for three days at 30°C. The yeast cotransformants were then streaked onto plates containing synthetic medium lacking leucine, tryptophan, and histidine and incubated at 30°C for 3–5 days and observed for their ability to grow and form colonies.
Construction and Purification of GST-ConnexinCT Fusion Proteins
The cDNAs encoding the glutathione-S-transferase (GST) fusion constructs for the cytoplasmic, carboxyl-terminal (CT) regions of Cx32 (GST-Cx32CT, amino acids 220–283), Cx40 (GST-Cx40CT, amino acids 197–356), and Cx45 (GST-Cx45CT, amino acids 265–396) were amplified by PCR from the mouse Cx32 cDNA (30), rat Cx40 cDNA (31) and the mouse Cx45 cDNA (32), respectively. The cDNA fragments encoding the carboxyl-terminal tails of Cx43wt (GST-Cx43CTwt, amino acids 236–382) (36), Cx43ΔI382 mutant (GST-Cx43CTΔI382, amino acids 222–381), and Cx43Δ378–382 mutant (GST-Cx43CTΔ5, amino acids 236–377) were PCR amplified from G2B. The PCR products were subcloned into the BamHI and EcoRI sites of the pGEX-KG expression vector (provided by J. Dixon, University of Michigan). The recombinant plasmids were sequenced to confirm that the cDNA inserts were cloned in frame and that no unwanted mutations were generated during the PCR amplification. The recombinant plasmids were transformed into E. coli strain BL21/DE3 (Novagen). The GST fusion proteins were induced with IPTG and affinity purified using glutathione-agarose beads as described (36).
Construction of Recombinant Transfer Vector and Baculovirus
The mouse full length ZO-1 cDNA in pBlue-script (provided by S. Tsukita, Kyoto University) was cloned into the EcoR1 site of pVL1393 resulting in pVL1393-ZO-1. The orientation of the cDNA insert in the vector was confirmed by restriction digestion with BamH1. Recombinant baculovirus was generated by cotransfecting a monolayer of Sf9 cells with recombinant transfer plasmid pVL1393-ZO-1 and BaculoGold baculovirus DNA (Pharmingen) as described (36). Recombinant baculoviruses were then subjected to further amplification by multiple rounds of infection into Sf9 cells. Sf9 cells were infected with recombinant baculovirus at a multiplicity of infection of 3–5 and harvested 48–72 h postinfection.
In Vitro Binding Assays
Sf9 cells were infected with recombinant baculovirus expressing ZO-1. The cells were harvested 72 h postinfection and lysed in NP-40 buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 1% NP-40, 1 mM dithiothreitol, 50 mM NaF, 160 μM Na3VO4, 1 mM phenylmethylsufonyl fluoride (PMSF), 50 μg/ml benzamidine, 1 μg/ml pepstatin, 1 μg/ml leupeptin). Untransfected MDCK cells were lysed in Lysis Buffer (20 mM Tris, pH 8, 0.5% Triton X-100, 0.5% NP-40, 150 mM NaCl, 2 mM EDTA, 1 mM DTT, 1 mg/ml BSA, 1 mM PMSF, 10 μg/ml leupeptin and aprotinin). Clarified supernatants containing 300 μg of protein were incubated with 10 μg of the purified appropriate GST fusion proteins bound to glutathione agarose beads at 4°C. After 2 h of incubation, the beads were washed three times with the appropriate lysis buffer. The bound proteins were denatured in SDS sample buffer by boiling for 5 min in the presence of β-mercaptoethanol, and resolved on SDS-containing 5–15% polyacrylamide gradient gels (37). Proteins were then electrotransferred to Immobilon P membranes (Millipore, Bedford, MA) and blocked in blocking buffer (5% BSA and 0.1% Tween 20 in PBS) for 1 h at room temperature. The membranes were probed with rat anti-ZO-1 antibody (R26.4C, Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA) followed by a horseradish peroxidase-coupled goat anti-rat secondary antibody (Amersham Corp. Arlington Heights, IL). The immune complexes were detected with ECL-Plus Western Blot Detection reagents (Amersham Corp. Arlington Heights, IL).
Stable Expression of Wild Type Cx43 and Cx43 Deletion Mutants in MDCK Cells
The full length Cx43wt gene was excised from the EcoRI site of G2B (28) and cloned into the mammalian expression vector pcDNA3 (Invitrogen) in the correct orientation. cDNA fragments encoding the full-length and the Cx43-deletion mutants, Cx43ΔI382 and Cx43Δ378–382, were PCR amplified from G2B and the PCR products were cloned into the BamH1 and EcoR1 sites of pcDNA3 (the mutations and the fidelity of the sequences were verified by DNA sequencing). The recombinant plasmids were used to transfect MDCK cells lacking endogenous Cx43 using Lipofectamine 2000 (GIBCO Life Science Technology). The transfected cells were grown in medium containing 800 μg/ml G418 (GIBCO) to select for stable transfectants. Single clones were isolated and examined for Cx43 expression by immunofluorescence microscopy.
Confocal and Deconvolution Microscopy
Untransfected MDCK cells or cells stably transfected with either Cx43wt or the deletion mutants Cx43ΔI382 or Cx43Δ378–382 were plated on coverslips and grown for approximately 18 h. The cells were then fixed with cold methanol at −20°C for 30 min and washed with PBS. Subsequently, the cells were permeabilized in 0.2% Triton X-100/PBS for 10 min at room temperature, washed twice in PBS and blocked in 1% bovine serum albumin (BSA) in PBS for 10 min. The cells were incubated for 1 h with a rabbit polyclonal peptide antibody against Cx43 (1:1000 dilution, CT368) and a rat monoclonal antibody against ZO-1 (1:200 dilution, R26.4C). After washing three times in PBS, the cells were then incubated for 1 h with Alexa 488-conjugated goat anti-rabbit (Molecular Probes, 1:500) and Alexa 568-conjugated goat anti-rat (Molecular Probes, 1:500) secondary antibodies. Following three washes in PBS, cell nuclei were stained with ToPro3 (1:1500 dilution, Molecular Probes) and mounted on glass slides in Prolong antifade solution (Molecular Probes).
Confocal images were sequentially collected on a Bio-Rad MRC1024 confocal system attached to a Zeiss Axiovert 100TV microscope. Images were collected using a 63× objective (NA 1.4). The krypton-argon mixed gas laser generated the 488 nm excitation line with which to view the FITC labeled cells using the 522-DF35 emission filter set, and the 568 nm laser line for the Texas red fluorophore using the HQ598-DF40 filter set. Pseudo-colored images were merged using Adobe Photoshop. For deconvolution microscopy, fluorescence images were documented using a Delta Vision Deconvolution microscope (API, Issaquah, WA) equipped with a Photometrics CH350L liquid-cooled CCD camera attached to an Olympus IX-70 inverted microscope. These data were collected using 100× oil immersion objective lens (NA 1.35) together with filter sets for FITC (Ex) 490/20 – (Em) 528/38 and Texas red (Ex) 555/28 – (Em) 617/73. All images were deconvolved using constrained iterative algorithms (10 iterations) of Delta Vision software (softWoRx, v2.5). The deconvolved images were subsequently processed (merged, made into movies and converted into tiff format) using softWoRx software.
Metabolic Labeling, Immunoprecipitation and Pulse Chase Analysis
For the analysis of Cx43 phosphorylation, MDCK cell clones stably transfected with either Cx43wt, Cx43ΔI382, or Cx43Δ378–382 were metabolically-labeled with either 32Pi (NEX-053, NEN) at 1 mCi/ml in phosphate-deficient medium with 4% dialyzed calf serum for 2 h at 37°C or (35S)-methionine/cysteine (AGQ-0080, Promix, Amersham) at 100 μCi/ml in methionine-deficient medium containing 4% dialyzed calf serum for 3 h at 37°C. At the end of the labeling period, cells were rinsed with PBS, lysed in RIPA buffer (150 mM NaCl, 1% sodium deoxycholate, 1% Triton X-100, 0.1% sodium dodecyl sulfate, 10 mM Tris-HCl pH7.2), clarified, and immunoprecipitated with either normal rabbit serum (control) or rabbit antiserum against the peptide (amino acids 241–254) of rat Cx43 (CT241) in antigen excess as described (38). The immunoprecipitates were resolved on SDS-containing 7.5–15% polyacrylamide gradient gels. The gels were stained, destained, dried, fluorographed for 35S-labeled proteins, and autoradiographed using Kodak X-Omat XAR-5 films at −70°C, or were exposed to phosphoimager screens and processed on a Cyclone phosphoimager (Packard Instruments). For the quantitation of the Cx43 phosphoisoforms in Figure 5B, immunoprecipitations were performed in antigen excess to isolate equal amounts of Cx43 protein for loading on SDS-PAGE gels. Quantification of the phosphorylated bands was accomplished by excising the Cx43 isoforms out of the gel followed by liquid scintillation counting or the autoradiograms were scanned and bands quantified using a Bio-Rad Fluor-S MultiImager and Quantity One program. For Figure 5A, the radioactivity of immunoprecipitated 35S- and 32P-labeled proteins in sample buffer was determined by liquid scintillation counting and the samples normalized in a 3 (35S) : 1 (32P) cpm ratio for gel loading. The stained gels were fluorographed before drying and exposure to film. This approach gave gel displays where both 35S- and 32P-labeled Cx43 bands were displayed simultaneously.
Figure 5.

Analysis of the phosphorylation patterns of the Cx43wt and Cx43 deletion mutants. (A) Cx43wt, Cx43ΔI382, and Cx43Δ378–382 were immunoprecipitated from 35S-met/cys or 32Pi-labeled MDCK cells under conditions of antigen excess and the immunoprecipitated proteins were scintillation counted and normalized in a 3 (35S) : 1 (32P) cpm ratio in order to show the banding patterns of the immunoprecipitated proteins resolved on SDS-polyacrylamide gradient gels. The radiolabeled proteins were visualized by autoradiography of the fluorographed gel using Kodak XOmat XAR-5 film. Lanes contain either 35S-labeled proteins (S) or 32P-labeled proteins (P). The NP, P1, P2, and P3 Cx43 isoforms are indicated for the Cx43wt in lane 3. (B) Analysis of the Cx43 phosphoisoforms from the Cx43wt and the Cx43 deletion mutant cell clones. Cx43 was immunoprecipitated from 32Pi-labeled MDCK cells in antigen excess and the entire immunoprecipitate was loaded on SDS-polyacrylamide gels. Gels were stained, destained, dried and autoradiographed. Cx43 phosphoisoform bands were excised from the gels and radioactivity of each band was quantitated by liquid scintillation counting. Bars indicate the percentage of each phosphoisoform (P1, P2 and P3) relative to the total Cx43 phosphoisoforms. Average percentage and error bars (SEM) for Cx43wt and Cx43ΔI382 were obtained from two different clones in two separate experiments (n = 4), while average percentage and error bars for the Cx43Δ378–382 cell clone were obtained in two separate experiments (SD, n = 2).
For coimmunoprecipitation of Cx43 with ZO-1, nonlabeled cells were frozen at −70°C, thawed and scraped into 2 ml of ice-cold lysis buffer (20 mM Tris, pH 8, 0.5% Triton X-100, 0.5% NP-40, 150 mM NaCl, 2 mM EDTA, 1 mM DTT, 1 mg/ml BSA, 1 mM PMSF, 10 μg/ml leupeptin and aprotinin). Lysates were clarified at 120,000 × g at 4°C for 20 min. The supernatant (∼2 ml) was added to tubes containing 100 μl protein A beads (Santa Cruz) and 15 μl rabbit anti-ZO-1 (25 μg/μl, Zymed) and rotated at 4°C for 1 h. Immunoprecipitates were washed three times with lysis buffer, denatured in SDS sample buffer, boiled for 5 min, and resolved on SDS-containing 7.5% polyacrylamide gels (37). Proteins were then electrotransferred to Immobilon P membranes (Millipore, Bedford, MA) and blocked in blocking buffer (5% nonfat dry milk “blotto” and 0.1% Tween 20 in PBS) for 1 h at room temperature. The membranes were probed with Cx43 monoclonal antibody (P2D12, Fred Hutchinson Cancer Research Center, Seattle, WA) followed by a horseradish peroxidase-coupled goat anti-mouse secondary antibody (Amersham Corp. Arlington Heights, IL). The immune complexes were detected with ECL Western Blot Detection reagents (Amersham Corp. Arlington Heights, IL).
For the control in vivo phosphorylation studies, stably transfected MDCK cells were allowed to attach to plates for 6 h in complete medium, after which they were transferred into medium containing 0.5% FCS and incubated overnight. Cells were radiolabeled in phosphate- and serum-deficient medium containing 1 mCi/ml 32Pi (NEX 053, Perkin Elmer) for 2 h. During the last 30 min EGF (100 ng/ml; USB), TPA (100 ng/ml; Sigma), or PBS (control) was added. Cells were lysed, immunoprecipitated with anti-Cx43 CT241, and proteins resolved by SDS-PAGE. Gels were stained, destained, dried, and autoradiographed. Bands were scanned and quantitated using Quantity One software (Bio-Rad).
For pulse chase experiments cells were labeled with (35S)-methionine (Amersham) at 200 μCi/ml for 15 min (short pulse) or 100 μCi/ml for 1 h (longer pulse), then chased for various lengths of time in cold complete medium supplemented with 5 mM methionine. At the appropriate times, cells were rinsed with PBS and frozen at −70°C. Cells were lysed in RIPA, immunoprecipitated and processed as described above.
Measurement of Gap Junctional Communication
Gap junctional communication was measured by counting the number of cells that became fluorescent after microinjection of Lucifer yellow (LY) dye into a single cell as described previously (38). MDCK cells, either untransfected or stably transfected with the Cx43wt, Cx43ΔI382 or Cx43Δ378–382 cDNAs, were grown to confluence. Single cells were then microinjected with LY (10% w/v in 0.33 M lithium chloride) using an Eppendorf micromanipulator pneumatic injector (Eppendorf, Fremont, CA) and observed using a phase-contrast inverted microscope (Zeiss, Thornwood, NJ) equipped with epifluorescence. Gap junctional communication was quantified by counting the number of fluorescent neighboring cells 1–2 min after microinjection. Only cells coupled to one or more neighboring cells were counted for the average values.
Results
Analysis of the Cx43 PDZ2 Binding Domain
Several reports have demonstrated that Cx43 interacts with ZO-1 through the PDZ2 domain of ZO-1 and the C-terminus of Cx43 (25, 26, 39). PDZ domains are known to selectively interact with specific sequences at the C-terminus of target proteins (40). The directed yeast two-hybrid assay was used to characterize the Cx43-ZO-1/PDZ2 interaction. LexA fusion constructs of Cx43CT mutants containing either deletions or substitutions of residues in the C-terminal region were cotransformed into yeast L40 along with pVP16-ZO-1aa163–310 (Figure 1A). The interaction of two fusion proteins was determined by the ability of the yeast to grow on histidine-minus plates after the yeast cotransformants were selected in synthetic minimal medium lacking tryptophan and leucine for coexpression of the LexA and the VP-16 fusion constructs. Yeast coexpressing pVP16-ZO-1aa163–310 and the LexA fusion of Cx43CT-wt or the Cx43CT-E381A mutant grew on the histidine-minus plates (Figure 1B). However, yeast coexpressing the LexA fusion of Cx43CT-D379A, Cx43CT-L380A, Cx43CTΔI382 (deletion of I382 at the C-terminus), or Cx43CTΔ378–382 (deletion of aa 378–382), along with pVP16-ZO-1aa163–310, did not grow on the histidine-minus plates (Figure 1B). These results indicated that the D379, L380, and I382 residues of Cx43 were required for Cx43 to bind ZO-1 and defined the connexin PDZ binding motif as D-L-X-I, where X is any amino acid. Although D379 and L380 are required for the interaction, loss of the terminal hydrophobic I382 was sufficient to completely disrupt the Cx43CT-PDZ2/ZO-1 interaction in the yeast system.
Figure 1.
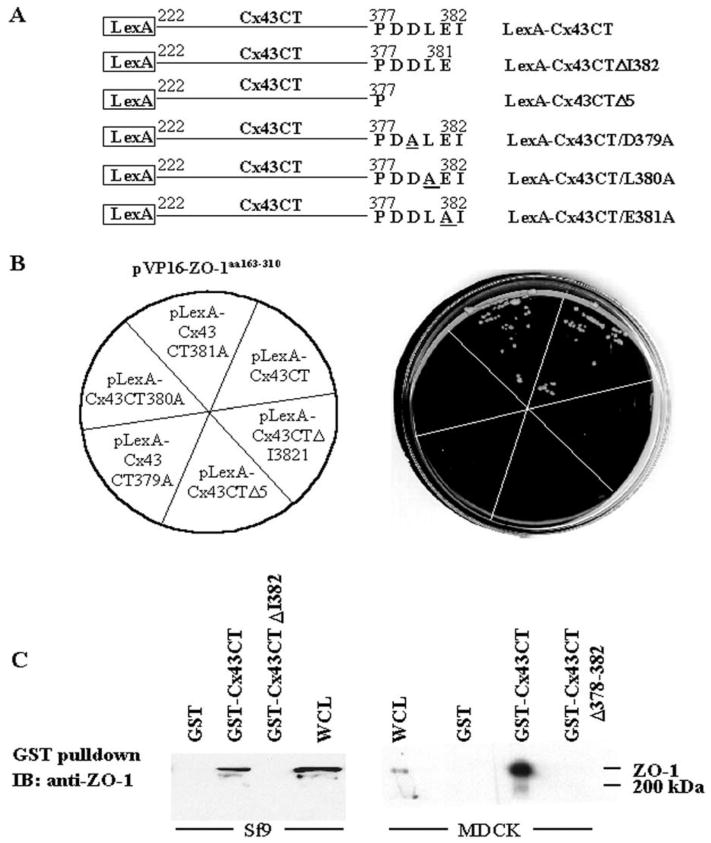
Characterization of the ZO-1-Cx43 interaction. (A) Schematic representation of LexA fusion constructs of wt, two carboxyl-terminal deletion mutants, and three substitution point mutants of Cx43CT. The cDNA fragments encoding the Cx43CT (carboxyl-terminal tail of Cx43wt, aa 222–382), Cx43CTΔI382 (deletion of I382), Cx43CTΔ378–382 (deletion of last five amino acids, Δ5), or point mutants D379A, L380A or E381A of Cx43 CT were subcloned into pBTM116 for expression as the corresponding LexA fusion proteins in the yeast strain L40. (B) Yeast strain L40 was cotransformed with pVP16-ZO-1aa163–310 (PDZ2 domain and flanking sequences) and pBTM116 containing the wt, deletion, or point mutants of Cx43CT. Yeast cotransformants were selected on plates containing synthetic medium lacking tryptophan and leucine (not shown). The individual Leu+Trp+ transformants was then streaked onto the corresponding section of synthetic medium plates without tryptophan, leucine, and histidine (shown on the left). The plates were incubated for three days at 30°C. Yeast expressing Cx43CT constructs that bound to the VP16-ZO-1aa163–310 grew on the LeuTrpHis− plates (shown on the right). (C) In vitro binding analysis of the full-length ZO-1 with the GST-Cx43CT, GST-Cx43ΔI382, or GST-Cx43Δ378–382 fusion proteins. Whole cell lysates (300 μg) of Sf9 cells infected with baculovirus expressing full length ZO-1 (left panel) or untransfected MDCK cells expressing endogenous ZO-1 (right panel) were incubated with 10 μg of GST, GST-Cx43CT, and GST-Cx43CTΔI382, or GST-Cx43Δ378–382 fusion proteins prebound to glutathione agarose beads. The beads were washed and eluted in SDS sample buffer. Whole cell lysate (WCL) and GST-fusion proteins eluates were resolved by SDS-PAGE, electrotransferred to an Immobilon P membrane and immunoblotted with ZO-1 antibody (R26.4C). The position of the ∼220 kDa ZO-1 protein and the 200 kDa molecular mass marker are shown on the right.
To confirm that deletion of I382 was sufficient to disrupt the Cx43-ZO-1 interaction, in vitro binding assays were performed by mixing lysates from Sf9 cells expressing full length ZO-1 with GST, GST-Cx43CT, or GST-Cx43ΔI382 fusion proteins bound to glutathione beads (Figure 1C, left panel). Binding of GST, GST-Cx43CT, or GST-Cx43Δ378–382 to ZO-1 in Cx43 negative MDCK cell lysates was also examined (Figure 1C, right panel). Binding of full length ZO-1 to the GST-fusion proteins was detected by immunoblotting with ZO-1 antisera (R26.4C). ZO-1 protein of approximately 220 kDa was detected in the samples incubated with GST-Cx43CT, but not in the samples containing GST-Cx43ΔI382, GST-Cx43Δ378–382, or GST alone (Figure 1C). These data confirmed that the Cx43 C-terminal I382 was required for association with the full length ZO-1 protein in vitro, and that the loss of this residue alone was sufficient to disrupt this protein-protein interaction.
Interaction of Other Connexins with ZO-1
PDZ domains are known to bind preferentially to transmembrane proteins that have a specific tetrapeptide motif ending in a hydrophobic carboxyl-terminus, usually valine, isoleucine, and sometimes, leucine (41, 42). Sequence analysis of the last 10 amino acids of 12 rodent connexins (43) was used to predict whether other connexins were likely to interact with the PDZ2 domain of ZO-1 (Table 1). Eleven of the 12 connexins have hydrophobic carboxyl-termini that could accommodate binding to a PDZ domain (43). Of these 12 connexins, only Cx32 lacks a hydrophobic terminus. Both Cx46 and Cx50 have the D-L-X-I motif identical to Cx43 (Table 1, underlined), and therefore, are likely to interact with the PDZ2 domain of ZO-1. Indeed, these connexins have been demonstrated recently to interact with ZO-1 (44). The directed yeast two-hybrid assay was utilized to determine whether Cx40, which has a valine at its C-terminus, and Cx45, which has an isoleucine, would interact with ZO-1 (Table 1, boxed). The schematic diagram in Figure 2A shows the connexin C-terminal regions (CTs) used in this assay. As shown in Figure 2B, yeast coexpressing VP16-ZO-1aa163–310 and LexA-Cx40CT, LexA-Cx45CT and LexA-Cx43CT grew on the histidine-minus plates, indicating that both the Cx40CT and Cx45CT proteins associated with the PDZ2 domain of ZO-1. However, yeast coexpressing the PDZ2 domain of ZO-1and Cx32CT did not grow on the histidine-minus plate. These results underscored the importance of a hydrophobic residue at the CT of the connexin molecule for its interaction with the PDZ2 domain of ZO-1 and indicated that Cx32 did not interact with the PDZ2 domain of ZO-1. These data confirm the earlier demonstration of the interaction of ZO-1 with Cx45 (27, 45).
TABLE 1.
Comparison or the C-termini of rodent connexins
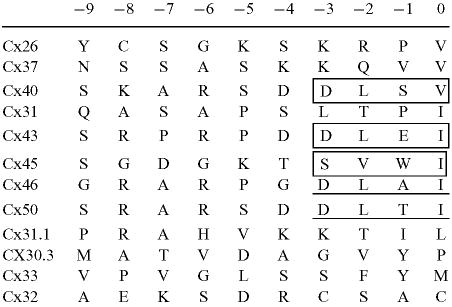 |
The carboxyl-terminal amino acids of the rodent connexins were obtained using the GCG “StringSearch” program against GenBank/EMB1 databases.
Figure 2.
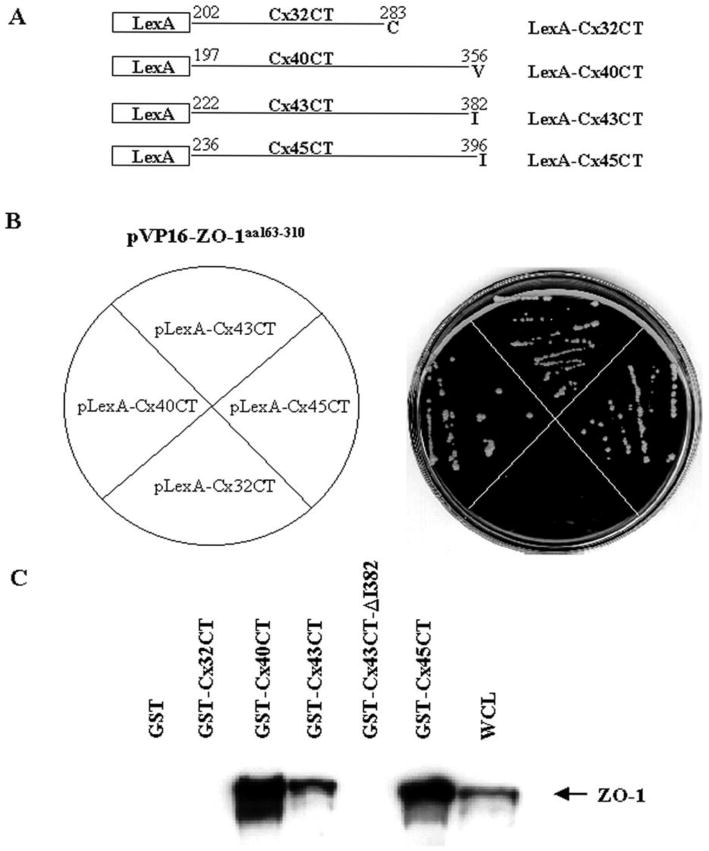
Interaction of ZO-1 with other connexins. (A) Schematic representation of LexA fusion constructs of the C-terminal tails of Cx32, Cx40, Cx43, and Cx45. (B) Yeast strain L40 was cotransformed with pVP16-ZO-1aa163–310 and the indicated plasmids as shown on the left. Cotransformed yeast cells were preselected by plating on synthetic medium lacking tryptophan and leucine. The individual Leu+Trp+ transformants were then streaked onto the corresponding section of synthetic medium plates without tryptophan, leucine, and histidine and the plates were incubated at 30°C for three days. Only yeast expressing constructs that bound to the VP16-ZO-1aa163–310 grew on the LeuTrpHis− plates (shown on the right). (C) Interaction of ZO-1 with different connexins. Whole cell lysate (300 μg) of Sf9 cells infected with baculovirus expressing full length ZO-1 was incubated with 20 μg of GST or GST-Cx32CT, GST-Cx40CT, GST-Cx45CT, GST-Cx43CT, or GST-Cx43CTΔI382 fusion proteins prebound to glutathione-agarose beads. Whole cell lysate (20 μg, WCL) and GST fusion proteins eluted from the beads in SDS sample buffer were resolved by SDS-PAGE, electrotransferred to an Immobilon P membrane and immunoblotted with ZO-1 antibody. The position of ZO-1 is shown at the right.
To determine whether full length ZO-1 interacted with the carboxyl-terminus of Cx40 and Cx45, in vitro binding assays were performed combining lysates of Sf9 cells expressing full length ZO-1 with GST alone, GST-Cx32CT, GST-Cx40CT, GST-Cx43CT, or GST-Cx45CT fusion proteins prebound to glutathione beads. ZO-1 bound to the GST fusion proteins was detected by immunoblotting with the rat monoclonal ZO-1 antibody R26.4C. As illustrated in Figure 2C, ZO-1 immunoreactive bands were detected in the lanes containing Sf9 whole cell lysate (WCL) and in lanes containing GST-Cx40CT, GST-Cx43CT, and GST-Cx45CT, but not in lanes containing GST alone, GST-Cx32, or GST-Cx43CT-I382. These data demonstrated that full length ZO-1 interacted with more than one connexin CT containing a PDZ binding domain that harbors a hydrophobic C-terminus, but did not interact with a connexin CT terminating with a non-hydrophobic amino acid.
Cx43Δ378–382 and Cx43ΔI382 do not Interact with ZO-1 In Vivo
To investigate the functional significance of the Cx43 interaction with ZO-1, two Cx43 deletion mutants, Cx43ΔI382 and Cx43Δ378–382, were expressed in the Madin-Darby canine kidney (MDCK) epithelial cell line. The MDCK cell line was chosen because the role of ZO-1 in the formation of tight junctions and adherens junctions has been well characterized in these cells (46, 47). In addition, an MDCK cell line lacking endogenous Cx43 was available (48), which made it possible to express the Cx43 mutants without interference by Cx43wt. The Cx43wt and the Cx43ΔI382 and Cx43Δ378–382 mutant genes were each cloned into the mammalian expression vector pcDNA3 and introduced into the parental MDCK cell line by transfection (see Materials and Methods). G418 resistant clones were isolated and analyzed for connexin expression by immunofluorescence microscopy. Two Cx43wt, two Cx43ΔI382, and one Cx43Δ378–382 expressing clones were independently isolated and used for further analysis. Coimmunoprecipitation studies were performed to determine whether disruption of the Cx43 PDZ2 binding domain would prevent the interaction of Cx43 with ZO-1 in the MDCK cells. ZO-1 immunoprecipitates showed the presence of Cx43wt by immunoblotting with Cx43 peptide antiserum (Figure 3, lanes 2 and 3), but the mutant Cx43 proteins were not detected in the immunoprecipitates (lanes 4, 5 and 6). These results confirmed that the interaction of full length Cx43 with full length ZO-1 required the C-terminal amino acids of Cx43, and in particular, the hydrophobic C-terminal I382.
Figure 3.
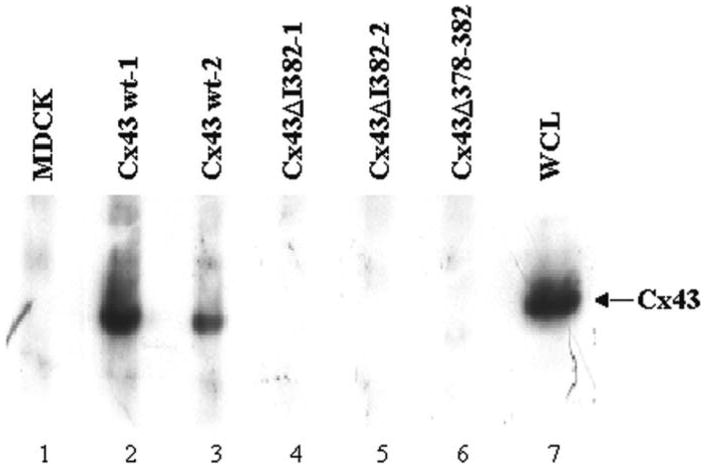
Interaction of full length Cx43 with full length ZO-1 in MDCK cells. ZO-1 binding proteins in MDCK whole cell lysates were immunoprecipitated with rabbit anti-ZO-1 antibody (Zymed) and separated on a 7.5% polyacrylamide gel. Proteins were transferred to an Immobilon P membrane and immunoblotted for Cx43. Whole cell lysate from Cx43wt-1 was used as a control for Cx43.
Cx43 Localizes to the Plasma Membrane Independent of its Interaction with ZO-1
A previous report suggested that the localization of Cx43 to the plasma membrane in HEK293 cells required an interaction with ZO-1 (25). To examine this requirement in MDCK cells, and to analyze the localization of Cx43wt, Cx43ΔI382, Cx43Δ378–382 and ZO-1 confocal and deconvolution immunofluorescence microscopy were employed. In all the clones observed, including the parental Cx43 negative cell line, the ZO-1 staining was present at the cell periphery, typical of ZO-1 associated with the tight junctions (Figure 4, ZO-1 panels). Cx43wt was observed at sites of cell-cell contact in a pattern that often followed the ZO-1 staining and in cytoplasmic regions. This cytoplasmic localization has been observed in other cell systems where Cx43 is exogenously expressed (Figure 4, Cx43 panels). Interestingly, the merged images revealed limited colocalization of Cx43wt and ZO-1 (yellow or orange color, see boxes containing enlarged areas) in very small plaques in the plasma membrane. Analysis using deconvolution microscopy confirmed that some of the Cx43wt was in the same plane as ZO-1 and that only a fraction of the Cx43 in the plasma membrane actually colocalized with ZO-1, and that this colocalization occurred primarily in very small plaques (see Supplemental Materials).
Figure 4.
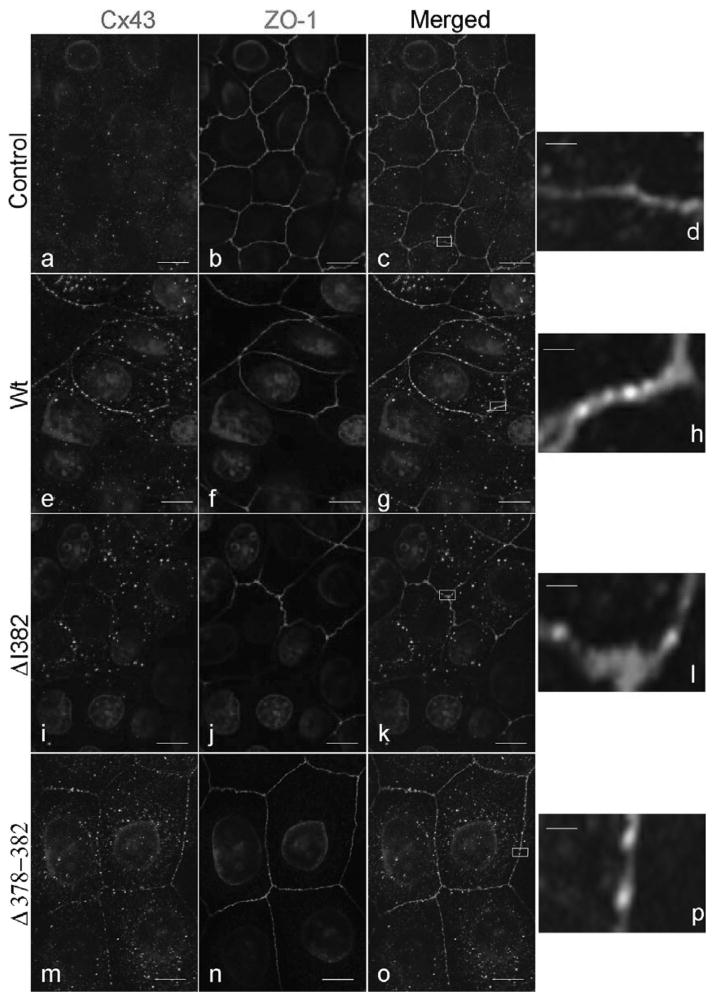
Subcellular localization of Cx43 and ZO-1 in MDCK cells. Untransfected MDCK cells lacking Cx43wt and MDCK cell clones expressing Cx43wt, Cx43ΔI382, or Cx43Δ378–382 were plated on glass coverslips, fixed in methanol, and stained for Cx43 using a rabbit polyclonal antibody (Cx43 panels, CT368 green) and costained for ZO-1 using a rat monoclonal antibody (ZO-1 panels, R26.4C red). Merged images of Cx43 and ZO-1 are shown in the panels at the far right (merged panels). Scale bars for these panels represent 3.2 μm. Significant areas of colocalization (orange or yellow color) are shown in the boxes in the merged panels. Enlargement of the boxed areas are shown in panels d, h, l, and p. Scale bars for the enlarged images represent 300 nm. (See Color Plate II at the end of this issue)
The localization of the Cx43Δ378–382 and Cx43ΔI382 (Figure 4, and Supplemental Materials) mutants was very similar to that of the Cx43wt expressing cells, although the Cx43ΔI382 showed overall a lower expression level, which indicated that these mutant connexins still were capable of translocating to the plasma membrane without the direct interaction with ZO-1. Interestingly, the confocal and deconvolution analysis showed that the mutant Cx43, which lacked the ability to bind to ZO-1 by directed yeast two-hybrid and coimmunoprecipitation studies, maintained a limited colocalization with ZO-1 in small gap junction plaques similarly to Cx43wt (Figure 4, merged panels k and o, and enlarged areas, panels l and p, and Supplemental Materials). Not all the cells in the Cx43ΔI382 clones appeared to express the mutant connexin, however, because expression was observed in nearly all the cells in the Cx43Δ378–382 clone we cannot attribute this difference in expression to loss of the ZO-1 interaction (data not shown). Taken together, these results indicate that Cx43 does not require an interaction with ZO-1 for its transport and localization to plasma membrane regions adjacent to the ZO-1 protein. In addition, it is clear from this study that in newly confluent MDCK cells the colocalization of Cx43 and ZO-1 in the plasma membrane is limited and is not greatly altered by the disruption of the direct interaction between the PDZ2 domain of ZO-1 and the C-terminal PDZ binding domain of Cx43.
The Cx43Δ378–382 and Cx43ΔI382 Mutants Form Functional Channels
Although the Cx43ΔI382 and Cx43Δ378–382 mutants were able to cluster into gap junction plaques, as observed by immunofluorescence, the loss of direct ZO-1 binding might affect the gating function of the channel. To test this possibility, cells expressing these mutants were examined for their ability to transfer dye. As shown in Table 2, the Cx43Δ378–382 and Cx43ΔI382 mutant expressing cell clones were capable of transferring LY dye to surrounding neighboring cells. Compared to the Cx43wt clones, the three mutant clones transferred dye to a slightly smaller number of neighboring cells. The average number of Cx43wt cells receiving LY was 14.8 while the average for the three mutant clones was 9.6. Because not all of the cells in the two Cx43ΔI382 clones expressed the Cx43-mutant proteins, some communicating cells would be adjacent to Cx43ΔI382 nonexpressing cells, and thus could not transfer the LY further, which would lower the average number of communicating cells. This was indirectly assessed by measuring the number of cells that communicated to at least one neighboring cell and reported as the incidence of coupling (IC) (Table 2). The IC value for the two Cx43ΔI382 clones was 88% and 63%. The Cx43Δ378–382 clone, lacking the five terminal residues, had an IC of 100% and communicated to a level similar to the Cx43wt cell clones. This suggested that the lower IC in the two Cx43ΔI382 clones was not due to the loss of the ZO-1 interaction, but more likely a consequence of not all the cells in the population expressing the Cx43ΔI382 protein. These data clearly demonstrated that the Cx43ΔI382 and Cx43Δ378–382 mutant proteins were not dependent on an interaction with ZO-1 to form functional gap junctional channels, as measured by the transfer of LY dye.
TABLE 2.
Measurement of gap junctional communication
| MDCK cell type | Average number of fluorescent cells | %ICa |
|---|---|---|
| Cx43 minus | 0 (n = 10) | 0 |
| Cx43wt (clone 1) | 14.7+/−3.7(n = 40) | 100 |
| Cx43wt (clone 2) | 14.9+/−2.1(n = 25) | 100 |
| Cx43ΔI382 (clone 1) | 6.3 +/− 2.3 (n = 45) | 88 |
| Cx43ΔI382 (clone 2) | 11.6+/−4.1 (n = 20) | 63 |
| Cx43Δ378–382 | 10.9+/−5.0 (n = 29) | 100 |
Gap junctional communication was measured by microinjecting Lucifer Yellow (LY) dye into a single cell. After 2 min the number of fluorescent neighboring cells was counted. Three or more plates of each cell type were injected. Only cells communicating to 1 or more cells were calculated in the average. n is the number of microinjected cells communicating to 1 or more neighboring cells.
IC (incidence of microinjected coupling) is the percent of cells that transferred LY dye to at least 1 or more neighboring cells.
Phosphorylation of Cx43 is Reduced in the Cx43Δ378–382 and Cx43ΔI382 Cell Clones
Phosphorylation of Cx43 is an important mechanism for regulating Cx43 processing and gating, and PDZ binding interactions are known to assemble components of intracellular signaling pathways. Therefore, we investigated the possibility that Cx43 phosphorylation might be affected by the lack of the intact Cx43 PDZ binding domain and the disrupted ZO-1 interaction in the cells expressing the Cx43 deletion mutants. 32P- and 35S-labeled Cx43 isoforms were immunoprecipitated from the Cx43wt, Cx43ΔI382, and Cx43Δ378–382 MDCK cell clones. Antibody directed against a peptide in the C-terminus of Cx43 that is upstream of the ZO-1 binding region (amino acids 241–255) was used in these immunoprecipitations. Four 35S-labeled protein bands were resolved on SDS-gels for Cx43wt, the fastest migrating band most likely corresponded to nonphosphorylated (NP) Cx43, while the three slower migrating forms represented the P1, P2, and P3 phosphoisoforms (Figure 5A, lanes 3 and 7). Interestingly, the 35S-labeled P2 isoform was reduced in the Cx43ΔI382 and Cx43Δ378–382 cell clones, as compared to the Cx43wt clones (Figure 5A, lanes 6, 10, and 11 compared to lanes 3 and 7). 32P-labeled Cx43 immunoprecipitated from the Cx43wt, Cx43ΔI382, and Cx43Δ378–382 cells did not reveal a NP band, confirming the nonphosphorylated nature of this isoform. Significantly, the Cx43ΔI382 and Cx43Δ378–382 mutants showed marked reductions in phosphorylation of most of the phosphoisoforms, particularly the P2 band, compared to Cx43wt (Figure 5A, compare lanes 5, 9, and 12 to lanes 4 and 8). A ratio of 1 (32P) : 3 (35S) cpm loaded was used for Cx43wt and the Cx43 deletion mutants (see Materials and Methods). In separate experiments where equivalent amounts of immunoprecipitated proteins were used, phosphorylation of the Cx43wt and the Cx43 deletion mutants was quantified by excising the 32P-labeled gel bands and measuring the radioactivity by liquid scintillation counting. The results from two independent experiments clearly indicated that the Cx43wt was more highly phosphorylated than either the Cx43ΔI382 or Cx43Δ378–382 mutant proteins (Figure 5B). In addition, the P2 isoform in the Cx43wt clones was 2-fold greater than the Cx43Δ378–382 protein. Analysis of each Cx43 phosphoisoform as a percentage of the total phosphoisoforms for each cell clone indicated that the P2 isoform in the Cx43wt cell clones was the predominant phosphoisoform, while P1 was the predominant phosphoisoform in the deletion mutants (Figure 5B). Experiments done by excising individual 35S-labeled Cx43 phosphoisoforms and plotting the band densities, showed a similar pattern (data not shown).
To exclude the possibility that the observed differences in phosphorylation of the mutant Cx43 were due to a nonspecific alteration in protein conformation induced by the deletions, we examined the phosphorylation of mutant Cx43 stably expressed in MDCK cells that were stimulated with either EGF or TPA. As shown in Figure 6A, EGF and TPA induced similar patterns of phosphorylation of the mutant Cx43 compared to the wt Cx43. Moreover, quantitation studies revealed that similar levels of phosphorylation were stimulated in the wt and mutant Cx43 proteins by EGF and TPA (Figure 6B). If anything, a somewhat greater increase in phosphorylation was observed with the Cx43 mutants treated with TPA, indicating that these mutants were readily targeted by activated protein kinases in vivo.
Figure 6.
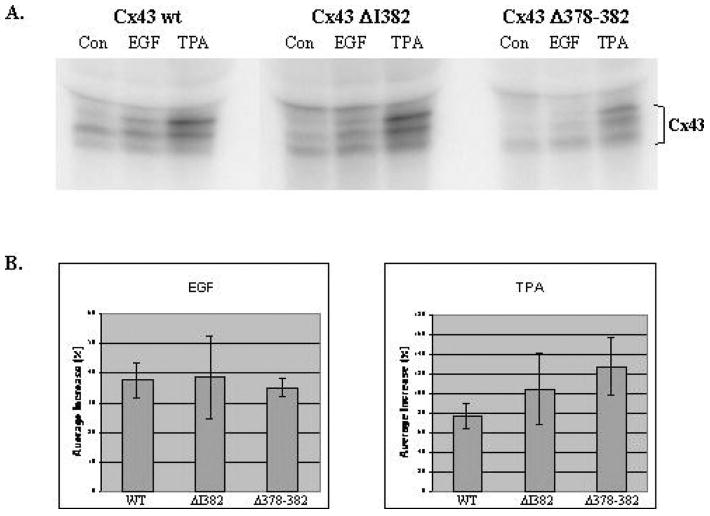
EGF and TPA stimulate similar levels and patterns of phosphorylation of wt and mutant Cx43. (A) MDCK cells, stably transfected with Cx43 wt, Cx43ΔI382, and Cx43Δ378–382, were plated, allowed to attach to dishes for 6 h, then serum-starved overnight in medium containing 0.5% FCS. Cells were labeled in phosphate- and serum-free medium containing 1 mCi/ml 32Pi for 2 h and treated with either EGF (100 ng/ml, USB) or TPA (100 ng/ml, Sigma) for the last 30 min. Cells were harvested, lysed, and immunoprecipitated with Cx43 CT241 polyclonal antibody. Immunoprecipitates were resolved by SDS-PAGE and the dried gel was autoradiographed to visualize wt Cx43 (exposed for 6 h) and the mutant Cx43 ΔI382 and Δ378–382 (exposed for 18 h). (B) Increases in phosphorylation of wt and mutant Cx43 stimulated by EGF or TPA relative to untreated controls were quantitated from the gel images of two experiments using the Quantity One software (Bio-Rad). Left panel: average increase in phosphorylation due to EGF treatment (wt Cx43–38%, Cx43ΔI382–39%, Cx43Δ378–382-35%). Right panel: average increase in phosphorylation due to TPA treatment (wt Cx43–78%, Cx43ΔI382–105%, Cx43Δ378–382-127%). Bars represent the standard deviation values.
Because not all of the cells transfected with Cx43ΔI382 expressed Cx43, it seemed plausible that the decreased phosphorylation of these Cx43 mutants might be the consequence of reduced docking with adjacent Cx43ΔI382 expressing cells. To test this possibility Cx43wt expressing cells were mixed with the parental Cx43 null cells in a 50:50 ratio to reduce the percentage of the cells that would come into contact with a Cx43 expressing cell. Analysis of the immunoprecipitated Cx43wt protein from this mix revealed a pattern of phosphoisoforms that was identical to the Cx43 isoforms obtained from the unmixed cells (data not shown). These results indicated that the differences observed in the phosphorylation were not likely due to differences in the numbers of Cx43 hemichannels that were docked. In addition, the Cx43Δ378–382 mutant clone, which had 100% incidence of coupling, also demonstrated decreased Cx43 phosphorylation. Taken together, these results clearly indicated that the phosphorylation of Cx43 was reduced in the Cx43ΔI382 and Cx43Δ378–382 mutants, which suggested that disruption of the Cx43 PDZ2 binding domain and the resultant inability of the mutant Cx43 proteins to interact with ZO-1 altered the phosphorylation of Cx43 in the MDCK cells.
Cx43 Turnover is not Altered by the Loss of Interaction with ZO-1
Activated c-Src was shown to disrupt the interaction of Cx43 with ZO-1 and to increase the rate of Cx43 turnover in HEK293 cells (49). To determine whether the direct disruption of the Cx43-ZO-1 interaction in MDCK cells would also affect Cx43 turnover in the absence of tyrosine phosphorylation on Cx43 induced by activated c-Src, pulse chase analysis was conducted with the Cx43wt and Cx43ΔI382 proteins. As shown in a representative pulse chase experiment (Figure 7A), no significant differences were observed in the turnover of the Cx43wt or Cx43ΔI382 proteins during a 3 h chase period. Quantitative analysis of Cx43wt and Cx43ΔI382 clones in two independent experiments confirmed that the degradation of Cx43ΔI382 was not significantly different from that of the Cx43wt protein [t½ = 2.4 h for wt and 1.9 h for ΔI382 (Figure 7B)]. Longer pulse-labeling and chase experiments were done, which also showed no significant differences in t½ values between the Cx43wt and the Cx43ΔI382 and Cx43Δ378–382 mutants (data not shown). These results indicated that disruption of the Cx43-ZO-1 interaction did not affect the turnover of newly translated Cx43 in MDCK cells. Furthermore, the pulse chase data demonstrated the appearance of the P2 phosphoisoform in the Cx43wt cells at 0.5 h of chase, and this phosphoisoform persisted throughout the 3 h chase (Figure 7A, left panel). In contrast, the P2 isoform of Cx43ΔI382 was not generated during this same time frame (Figure 7A, right panel). These data further supported the conclusion that Cx43 phosphorylation is altered by the loss of interaction with ZO-1.
Figure 7.
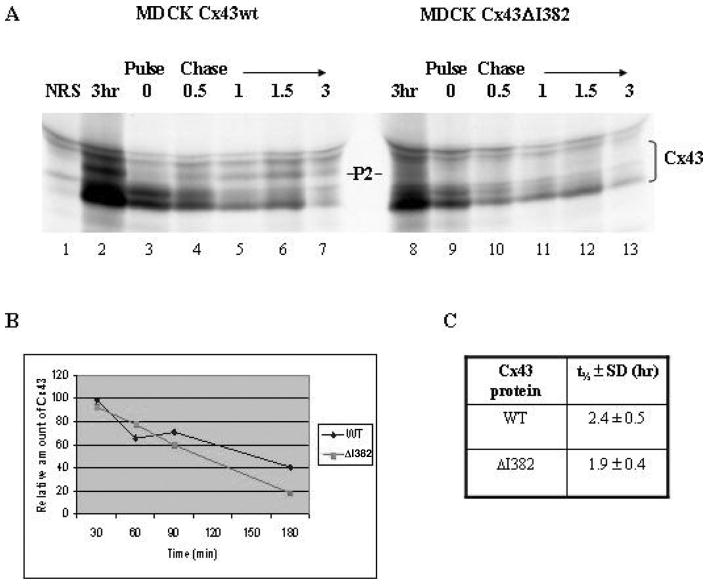
Analysis of the turnover of the Cx43wt and Cx43ΔI382 proteins. (A) MDCK clones stably transfected with either Cx43wt or Cx43ΔI382 were metabolically-labeled with (35S)-met/cys for 15 min and then chased for various lengths of time in nonradioactive, complete medium supplemented with 5 mM methionine. Cx43 was immunoprecipitated and resolved on a SDS polyacrylamide gradient gel. The gel was fluorographed and radiolabeled proteins were visualized by autoradiography using Kodak XOmat XAR-5 film. Lane 1, normal rabbit serum (NRS) immunoprecipitation control; lanes 2 and 8, Cx43 isoforms after a 3-h labeling; lanes 7 and 13, Cx43 isoforms after a 3 h chase. The P2 isoform of Cx43 is marked. (B) Graphical representation of averaged (n = 2) pulse chase band densities for Cx43wt and Cx43ΔI382 obtained from Quantity One software (Bio-Rad). (C) Bands densities were used to calculate Cx43 protein t½ values (±SD).
Discussion
Specific serine and tyrosine kinase phosphorylation sites and SH3 and SH2 interacting domains (50) in the C-terminal region of Cx43 indicate that this portion of Cx43 is a target for protein kinases regulating Cx43 function and turnover (19–24, 51, 52) and is engaged in dynamic protein-protein interactions (53). Using the Cx43 C-terminal domain in a yeast two-hybrid screen, it has been shown that the PDZ2 and flanking regions of ZO-1 interact with Cx43 (25, 39 and data presented here). In this study, we have characterized the Cx43 PDZ binding domain and analyzed the biochemical and functional consequences of disrupting this domain and abolishing the interaction of Cx43 with ZO-1.
PDZ domains are protein domains that interact primarily, although not exclusively, with recognition sequences in the carboxyl-termini of transmembrane proteins terminating with hydrophobic amino acids, usually valine or isoleucine (54). Three classes of PDZ domains have been characterized, Class I, Class II, and Class III. In Class I PDZ domains, the −2 residue is particularly important and requires a serine or threonine residue, while a hydrophobic valine or leucine is typically present in the 0 position. Class II PDZ binding domains have no particular amino acid preference at the −2 position, but require a hydrophobic residue at the 0 and −2 positions. Characterization of the Cx43 PDZ binding motif using the directed two-hybrid assay with Cx43 CT point mutations indicated that the 0, −2 and −3 positions, which correspond to I382, L380 and D379 of Cx43, were required for the in vitro interaction with the ZO-1 PDZ2 domain. These results suggest that Cx43 interacts with the PDZ2 domain of ZO-1 through a typical Class II PDZ binding motif. Furthermore, minimally mutated Cx43 deletion mutants with disrupted PDZ binding regions, Cx43ΔI382 and Cx43Δ378–382, failed to interact with ZO-1 when expressed in Cx43 deficient MDCK cells. These results confirmed that the Cx43 Class II PDZ binding domain was responsible for the in vivo interaction with ZO-1. The requirement for D379, at the −3 position, for the Cx43 PDZ interaction is consistent with structural studies that indicate that the amino acid at the −3 position makes contact with specific residues in the PDZ domain.
Cx32 has previously been shown to colocalize with ZO-1 and to associate with ZO-1 in coimmunoprecipitation experiments using primary rat hepatocytes (55). Results from this study indicate that the basis for the interaction of Cx32 with ZO-1 differs from that of the Cx43, Cx40 and Cx45 interactions in that it does not involve a PDZ binding domain at the Cx32 C-terminus. Cx46, Cx50, and Cx31.9 demonstrate the same PDZ binding motif as Cx43 (D-L-X-I) and have also been shown to interact with ZO-1 (44, 56), further supporting the Class II PDZ binding motif in the interaction of these connexins with ZO-1. Although this consensus sequence is consistent with binding to other PDZ2 domains, Cx43CT did not bind PDZ1 or PDZ3 in two-hybrid assays (data not shown).
It has been reported that phosphorylation of Tyr265 in Cx43 by activated c-Src inhibited the interaction of Cx43 and ZO-1 in cardiomyocytes and increased Cx43 turnover (49). However, in the current study, disruption of the Cx43-ZO-1 interaction had no apparent effect on Cx43 degradation. This result is consistent with other studies using Cx43 knockout mouse fibroblasts induced to express Cx43 and the v-Src kinase, or v-Src-transformed fibroblasts (24, 38). In these cells, a loss of Cx43 from gap junction plaques was not observed, even though the interaction of Cx43 with ZO-1 would have presumably been disrupted by the phosphorylation of Cx43 on Tyr265. Taken together these results suggest that disruption of the Cx43-ZO-1 interaction is either not sufficient to affect Cx43 turnover, or that upregulation of Cx43 turnover through the disruption of the Cx43-ZO-1 interaction is cell type specific.
PDZ domain interactions may provide the means for targeting proteins to or maintaining proteins at the plasma membrane (57). The significance of the Cx43-ZO-1 interaction for the localization of connexins to the plasma membrane has not been clear. The Toyofuku et al. (25) study suggested that an interaction with ZO-1 was required for the transfer of Cx43 from the cytoplasm to the intercalated discs in cardiomyocytes. However, earlier studies demonstrated that a truncated Cx43 mutant lacking the C-terminal domain (aa 258–382) localized to the plasma membrane despite a lack of the C-terminus, including the ZO-1 PDZ2 binding domain (58). In our study in MDCK cells, where ZO-1 is associated with tight junctions, the Cx43ΔI382 and Cx43Δ378–382 mutants formed functional gap junction plaques that were located at the plasma membrane, as assessed by LY dye transfer and confocal microscopy. Taken together, these data suggest that the localization of Cx43 to the plasma membrane is not dependent on an interaction with ZO-1 in a number of cell types.
Our confocal microscopy studies further indicated that in the MDCK cells Cx43wt and ZO-1 demonstrated limited colocalization that was observed primarily in small gap junction plaques. A similar limited colocalization of Cx43 and ZO-1 was also observed within intercalated discs of the myocardium (59). However, in this study of Barker et al., the limited colocalization of Cx43 and ZO-1 was greatly increased after enzymatic dissociation of the myocytes from the intact ventricle, and was associated with changes in Cx43 phosphorylation (59). The limited colocalization of Cx43 and ZO-1 to small gap junction plaques in the MDCK cells may indicate that the Cx43-ZO-1 interaction is a transient interaction occurring in newly-formed plaques, that the interaction is cell type or tissue type specific, or that a more significant interaction may occur under conditions that were not examined in this study.
Furthermore, our confocal and deconvolution microscopy analysis revealed that loss of the PDZ2 binding domain at the C-terminus of Cx43 did not alter the subcellular localization of Cx43, or the ability of Cx43 and ZO-1 to colocalize. The deconvolution microscopy studies showed no obvious basal-apical polarity differences in the localization of wild type or mutant Cx43. These observations indicated that the subtle positioning of Cx43 at the plasma membrane was not dependent upon the Cx43-ZO-1 interaction.
The apparent colocalization of the Cx43 PDZ2-domain binding mutants with ZO-1 may seem paradoxical given our biochemical data indicating their lack of interaction. However, these colocalization data should be considered in light of the known resolution of this technique, which is around 200–300 nm. Employing higher resolution techniques, such as immuno-EM or FRET, would be required to confirm that the observed colocalization of the Cx43 PDZ2 domain mutants with ZO-1 is an indication of a direct interaction rather than close positioning of the two proteins. This apparent similar colocalization and the recognition of the mutants by the C-terminal peptide antibody (aa 368–382) suggests that the minimal mutation of the PDZ2 binding domain of Cx43 did not grossly perturb Cx43 structure or the spatial positioning of Cx43 or ZO-1 in the plasma membrane.
Proteins containing PDZ domains are believed to serve as molecular scaffolds that promote assembly of multiprotein signaling complexes (57, 60). Cx43 has also been proposed to function as a scaffold protein to assemble a complex or a “nexus” of intracellular proteins (61). One of the major mechanisms for signal transduction is through protein phosphorylation events. Cx43 is a phosphoprotein and serine phosphorylation has been implicated in its transport, processing and gating (16, 18). The minimal mutations of the Cx43 PDZ binding domain, which led to the disruption of its direct binding interaction with ZO-1, was sufficient to reduce the level of serine phosphorylation of Cx43, as determined by phosphoamino acid analysis (data not shown). The reduced levels of phosphorylation in the Cx43ΔI382 and Cx43Δ378–382 mutants was particularly apparent in the decreased levels of the P2 isoform, when compared to the Cx43wt protein. Control experiments showed that the Cx43ΔI382 and Cx43Δ378–382 mutants exhibited similar levels and patterns of phosphorylation as wt Cx43 following stimulation of cells with either EGF or TPA, which indicated that the phosphorylation differences did not result from nonspecific changes in conformation induced by the deletions. Moreover, this conclusion is substantiated by the observations that: (1) the engineered deletions are minimal (especially Cx43ΔI382); (2) peptide antibody against the C-terminal domain of wt Cx43 still recognizes the deletion mutants; and (3) the Cx43 deletion mutants not only localize to the plasma membrane, but create functional gap junction channels.
Phosphorylation to the P2 isoform was originally associated with insertion of Cx43 into the plasma membrane and formation of functional channels (62). Additional studies have suggested that Cx43 in NRK cells is transported to the membrane mainly in the nonphosphorylated form and then converted at the plasma membrane to a Triton-insoluble, more highly phosphorylated P2 isoform. This P2 isoform was not exclusively associated with communication competent cells, but was observed in uncoupled cells as well (63). However, in the NRK cells studies, the P2 isoform was linked to the accumulation of connexin 43 into gap junctional plaques (63). The decrease in the P2 isoform in Cx43 ZO-1 binding mutants in MDCK cells also was not associated with membrane insertion or altered function in our studies. In addition, we did not observe any obvious differences in gap junctional plaques in the Cx43 ZO-1 binding mutants.
This novel observation of diminished Cx43 phosphorylation in the Cx43 PDZ binding mutants, suggests that the interaction between Cx43wt and ZO-1 either mediates the association of Cx43 with a protein kinase, or perhaps protects Cx43 from a phosphatase in the gap junctional plaque. The SH3 domain of ZO-1 is known to interact with a serine protein kinase that phosphorylates ZO-1 (64). It is plausible that the altered phosphorylation of the connexin protein in the gap junctional plaque may affect its interactions with other localized proteins, and thus affect an as yet uncharacterized connexin-mediated signaling process—a scenario consistent with the molecular scaffolding functions of ZO-1 and Cx43.
Supplementary Material
Acknowledgments
We thank S. Hollenberg for generously providing the two-hybrid plasmids, D. Laird for the MDCK cells, J. Dixon for the pGEX-KG vector, S. Tsukita for the ZO-1 cDNA, and Megan Brown for technical advice. We also thank M. Woods for help with the confocal and deconvolution microscopy. This research was supported in part by Grant CA052098 from the National Cancer Institute, NIH (to A. F. L.) and Predoctoral Fellowship (HIFW 029-96) from the American Heart Association, Hawaii Affiliate (to C. J.).
References
- 1.Loewenstein WR. Junctional intercellular communication: The cell-to-cell membrane channel. Physiol Rev. 1981;61:829–913. doi: 10.1152/physrev.1981.61.4.829. [DOI] [PubMed] [Google Scholar]
- 2.Kumar NM, Gilula NB. The gap junction communication channel. Cell. 1996;84:381–388. doi: 10.1016/s0092-8674(00)81282-9. [DOI] [PubMed] [Google Scholar]
- 3.White TW, Bruzzone R. Multiple connexin proteins in single intercellular channels: Connexin compatibility and functional consequences. J Bioenerg Biomembr. 1996;28:339–350. doi: 10.1007/BF02110110. [DOI] [PubMed] [Google Scholar]
- 4.Veenstra RD. Size and selectivity of gap junction channels formed from different connexins. J Bioenerg Biomembr. 1996;28:327–337. doi: 10.1007/BF02110109. [DOI] [PubMed] [Google Scholar]
- 5.Spray DC, Burt JM. Structure-activity relations of the cardiac gap junction channel. Am J Physiol. 1990;258:C195–C205. doi: 10.1152/ajpcell.1990.258.2.C195. [DOI] [PubMed] [Google Scholar]
- 6.Brink PR, Cronin K, Ramanan SV. Gap junctions in excitable cells. J Bioenerg Biomembr. 1996;28:351–358. doi: 10.1007/BF02110111. [DOI] [PubMed] [Google Scholar]
- 7.Meda P. The role of gap junction membrane channels in secretion and hormonal action. J Bioenerg Biomembr. 1996;28:369–377. doi: 10.1007/BF02110113. [DOI] [PubMed] [Google Scholar]
- 8.Reaume AG, de Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, Juneja SC, Kidder GM, Rossant J. Cardiac malformation in neonatal mice lacking connexin 43. Science. 1995;267:1831–1834. doi: 10.1126/science.7892609. [DOI] [PubMed] [Google Scholar]
- 9.Lo CW. The role of gap junction membrane channels in development. J Bioenerg Biomembr. 1996;28:379–385. doi: 10.1007/BF02110114. [DOI] [PubMed] [Google Scholar]
- 10.Yamasaki H, Naus CC. Role of connexin genes in growth control. Carcinogenesis. 1996;17:1199–1213. doi: 10.1093/carcin/17.6.1199. [DOI] [PubMed] [Google Scholar]
- 11.Loewenstein WR, Rose B. The cell-cell channel in the control of growth. Semin Cell Biol. 1992;3:59–79. doi: 10.1016/s1043-4682(10)80008-x. [DOI] [PubMed] [Google Scholar]
- 12.Musil LS, Le AC, VanSlyke JK, Roberts LM. Regulation of connexin degradation as a mechanism to increase gap junction assembly and function. J Biol Chem. 2000;275:25207–25215. doi: 10.1074/jbc.275.33.25207. [DOI] [PubMed] [Google Scholar]
- 13.Rose B, Loewenstein WR. Permeability of cell junction depends on local cytoplasmic calcium activity. Nature. 1975;254:350–352. doi: 10.1038/254250a0. [DOI] [PubMed] [Google Scholar]
- 14.Ek-Vitorin JF, Calero G, Morley GE, Coombs W, Taffet SM, Delmar M. PH regulation of connexin43: Molecular analysis of the gating particle. Biophys J. 1996;71:1273–1284. doi: 10.1016/S0006-3495(96)79328-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennett MV, Verselis VK. Biophysics of gap junctions. Semin Cell Biol. 1992;3:29–47. doi: 10.1016/s1043-4682(10)80006-6. [DOI] [PubMed] [Google Scholar]
- 16.Lampe PD, Lau AF. Regulation of gap junctions by phosphorylation of connexins. Arch Biochem Biophys. 2000;384:205–215. doi: 10.1006/abbi.2000.2131. [DOI] [PubMed] [Google Scholar]
- 17.Goodenough DA, Goliger JA, Paul DL. Connexins, connexons, and intercellular communication. Annu Rev Biochem. 1996;65:475–502. doi: 10.1146/annurev.bi.65.070196.002355. [DOI] [PubMed] [Google Scholar]
- 18.Musil LS. Structure and assembly of gap junctions. In: Citi S, editor. Molecular mechanisms of epithelial cell junctions: From development to disease. RG Landes Co.; 1994. pp. 173–194. [Google Scholar]
- 19.Xie H, Laird DW, Chang TH, Hu VW. A mitosis-specific phosphorylation of the gap junction protein connexin43 in human vascular cells: Biochemical characterization and localization. J Cell Biol. 1997;137:203–210. doi: 10.1083/jcb.137.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanemitsu MY, Jiang W, Eckhart W. Cdc2-mediated phosphorylation of the gap junction protein, connexin43, during mitosis. Cell Growth Differ. 1998;9:13–21. [PubMed] [Google Scholar]
- 21.Lampe PD, Kurata WE, Warn-Cramer BJ, Lau AF. Formation of a distinct connexin43 phosphoisoform in mitotic cells is dependent upon p34cdc2 kinase. J Cell Sci. 1998;111:833–841. doi: 10.1242/jcs.111.6.833. [DOI] [PubMed] [Google Scholar]
- 22.Warn-Cramer BJ, Cottrell GT, Burt JM, Lau AF. Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J Biol Chem. 1998;273:9188–9196. doi: 10.1074/jbc.273.15.9188. [DOI] [PubMed] [Google Scholar]
- 23.Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol. 2000;149:1503–1512. doi: 10.1083/jcb.149.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin R, Warn-Cramer BJ, Kurata WE, Lau AF. v-Src phosphorylation of connexin43 on Tyr247 and Tyr265 disrupts gap junctional communication. J Cell Biol. 2001;154:815–827. doi: 10.1083/jcb.200102027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toyofuku T, Yabuki M, Otsu K, Kuzuya T, Hori M, Tada M. Direct association of the gap junction protein connexin43 with ZO-1 in cardiac myocytes. J Biol Chem. 1998;273:12725–12731. doi: 10.1074/jbc.273.21.12725. [DOI] [PubMed] [Google Scholar]
- 26.Giepmans BN, Moolenaar WH. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr Biol. 1998;8:931–934. doi: 10.1016/s0960-9822(07)00375-2. [DOI] [PubMed] [Google Scholar]
- 27.Laing JG, Manley-Markowski RN, Koval M, Civitelli R, Steinberg TH. Connexin45 interacts with zonula occludens-1 and connexin43 in osteoblastic cells. J Biol Chem. 2001;276:23051–23055. doi: 10.1074/jbc.M100303200. [DOI] [PubMed] [Google Scholar]
- 28.Beyer EC, Paul DL, Goodenough DA. Connexin43: A protein from rat heart homologous to a gap junction protein from liver. J Cell Biol. 1987;105:2621–2629. doi: 10.1083/jcb.105.6.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vojtek AB, Hollenberg SM, Cooper JA. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell. 1993;74:205–214. doi: 10.1016/0092-8674(93)90307-c. [DOI] [PubMed] [Google Scholar]
- 30.Hennemann H, Kozjek G, Dahl E, Nicholson B, Willecke K. Molecular cloning of mouse connexins26 and -32: Similar genomic organization but distinct promoter sequences of two gap junction genes. Eur J Cell Biol. 1992;58:81–89. [PubMed] [Google Scholar]
- 31.Haefliger JA, Bruzzone R, Jenkins NA, Gilbert DJ, Copeland NG, Paul DL. Four novel members of the connexin family of gap junction proteins. Molecular cloning, expression, and chromosome mapping. J Biol Chem. 1992;267:2057–2064. [PubMed] [Google Scholar]
- 32.Hennemann H, Schwarz HJ, Willecke K. Characterization of gap junction genes expressed in F9 embryonic carcinoma cells: Molecular cloning of mouse connexin31 and -45 cDNAs. Eur J Cell Biol. 1992;57:51–58. [PubMed] [Google Scholar]
- 33.Rose MD, Winston F, Heiter P. Laboratory course manual for methods in yeast genetics. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1990. [Google Scholar]
- 34.Zakian VA, Scott JF. Construction, replication, and chromatin structure of TRP1 RI circle, A multiple-copy synthetic plasmid derived from Saccharomyces cerevisiae chromosomal DNA. Mol Cell Biol. 1982;2:221–232. doi: 10.1128/mcb.2.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jin C, Lau AF, Martyn KD. Identification of connexin-interacting proteins: Application of the yeast two-hybrid screen. Methods. 2000;20:219–231. doi: 10.1006/meth.1999.0939. [DOI] [PubMed] [Google Scholar]
- 36.Loo LW, Berestecky JM, Kanemitsu MY, Lau AF. pp60src-mediated phosphorylation of connexin 43, a gap junction protein. J Biol Chem. 1995;270:12751–12761. doi: 10.1074/jbc.270.21.12751. [DOI] [PubMed] [Google Scholar]
- 37.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 38.Crow DS, Beyer EC, Paul DL, Kobe SS, Lau AF. Phosphorylation of connexin43 gap junction protein in uninfected and Rous sarcoma virus-transformed mammalian fibroblasts. Mol Cell Biol. 1990;10:1754–1763. doi: 10.1128/mcb.10.4.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giepmans BN, Verlaan I, Hengeveld T, Janssen H, Calafat J, Falk MM, Moolenaar WH. Gap junction protein connexin43 interacts directly with microtubules. Curr Biol. 2001;11:1364–1368. doi: 10.1016/s0960-9822(01)00424-9. [DOI] [PubMed] [Google Scholar]
- 40.Cowburn D. Peptide recognition by PTB and PDZ domains. Curr Opin Struct Biol. 1997;7:835–838. doi: 10.1016/s0959-440x(97)80155-8. [DOI] [PubMed] [Google Scholar]
- 41.Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science. 1997;275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 42.Saras J, Heldin CH. PDZ domains bind carboxy-terminal sequences of target proteins. Trends Biochem Sci. 1996;21:455–458. doi: 10.1016/s0968-0004(96)30044-3. [DOI] [PubMed] [Google Scholar]
- 43.Bruzzone R, White TW, Paul DL. Connections with connexins: The molecular basis of direct intercellular signaling. Eur J Biochem. 1996;238:1–27. doi: 10.1111/j.1432-1033.1996.0001q.x. [DOI] [PubMed] [Google Scholar]
- 44.Nielsen PA, Baruch A, Shestopalov VI, Giepmans BN, Dunia I, Benedetti EL, Kumar NM. Lens connexins alpha3Cx46 and alpha8Cx50 interact with zonula occludens protein-1 (ZO-1) Mol Biol Cell. 2003;14:2470–2481. doi: 10.1091/mbc.E02-10-0637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kausalya PJ, Reichert M, Hunziker W. Connexin45 directly binds to ZO-1 and localizes to the tight junction region in epithelial MDCK cells. FEBS Lett. 2001;505:92–96. doi: 10.1016/s0014-5793(01)02786-7. [DOI] [PubMed] [Google Scholar]
- 46.Anderson JM, Stevenson BR, Jesaitis LA, Goodenough DA, Mooseker MS. Characterization of ZO-1, A protein component of the tight junction from mouse liver and Madin-Darby canine kidney cells. J Cell Biol. 1988;106:1141–1149. doi: 10.1083/jcb.106.4.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajasekaran AK, Hojo M, Huima T, Rodriguez-Boulan E. Catenins and zonula occludens-1 form a complex during early stages in the assembly of tight junctions. J Cell Biol. 1996;132:451–463. doi: 10.1083/jcb.132.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jordan K, Solan JL, Dominguez M, Sia M, Hand A, Lampe PD, Laird DW. Trafficking, assembly, and function of a connexin43-green fluorescent protein chimera in live mammalian cells. Mol Biol Cell. 1999;10:2033–2050. doi: 10.1091/mbc.10.6.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Toyofuku T, Akamatsu Y, Zhang H, Kuzuya T, Tada M, Hori M. c-Src regulates the interaction between connexin43 and ZO-1 in cardiac myocytes. J Biol Chem. 2001;276:1780–1788. doi: 10.1074/jbc.M005826200. [DOI] [PubMed] [Google Scholar]
- 50.Kanemitsu MY, Loo LW, Simon S, Lau AF, Eckhart W. Tyrosine phosphorylation of connexin 43 by v-Src is mediated by SH2 and SH3 domain interactions. J Biol Chem. 1997;272:22824–22831. doi: 10.1074/jbc.272.36.22824. [DOI] [PubMed] [Google Scholar]
- 51.Lau AF, Kurata WE, Kanemitsu MY, Loo LW, Warn-Cramer BJ, Eckhart W, Lampe PD. Regulation of connexin43 function by activated tyrosine protein kinases. J Bioenerg Biomembr. 1996;28:359–368. doi: 10.1007/BF02110112. [DOI] [PubMed] [Google Scholar]
- 52.Leithe E, Rivedal E. Epidermal growth factor regulates ubiquitination, internalization and proteasome-dependent degradation of connexin43. J Cell Sci. 2004;117:1211–1220. doi: 10.1242/jcs.00951. [DOI] [PubMed] [Google Scholar]
- 53.Giepmans BNG. Gap junctions and connexin-interacting proteins. Cardiovasc Res. 2004;62:233–245. doi: 10.1016/j.cardiores.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 54.Sheng M, Sala C. PDZ domains and the organization of supramolecular complexes. Annu Rev Neurosci. 2001;24:1–29. doi: 10.1146/annurev.neuro.24.1.1. [DOI] [PubMed] [Google Scholar]
- 55.Kojima T, Kokai Y, Chiba H, Yamamoto M, Mochizuki Y, Sawada N. Cx32 but not Cx26 is associated with tight junctions in primary cultures of rat hepatocytes. Exp Cell Res. 2001;263:193–201. doi: 10.1006/excr.2000.5103. [DOI] [PubMed] [Google Scholar]
- 56.Nielsen PA, Beahm DL, Giepmans BN, Baruch A, Hall JE, Kumar NM. Molecular cloning, functional expression, and tissue distribution of a novel human gap junction-forming protein, connexin-31.9. Interaction with zona occludens protein-1. J Biol Chem. 2002;277:38272–38283. doi: 10.1074/jbc.M205348200. [DOI] [PubMed] [Google Scholar]
- 57.Fanning AS, Anderson JM. PDZ domains: Fundamental building blocks in the organization of protein complexes at the plasma membrane. J Clin Invest. 1999;103:767–772. doi: 10.1172/JCI6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dunham B, Liu S, Taffet S, Trabka-Janik E, Delmar M, Petryshyn R, Zheng S, Perzova R, Vallano ML. Immunolocalization and expression of functional and nonfunctional cell-to-cell channels from wild-type and mutant rat heart connexin43 cDNA. Circ Res. 1992;70:1233–1243. doi: 10.1161/01.res.70.6.1233. [DOI] [PubMed] [Google Scholar]
- 59.Barker RJ, Price RL, Gourdie RG. Increased association of ZO-1 with connexin43 during remodeling of cardiac gap junctions. Circ Res. 2002;90:317–324. doi: 10.1161/hh0302.104471. [DOI] [PubMed] [Google Scholar]
- 60.Gonzalez-Mariscal L, Betanzos A, Avila-Flores A. MAGUK proteins: Structure and role in the tight junction. Semin Cell Dev Biol. 2000;11:315–324. doi: 10.1006/scdb.2000.0178. [DOI] [PubMed] [Google Scholar]
- 61.Duffy HS, Delmar M, Spray DC. Formation of the gap junction nexus: Binding partners for connexins. J Physiol Paris. 2002;96:243–249. doi: 10.1016/s0928-4257(02)00012-8. [DOI] [PubMed] [Google Scholar]
- 62.Musil LS, Cunningham BA, Edelman GM, Goodenough DA. Differential phosphorylation of the gap junction protein connexin43 in junctional communication-competent and -deficient cell lines. J Cell Biol. 1990;111:2077–2088. doi: 10.1083/jcb.111.5.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol. 1991;115:1357–1374. doi: 10.1083/jcb.115.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Balda MS, Anderson JM, Matter K. The SH3 domain of the tight junction protein ZO-1 binds to a serine protein kinase that phosphorylates a region C-terminal to this domain. FEBS Lett. 1996;399:326–332. doi: 10.1016/s0014-5793(96)01352-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.