

Abstract
The Myc oncogene regulates the expression of multiple components of the protein synthetic machinery, including ribosomal proteins, initiation factors of translation, Pol III, and rDNA1,2. An outstanding question is whether and how increasing the cellular protein synthesis capacity can affect the multi-step process leading to cancer. We utilized ribosomal protein heterozygote mice as a genetic tool to restore increased protein synthesis in Eμ–Myc/+ transgenic mice to normal levels and show that in this context Myc's oncogenic potential is suppressed. Our findings demonstrate that the ability of Myc to increase protein synthesis directly augments cell size and is sufficient to accelerate cell cycle progression independently of known cell cycle targets transcriptionally regulated by Myc. In addition, when protein synthesis is restored to normal levels, Myc overexpressing precancerous cells are more efficiently eliminated by programmed cell death. Our findings reveal a novel paradigm that links increases in general protein synthesis rates downstream of an oncogenic signal to a specific molecular impairment in the modality of translation initiation employed to regulate the expression of selective mRNAs. We show that an aberrant increase in cap-dependent translation downstream Myc hyperactivation specifically impairs the translational switch to internal ribosomal entry site (IRES)-dependent translation required for accurate mitotic progression. Failure of this translational switch results in reduced mitotic-specific expression of the endogenous IRES-dependent form of Cdk11 (p58-PITSLRE)3-5, which leads to cytokinesis defects and is associated with increased centrosome numbers and genome instability in Eμ–Myc/+ mice. When accurate translational control is re-established in Eμ–Myc/+ mice, genome instability is suppressed. Our findings reveal how perturbations in translational control provide a highly specific outcome on gene expression, genome stability, and cancer initiation that have important implications for understanding the molecular mechanism of cancer formation at the post-genomic level.
Deregulation of Myc activity is one of the most frequent oncogenic lesions underlying human cancers6,7. Myc plays an evolutionarily conserved role in control of cell size and protein synthesis rates, which in Drosophila confers a cell survival advantage1,8,9. When dMyc induced protein synthesis is restrained in a minute mutant background, haploinsufficient for ribosomal protein function, cells no longer possess a competitive advantage10,11. To date, the relevance of Myc-dependent increases in protein synthesis and cell growth in the multi-step process leading to cancer remain unknown.
To restore protein synthesis rates in Myc overexpressing cells to normal, we employed mouse minute mutants, haploinsufficient for ribosomal protein function. Haploinsufficiency in certain ribosomal proteins decreases overall protein synthesis rates to an extent that is compatible with overall cellular and tissue homeostasis. L24+/− mice are viable12 and do not display any overt differences in B-lymphocyte development, growth, and cell division (Supplementary Fig. 1 and Fig. 1). We intercrossed Myc transgenic mice, in which Myc is overexpressed in the B-cell compartment (Eμ–Myc/+)13, with L24+/− mice (Supplementary Fig. 2). By lowering the threshold of protein production in L24+/− mice, the increased protein synthesis rates and cell size in Eμ–Myc/+ cells14 were restored to normal levels in Eμ–Myc/+;L24+/− mice (Fig. 1a,b). Therefore, this genetic approach reveals that Myc-induced increases in general protein synthesis rates are responsible for augmented cell growth. Moreover, Eμ–Myc/+;L24+/−mice are an important genetic model for selectively rescuing increased protein synthesis rates and cell growth downstream of oncogenic Myc signaling.
Figure 1. Myc-induced increases in protein synthesis regulate B lymphocyte size, division and apoptosis prior lymphomagenesis.
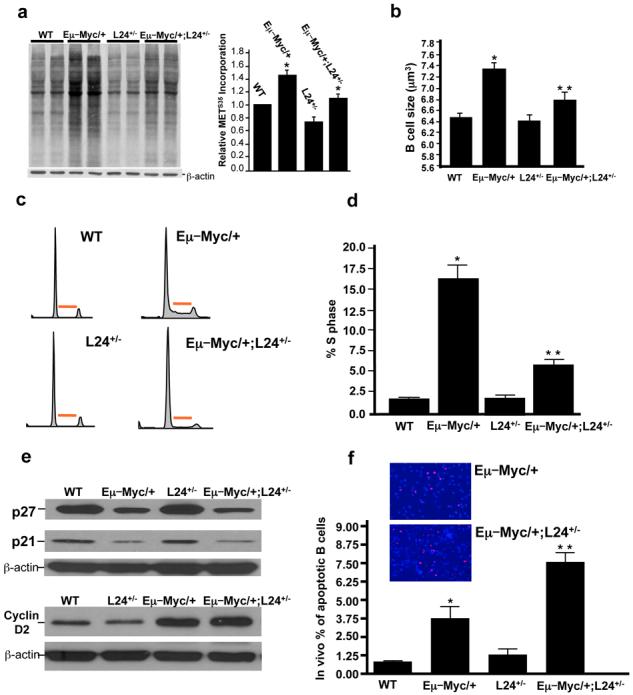
a, Protein synthesis rates assessed by S35 methionine incorporation and densitometry analysis (n=3) * p < 0.01. b, Cell size analysis (n=3) * p < 0.001 for Eμ–Myc/+ versus WT, ** p < 0.01 for Eμ–Myc/+ versus Eμ–Myc/+;L24+/−. c, d, Cell cycle distribution and quantification of the percentage of cells in S phase (n=3). Red bar indicates S-phase, * p < 0.01, ** p < 0.05. e, Western blot analysis for cell cycle targets transcriptionally regulated by Myc f, In situ Tunnel analysis (n=3). Inserts are representative pictures of Tunnel analysis comparing Eμ–Myc/+ and Eμ–Myc/+;L24+/− samples * p < 0.05, ** p < 0.05. a-e, all experiments were performed on freshly isolated B-lymphocytes. Error bars, s.d.
During cell cycle progression, cell growth normally precedes cell division and it has been suggested that cells must reach a critical cell size or “setpoint” in order to facilitate G1-S progression15. In mammalian cells, it remains undetermined whether an increase in cell growth is coupled to an increase in cell division following Myc activation. The percentage of Eμ–Myc/+ cells in S phase is markedly increased compared to WT cells (Fig. 1c,d). Strikingly, in Eμ–Myc/+;L24+/− mice the augmented number of cells in S phase is restored to normal levels (Fig. 1 c,d). The rate of cell cycle progression in Eμ–Myc/+ B-lymphocytes was also monitored by BrdU incorporation, [WT 0.16±0.04 vs Myc 5.77±1.78 BrdU+cells/hour, P<0.005] and was similarly restored to normal levels in Eμ–Myc/+;L24+/− cells [0.11±0.02 BrdU+cells/hour, P<0.02]. Key cell cycle targets that are transcriptionally regulated by Myc such as p2716, p217, and Cyclin D218, were expressed at similar levels when Eμ–Myc/+ and Eμ–Myc/+;L24+/− cells were compared (Fig. 1e and Supplementary Fig. 3). Thereby, the overall protein synthetic capacity of the cell may dictate cell cycle progression independently from the cell cycle program established at the transcriptional level by Myc hyperactivation. These results strongly suggest that Myc-induced cell growth is dependent on Myc's ability to regulate protein synthesis and is coupled to uncontrolled cell cycle progression in cancer.
Non-immortalized cells counteract Myc overexpression by undergoing programmed cell death as a tumor suppressive response, and inhibition of cell death downstream of Myc activation accelerates lymphoma initiation7. In Eμ–Myc/+;L24+/− mice the percentage of dying cells was more than double that of Eμ–Myc/+ mice prior to lymphoma onset (Fig. 1f). Therefore, the tumor suppressive response elicited by Myc overexpression is strongly enhanced in the background of normal protein synthesis and these findings suggest that clonal derivatives of precancerous cells may be more efficiently eliminated by programmed cell death in Eμ–Myc/+;L24+/− mice.
The restoration of normal protein synthesis downstream of Myc in Eμ–Myc/+;L24+/− mice is first associated with a dramatic reduction in splenomegaly, the earliest manifestation of Myc-induced lymphomagenesis (Fig. 2a). We next scored for lymphoma formation in each cohort of mice (Fig. 2b). The onset of lymphomas in the Eμ–Myc/+;L24+/− mice is dramatically delayed compared to Eμ–Myc/+ mice (Fig. 2b). In addition, a significant percentage of Eμ-Myc/+;L24+/− mice do not develop lymphomas even after 1.5 years of age (Fig. 2b). A second mouse minute line, heterozygous for the ribosomal protein L38 (manuscript in preparation, Barna Lab), was intercrossed with Eμ–Myc/+ mice. Eμ–Myc/+;L38+/− mice also display a marked rescue in cell growth and cell division as well as increased cell death of precancerous cells compared to Eμ–Myc/+ cells (Supplementary Fig. 1 and 4). Importantly, lymphoma initiation in Eμ–Myc/+;L38+/− mice was markedly suppressed, as in Eμ–Myc/+;L24+/− mice (Supplementary Fig. 5). The suppression of lymphomagenesis was specific to the direct effect of Myc signaling on protein synthesis, as ribosomal protein haploinsufficiency in the context of the p53−/− background did not have any effect on tumor formation (Figure 2c). These genetic results demonstrate that Myc's ability to augment protein synthesis is necessary for its oncogenic potential.
Figure 2. The ability of Myc to augment protein synthesis is necessary for its oncogenic potential.
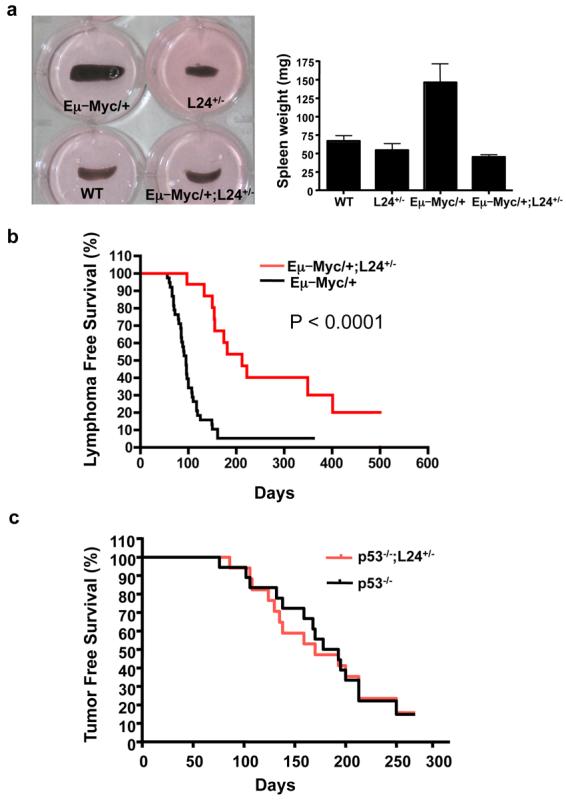
a, Representative photographs of spleens and average spleen weight (n=6 per genotype, 4 weeks of age). Error bars, s.d. b, Kaplan-Meier curves showing lymphoma-free survival (LFS) of Eμ–Myc/+ and Eμ–Myc/+;L24+/− mice (n=30 per genotype). c, Kaplan-Meier curves showing tumor-free survival (TFS) of p53−/− and p53−/−;L24+/− mice (n=25 per genotype).
To further understand the molecular mechanisms by which unrestrained increases in global protein synthesis can lead to tumorigenesis, we analyzed protein synthesis control during specific phases of the cell cycle in Eμ–Myc/+ B-lymphocytes. Unexpectedly, WT and Eμ–Myc/+ cells synchronized in S phase do not show a difference in protein synthesis rates (Fig. 3a). During mitosis, cap-dependent protein synthesis is normally decreased to facilitate cap-independent translation of a subset of mRNA required for accurate mitotic progression19. On the contrary, Eμ–Myc/+ cells show elevated protein synthesis rates during mitosis, which is cap-dependent as it is restored to normal levels upon Rapamycin treatment (Supplementary Fig.6) and in Eμ–Myc/+; L24+/− cells (Fig. 3 a,b) . Moreover, elevated activity of a cap-dependent luciferase reporter gene is observed in Myc overexpressing cells synchronized in mitosis and restored to normal when Myc is overexpressed in the L24+/− background (Fig. 3c). Accurate mitotic progression relies on a very precise and orderly switch in translational control through a general decrease in cap-dependent translation and a switch to IRES-dependent translation initiation19. We next asked whether a persistent enhancement of cap-dependent translation during mitosis downstream of Myc overexpression would be unfavorable to an IRES-dependent translational switch. The expression of a bicistronic reporter construct harboring the Hepatitis C Virus (HCV) IRES element, a molecular readout of IRES-dependent translation20, is impaired in Myc overexpressing cells and restored to normal when Myc is overexpressed in the L24+/− background (Fig. 3d). Importantly, L24+/− cells do not show differences in IRES-dependent translation compared to WT cells (Fig. 3d), suggesting that the rescue in IRES-dependent translation downstream Myc hyperactivation in the L24+/− background is the result of restoring cap-dependent translation to normal levels (Fig. 3c). Taken together, these data demonstrate that aberrant and continuous stimulation of cap-dependent protein synthesis by Myc perturbs the mitotic switch to IRES-dependent translation.
Figure 3. Myc hyper-activation impairs the translational switch from cap to IRES-dependent translation control during mitosis and blocks mitotic translation of the Cdk11/p58 PITSLRE kinase.

a,b, S35 methionine incorporation in B-lymphocytes synchronized in S phase and mitosis. Densitometry analysis (n=3) comparing protein synthesis levels in S-phase and mitotically arrested B-lymphocytes. c, Cap-dependent activity of the Renilla luciferase reporter mRNA in asynchronous and mitotic synchronized MEFs expressing a tamoxifen inducible Myc vector (n=6 experiments performed in triplicate) *(p < 0.001) compared to WT, **(p < 0.005) compared to asynchronous values. d, HCV-IRES dependent activity of the Firefly luciferase reporter mRNA (n=6 experiments performed in triplicate) in asynchronous and mitotic synchronized MEFs ** (p < 0.001) compared to asynchronous values. e, Representative western blot of the endogenous Cdk11/p58 PITSLRE kinase in asynchronous and mitotic synchronized primary B-lymphocytes. Note that expression of Cdk11/p58 PITSLRE is only present in mitotically synchronized cells (left), and not asynchronous cells (right) via an IRES-element positioned in its 5′UTR. Densitometry analysis (n=3). f, Cdk11/p58-IRES dependent activity of Firefly luciferase reporter mRNA (n=4 experiments performed in triplicate) transfected in asynchronous and mitotic synchronized cells ** (p < 0.001) compared to asynchronous values. (c,d,f) Average steady state WT values were set to 1. The Y-axes show fold change. Error bars, s.d.
We next monitored the expression of a well-characterized endogenous mRNA that is only translated during mitosis via an IRES element. PITSLRE/Cdk11 is a member of the Cdc-2 like protein kinase family that undergoes cap-independent translation from an IRES element during mitosis to produce a Cdk11/58-kDa isoform that facilitates accurate mitotic progression3,4. Importantly, deletions containing the PITSLRE/Cdk11 locus are found in non-Hodgkin lymphoma21 and other cancers22,23, strongly suggesting that Cdk11/p58 may act as a tumor suppressor gene24. The expression of Cdk11/p58 was markedly reduced in mitotically synchronized Eμ–Myc/+ cells (Fig. 3e and Supplementary Fig. 7) but was rescued to normal levels in Eμ–Myc/+; L24+/− cells (Fig. 3d). Moreover, expression of a reporter gene directed by the Cdk11/p58 IRES element is also impaired in Myc overexpressed cells and restored to normal levels when Myc is overexpressed in the L24+/− background (Fig. 3f). These findings indicate that the general increase in cap-dependent translation downstream of Myc activation prevents the accurate mitotic switch to IRES-dependent translation that regulates Cdk11/p58 expression.
Decreased Cdk11/p58 expression during mitosis impairs accurate cytokinesis, resulting in a binucleated cell phenotype5 that is associated with aneuploidy25. We observed that Myc overexpressing MEFs display cytokinesis defects and show a significant increase in number of binucleated cells, a hallmark of Cdk11/p58 loss of function5 (Fig. 4a). Importantly, restoring accurate mitotic Cdk11/p58 expression in Myc overexpressing cells was sufficient to revert these cytokinesis defects (Fig. 4a,b,c). These findings strongly suggest that decreased IRES-dependent translation of Cdk11/p58 downstream of oncogenic Myc signaling may be an early event in tumorigenesis that underlies the subsequent development of genomic instability26.
Figure 4. Aberrant translation control downstream of Myc activation underlies cytokinesis defects and genome instability.
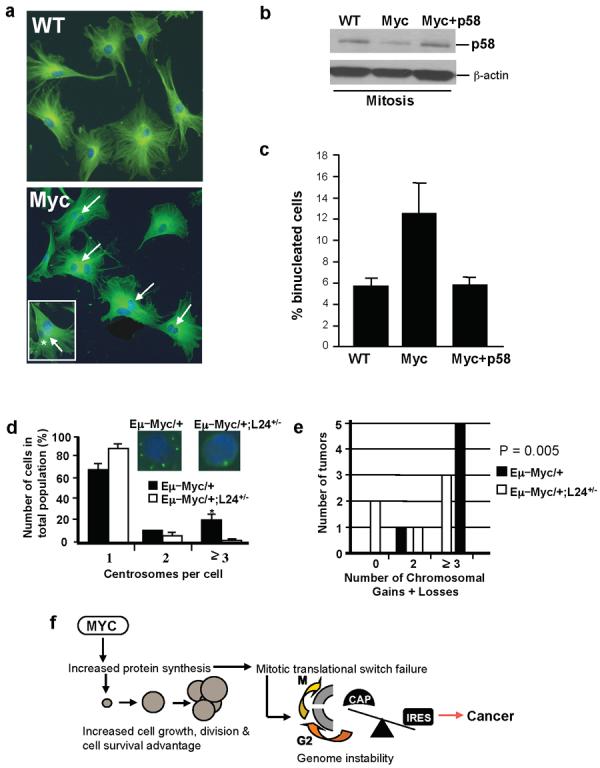
a, Myc overexpressing MEFs show increased numbers of binucleated cells (arrows). Insert illustrates dysmorphic and variably sized nuclei frequently observed in Myc overexpressing MEFs. b, Western blot of mitotic Cdk11/p58 expression showing decreased expression in MEFs expressing a Myc inducible vector. Myc+p58 cells express a retroviral Cdk11/p58 cDNA. c, The increased number of binucleated cells in Myc overexpressing cells is restored to normal level when Cdk11/p58 is reintroduced (n=4, at least 500 cells were scored per experiment) Error bars, s.d. d, Percentages of cells with normal (1 and 2) and aberrant (3) centrosome numbers in freshly isolated B-lymphocytes (n=6, at least 300 cells per experiment), *p < 0.001. Insert shows representative immunofluorescence staining with a centrosome marker. e, Comparative genomic hybridization (CGH) analysis of tumors (n=6). Error bars, s.d. f, Proposed model for how deregulations in translation control downstream of Myc activation lead to cancer initiation. Myc dependent increases in protein synthesis augment cell growth and this effect is coupled to increased cell cycle progression and a cell survival advantage. Increasing cap-dependent translation downstream of Myc-activation also gives rise to a specific molecular impairment in the modality of translation initiation employed during mitosis that leads to cytokinesis defects associated with genome instability.
We therefore assessed whether Eμ–Myc/+ lymphocytes display supernumerary centrosomes, an early hallmark of genome instability. We observed a large percentage of Eμ–Myc/+ cells that show centrosome duplications, which are the result of aberrant protein synthesis control downstream of Myc activation as Eμ–Myc/+;L24+/− cells show normal centrosome numbers (Fig. 4d). We next directly monitored genomic instability by employing comparative genomic hybridization (CGH) analysis. All of the lymphomas analyzed from Eμ–Myc/+ mice had chromosomal abnormalities, but Eμ–Myc/+;L24+/− tumors showed either no chromosomal abnormalities or showed them at a lower frequency (Fig. 4e). These findings establish a direct and previously unrecognized molecular connection between aberrant control of protein synthesis downstream of Myc activation and the accumulation of genetic lesions in tumors.
In our study, we have utilized ribosomal protein haploinsufficiency as a genetic tool to restore protein synthesis to normal levels downstream of oncogenic Myc activation. It is worth noting that subsets of ribosomal proteins act as tumor suppressors in Zebrafish27. However, L24+/− and L38+/− mice, employed in this study, do not show cancer susceptibility (unpublished observations). Our findings provide genetic evidence that increased global protein synthesis downstream of Myc activation is a rate-limiting determinant of cancer initiation and delineate how deregulations in protein synthesis control confer oncogenic potential (Fig. 4f). These findings strongly suggest that oncogenic signals may monopolize the translational machinery to elicit cooperative effects on cell growth, cell cycle progression, and cell survival (Fig. 4f). Moreover, Myc overexpressing cells display cytokinesis defects, supernumery centrosomes and genomic instability as a consequence of augmented cap-dependent translation, demonstrating a previously unrecognized molecular connection between aberrant protein synthesis control and genome instability in cancer (Fig. 4f).
We have identified a specific translational impairment as a consequence of increasing protein synthesis downstream of oncogenic signaling (Fig. 4f). The failure to suppress cap-dependent translation during mitosis in Myc overexpressing cells prevents the critical switch to IRES-dependent translation that is required for accurate expression of mitotically expressed mRNAs and suggests that many IRES-containing mRNAs, such as Cdk11/p58, may be deregulated at the translational level in Myc overexpressing cells. Defects in the mitotic translational switch are directly relevant for tumorigenesis, as we have shown that impairments in IRES-dependent translation of Cdk11/p58 result in cytokinesis failure, an early event in cancer that underlies the subsequent development of genomic instability5,25,28 and which can be reverted in Myc overexpressing cells by restoring accurate Cdk11/p58 mitotic expression (Fig. 4 a, b, c). Why would an increase in cap-dependent translation downstream of Myc activation decrease IRES-dependent translation? An aberrant increase in cap-dependent translation during mitosis may cause preferential recruitment of translational components (ie., ribosomes, translation initiation factors) to the cap structure at the expense of IRES elements that govern accurate expression of a subset of mRNAs29. Interestingly, multiple tumor suppressor genes possess an IRES-element2 and defects in IRES-dependent translation underlie the cancer susceptibility syndrome Dyskeratosis Congenita30. Therefore, IRES-containing mRNAs may be preferentially found deregulated at the translation level in cancer and contribute to tumorigenesis. Our data strongly suggest that alterations in quantitative as well as qualitative translational control downstream of oncogenic signaling provide a highly specific and rapid response that may overshadow the effect of the transcriptosome towards cellular transformation.
Methods Summary
Mice
Eμ Myc/+, L24+/−, L38+/− and p53−/− mice were all maintained on a C57/BL6 background. Mice were monitored twice a week for signs of morbidity and tumor development. Myc tumor initiation was scored by peripheral lymph node palpation. Moribund mice or mice with obvious tumors were sacrificed, and tumors and different organs were analyzed by histology or processed for further analysis.
Cell culture and analysis of IRES-dependent translation in mitosis
Primary B-lymphocytes were isolated from spleen or bone marrow from 4-5 week old mice utilizing an autoMACS separator (Miltenyi Biotec). Primary B-lymphocytes and MEFs were synchronized in mitosis by thymidine and aphidicolin block, respectively. Bicistronic vectors, HCV-IRES and CDK11/p58 IRES were transfected as RNAs. IRES-dependent expression of the endogenous CDK11/p58 was assessed by western blot utilizing anti-CDK11/p58 (Abcam).
Analysis of global protein synthesis
Equal numbers of freshly isolated or cultured primary B-lymphocytes synchronized in S phase or mitosis were incubated in methionine-free DMEM and then 50 μCi/well (25 uCi/mL) of [35S] methionine was added to the cultures for 35 minutes. Radiolabeled proteins were visualized by exposure to X-ray film and quantified by densitometry analysis.
Cellular and molecular analysis of B-lymphocytes
Freshly isolated and cultured B-lymphocytes from 4-5 week old mice were fixed and stained with the following combination of mAbs conjugated with FITC or PE: CD19-PE/CD3-FITC, CD4-PE/CD8-FITC, CD43-PE/CD45R-B220-FITC. Cell volume measurements were performed using a Coulter Model Z2 (Coulter).
CGH and Cytokinesis analysis
Genomic DNA extracted from lymphomas of Eμ–Myc/+ mice, and Eμ–Myc/+;L24+/− mice was subjected to CGH analysis by standard methods. For cytokinesis analysis, primary MEFs were stably transfected with MycER harboring puromycin resistance or P58 cDNA plus MycER via a Phoenix viral vector and cultured. Upon release from aphidicolin, Myc was activated by the addition of hydroxytamoxifen. At the 20 hour time point, cells were fixed and stained. Binucleated cells were scored utilized automated segmentation routines.
Supplementary Material
Acknowledgements
We thank Drs. Frank McCormick, Gerard Evan, and Pat O'Farrell for critically reading the manuscript; Dr. Joseph Testa for support and critical discussion during early stages of this work; Wei Xu and Richard Adamo for technical assistance; Joanna Copley for editing the manuscript, Dr. Cornelis for the Cdk11/p58 IRES bicistronic vector. This work was supported by the NIH (D.R.) and the Sandler Foundation (M.B.).
References
- 1.Gomez-Roman N, et al. Activation by c-Myc of transcription by RNA polymerases I, II and III. Biochemical Society symposium. 2006:141–154. doi: 10.1042/bss0730141. [DOI] [PubMed] [Google Scholar]
- 2.Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3:179–192. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- 3.Cornelis S, et al. Identification and characterization of a novel cell cycle-regulated internal ribosome entry site. Mol Cell. 2000;5:597–605. doi: 10.1016/s1097-2765(00)80239-7. [DOI] [PubMed] [Google Scholar]
- 4.Petretti C, et al. The PITSLRE/CDK11p58 protein kinase promotes centrosome maturation and bipolar spindle formation. EMBO Rep. 2006;7:418–424. doi: 10.1038/sj.embor.7400639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilker EW, et al. 14-3-3sigma controls mitotic translation to facilitate cytokinesis. Nature. 2007;446:329–332. doi: 10.1038/nature05584. [DOI] [PubMed] [Google Scholar]
- 6.Boxer LM, Dang CV. Translocations involving c-myc and c-myc function. Oncogene. 2001;20:5595–5610. doi: 10.1038/sj.onc.1204595. [DOI] [PubMed] [Google Scholar]
- 7.Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2:764–776. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- 8.Grewal SS, Li L, Orian A, Eisenman RN, Edgar BA. Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat Cell Biol. 2005;7:295–302. doi: 10.1038/ncb1223. [DOI] [PubMed] [Google Scholar]
- 9.Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P. Drosophila myc regulates cellular growth during development. Cell. 1999;98:779–790. doi: 10.1016/s0092-8674(00)81512-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moreno E, Basler K. dMyc transforms cells into super-competitors. Cell. 2004;117:117–129. doi: 10.1016/s0092-8674(04)00262-4. [DOI] [PubMed] [Google Scholar]
- 11.de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA. Drosophila myc regulates organ size by inducing cell competition. Cell. 2004;117:107–116. doi: 10.1016/s0092-8674(04)00214-4. [DOI] [PubMed] [Google Scholar]
- 12.Oliver ER, Saunders TL, Tarle SA, Glaser T. Ribosomal protein L24 defect in belly spot and tail (Bst), a mouse Minute. Development. 2004;131:3907–3920. doi: 10.1242/dev.01268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris AW, et al. The E mu-myc transgenic mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J Exp Med. 1988;167:353–371. doi: 10.1084/jem.167.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iritani BM, Eisenman RN. c-Myc enhances protein synthesis and cell size during B lymphocyte development. Proc Natl Acad Sci U S A. 1999;96:13180–13185. doi: 10.1073/pnas.96.23.13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas G. An encore for ribosome biogenesis in the control of cell proliferation. Nat Cell Biol. 2000;2:E71–72. doi: 10.1038/35010581. [DOI] [PubMed] [Google Scholar]
- 16.Yang W, et al. Repression of transcription of the p27(Kip1) cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene. 2001;20:1688–1702. doi: 10.1038/sj.onc.1204245. [DOI] [PubMed] [Google Scholar]
- 17.Wu S, et al. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene. 2003;22:351–360. doi: 10.1038/sj.onc.1206145. [DOI] [PubMed] [Google Scholar]
- 18.Bouchard C, et al. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. The EMBO journal. 1999;18:5321–5333. doi: 10.1093/emboj/18.19.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pyronnet S, Sonenberg N. Cell-cycle-dependent translational control. Curr Opin Genet Dev. 2001;11:13–18. doi: 10.1016/s0959-437x(00)00150-7. [DOI] [PubMed] [Google Scholar]
- 20.Hellen CU, Sarnow P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes & development. 2001;15:1593–1612. doi: 10.1101/gad.891101. [DOI] [PubMed] [Google Scholar]
- 21.Dave BJ, et al. Deletion of cell division cycle 2-like 1 gene locus on 1p36 in non-Hodgkin lymphoma. Cancer genetics and cytogenetics. 1999;108:120–126. doi: 10.1016/s0165-4608(98)00138-1. [DOI] [PubMed] [Google Scholar]
- 22.Nelson MA, et al. Abnormalities in the p34cdc2-related PITSLRE protein kinase gene complex (CDC2L) on chromosome band 1p36 in melanoma. Cancer genetics and cytogenetics. 1999;108:91–99. doi: 10.1016/s0165-4608(98)00122-8. [DOI] [PubMed] [Google Scholar]
- 23.Lahti JM, et al. Alterations in the PITSLRE protein kinase gene complex on chromosome 1p36 in childhood neuroblastoma. Nature genetics. 1994;7:370–375. doi: 10.1038/ng0794-370. [DOI] [PubMed] [Google Scholar]
- 24.Chandramouli A, et al. Haploinsufficiency of the cdc2l gene contributes to skin cancer development in mice. Carcinogenesis. 2007;28:2028–2035. doi: 10.1093/carcin/bgm066. [DOI] [PubMed] [Google Scholar]
- 25.Fujiwara T, et al. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–1047. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- 26.Wade M, Wahl GM. c-Myc, genome instability, and tumorigenesis: the devil is in the details. Current topics in microbiology and immunology. 2006;302:169–203. doi: 10.1007/3-540-32952-8_7. [DOI] [PubMed] [Google Scholar]
- 27.Amsterdam A, et al. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2004;2:E139. doi: 10.1371/journal.pbio.0020139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev. 2007;17:157–162. doi: 10.1016/j.gde.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 29.Svitkin YV, et al. Eukaryotic translation initiation factor 4E availability controls the switch between cap-dependent and internal ribosomal entry site-mediated translation. Molecular and cellular biology. 2005;25:10556–10565. doi: 10.1128/MCB.25.23.10556-10565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoon A, et al. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science. 2006;312:902–906. doi: 10.1126/science.1123835. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.