

Abstract
Whole-genome resequencing technology has improved rapidly during recent years and is expected to improve further such that the sequencing of an entire human genome sequence for $1000 is within reach. Our main aim here is to use whole-genome sequence data for the prediction of genetic values of individuals for complex traits and to explore the accuracy of such predictions. This is relevant for the fields of plant and animal breeding and, in human genetics, for the prediction of an individual's risk for complex diseases. Here, population history and genomic architectures were simulated under the Wright–Fisher population and infinite-sites mutation model, and prediction of genetic value was by the genomic selection approach, where a Bayesian nonlinear model was used to predict the effects of individual SNPs. The Bayesian model assumed a priori that only few SNPs are causative, i.e., have an effect different from zero. When using whole-genome sequence data, accuracies of prediction of genetic value were >40% increased relative to the use of dense ∼30K SNP chips. At equal high density, the inclusion of the causative mutations yielded an extra increase of accuracy of 2.5–3.7%. Predictions of genetic value remained accurate even when the training and evaluation data were 10 generations apart. Best linear unbiased prediction (BLUP) of SNP effects does not take full advantage of the genome sequence data, and nonlinear predictions, such as the Bayesian method used here, are needed to achieve maximum accuracy. On the basis of theoretical work, the results could be extended to more realistic genome and population sizes.
GENOME resequencing technologies are currently developing at a very rapid rate, which we for simplicity call genome sequencing even though it is used on a species with a reference sequence. The current generation sequencing technology is two orders of magnitude faster and more cost effective than the technologies used for the sequencing of the human genome (Shendure and Ji 2008; TenBosch and Grody 2008). Future technologies are expected to reduce cost by another 100-fold so that sequencing an entire human genome for $1000 is considered achievable in the near future (Mardis 2008). The question arises: How can we make best use of entire genome sequence data on many individuals? One use will be the ability to predict the genetic value of an individual for complex traits. In the fields of animal and plant breeding, this would be of great practical benefit because most important traits are complex, quantitative traits, i.e., traits that are affected by many genes and by the environment. In humans the promise of personalized medicine relies on the ability to predict an individual's genetic risk for complex, multifactorial diseases, such as Crohn's disease (Barrett et al. 2008), and the ability to predict response to alternative treatments. The first aim of this article is to explore the accuracy of this prediction using the full genome sequence of the individual.
The use of high-density SNP genotype data to predict genetic value, called genomic selection, was first proposed by Meuwissen et al. (2001). In its most sophisticated form, a Bayesian model was used to predict the effects of thousands of SNPs on the total genetic value simultaneously, where a priori it was assumed that only few SNPs were useful for predicting the trait [because they were in linkage disequilibrium (LD) with mutations causing variation in the trait], while many SNPs were not useful. Even among the SNPs that were useful for prediction, it was assumed that the distribution of effects was not normal because there were occasionally SNPs in LD with quantitative trait loci (QTL) that may occasionally have very large effect. To model this, the distribution of SNP effects was assumed to follow a distribution with thicker tails than the normal distribution (e.g., the t-distribution is often used). In the case of whole-genome sequence data, the polymorphisms that are causing the genetic differences between the individuals are among those being analyzed. For the sake of simplicity we call all polymorphisms in the sequence data SNPs while recognizing that other types of polymorphisms such as indels will be included. Assuming that the causal SNPs are included in the analysis simplifies the prior distribution of the SNP effects, because the effects of all the other SNPs, even if they are in LD with the causal SNPs, are expected to disappear. Thus, the prior distribution simplifies to the fact that some SNPs are expected to be causative and have an effect drawn from the distribution of the gene effects. The distribution of gene effects is investigated extensively in the evolutionary and other literature and is reported to be gamma (Hayes and Goddard 2001) or exponentially distributed (Erickson et al. 2004; Rocha et al. 2004), where the latter is a special form of the gamma distribution. On the downside, whole-genome sequence data will contain millions of SNPs and it may be difficult for genomic selection to separate the relatively few causative SNPs from all the others.
Meuwissen et al. (2001) also investigated a model in which all SNPs were assumed to have an effect drawn from the same normal distribution [the so-called genome-wide best linear unbiased prediction (GWBLUP) model]. Although this model seems biologically implausible, it has been found to perform well in data from dairy cattle (VanRaden et al. 2009). However, we hypothesize that with sequence level data the BLUP model will not perform as well as models that assume that only some causal SNPs need to be included in the model.
The aims here are to investigate the following: how accurately genetic values for complex traits can be predicted by genomic selection when whole-genome sequence data are available on a large number of individuals; whether it makes a difference to have the whole-genome sequence available, including the causative mutations, vs. very dense SNP marker genotypes; whether the estimates of the SNP effects can be used on individuals that are many generations separated from the data set in which they were estimated; the effect of the statistical model used on accuracy of prediction; and how accurately causative mutations can be detected and mapped. Because whole-genome sequence data on many individuals are not yet available, and because we needed to know the true genetic values of the individuals, the aforementioned questions were investigated by computer simulations of whole-genome sequence data.
METHODS
Computer simulation of whole-genome sequence data:
Sequence data are efficiently simulated by the coalescence process, which simulates the coalescence of the current population backward in time (Kingman 1982; Hudson 2002). Although coalescence simulations are remarkably efficient for small chromosome segments, their computational requirements increase exponentially with the size of the segment being simulated. For forward in time simulations, the computational requirements increase only linearly with the size of the genome (Hoggart et al. 2007), and thus forward simulations were used here. The assumptions about the population model followed those of coalescence theory: the Fisher–Wright idealized population model (Falconer and Mackay 1996) and the infinite-sites mutation model were assumed (Kimura 1969) with a mutation frequency of 2 × 10−8 per nucleotide per generation. The historical effective size of the population was Ne = 1000, and the forward simulations were conducted for 10,000 generations to achieve a steady-state population in mutation–drift balance. Recombinations were sampled according to the Haldane mapping function, assuming a recombination frequency of 10−8 per nucleotide per generation. After these 10,000 generations, the population was simulated for 20 more generations, called G1–G20, at an effective size of 10,000 to further reduce the probability of sampling close relatives. Because the population had Ne = 10,000 only for a very short time, its mutation–drift balance and the linkage disequilibria between the SNPs are very much like that of a population of Ne = 1000, i.e., its historical size. Two samples were taken from generation G10, namely a training sample of size T individuals (TRAIN) and a test sample of size 500 (TEST1). The training and test samples were nonoverlapping. To test whether the predictions also hold for distantly related individuals, a second test sample was taken 10 generations later, generation G20, with a size of 500 (TEST2).
For the GWBLUP model, Daetwyler et al. (2008) and Goddard (2009) predicted that the accuracy of genomic selection is dependent on the parameter λ = Th2/ML, where h2 is the heritability of the trait, T is the number of records in the training data, M is the effective number of loci per morgan (∼2Ne), and L is the genome size in morgans. The number of loci per morgan may be defined as the number of chromosome segments that are inherited identical-by-descent from a common ancestor without a recombination. The effective number of loci also accounts for the variation in size of these segments; i.e., the actual number of segments behaves like M segments of equal size. This relationship predicts that accuracy will be the same for all cases where λ is the same. We show that this also holds for the data and analysis used here and use this relationship to reduce the computer time needed to simulate many replicates. In most simulations we use a genome with 1 chromosome of L = 1 M and realize that for larger genome sizes we would need to increase the number of records (T) to keep λ constant. Thus, a genome of 30 chromosomes of 1 M each requires 30 times as many training records to achieve the same accuracy.
Simulation of the QTL and phenotypes:
After the 10,000 generations at Ne = 1000, 30 SNPs were randomly sampled (without replacement) among all the simulated SNPs and were designated to be causative; i.e., they were QTL. For every QTL, the “1” allele obtained an additive effect (aj′) sampled from the double-exponentional distribution, which is also called the Laplace distribution. After all 30-QTL effects were sampled, their effect was standardized to achieve a total genetic variance of 1, i.e.,  , where subscripts i (j) denote the ith (jth) QTL; the summation is over all QTL, and pi is the frequency of the 1 allele of the ith QTL. The total genetic value of individual i was calculated as
, where subscripts i (j) denote the ith (jth) QTL; the summation is over all QTL, and pi is the frequency of the 1 allele of the ith QTL. The total genetic value of individual i was calculated as
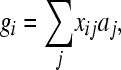 |
where xij is the number of 1 alleles individual i carries at locus j. Phenotypic values were obtained by adding an environmental effect sampled from the N(0, 1) distribution to these total genetic values, resulting in a heritability of 0.5. The resulting data set is called the “30-QTL” data.
Thirty QTL per morgan may lead to a lot of QTL for realistic genome sizes, e.g., 900 QTL for a 30-M genome. Thus, also a data set with 10-fold fewer QTL was obtained, called the “3-QTL” data set. The 3 QTL were randomly picked among all the SNPs with minor allele frequency (MAF) > 0.05 instead among all SNPs, to avoid the situation that all genetic variance was explained by 3 very rare QTL. If the causative SNPs were included in the SNP genotypes data, the data sets are called “3 QTL+” (see supporting information, File S1) and “30 QTL+” (see File S2), and if the causative SNPs were excluded from the data, they are called “3 QTL−” and “30 QTL−”. The latter reflects the situation where we have very dense SNP genotype data, but not the complete sequence; i.e., the causative mutations are not expected to be among the genotyped SNPs. All simulated data sets were replicated 20 times, unless stated otherwise. The mutation–drift process resulted on average in 33,066 SNPs per morgan, and the standard deviation of this number was 439. To simulate also a situation with an increased genome size, the 20 (replicates of the) chromosomes of the 3-QTL+ and 30-QTL+ data sets were divided into groups (genomes) of 2 chromosomes each, resulting in 10 replicates with 2 chromosomes; i.e., L = 2. The latter data sets are denoted 6 QTL+ (6 QTL−) and 60 QTL+ (60 QTL−), since there are 6 or 60 QTL in these genomes of 2 chromosomes.
Analysis of the data:
Estimates of the SNP effects were obtained from the TRAIN data using the BayesB method (Meuwissen et al. 2001). The model assumes additive inheritance and is
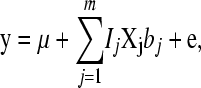 |
where y is a (T × 1) vector of phenotypes with T records; μ is overall mean; m is total number of genotyped SNPs; Ij is an indicator whether the SNP is included in the model (Ij = 1) or not (Ij = 0); Xj is a (T × 1) vector denoting the genotype of the individuals for marker j, where 0 denotes homozygous for the first allele; 1/√Hj denotes heterozygous; 2/√Hj denotes homozygous for the second allele, and the division by √Hj standardizes the variance of the marker genotypes with Hj being the marker heterozygosity calculated as 2pj(1 − pj) with pj being the frequency of the jth marker; bj are standardized effects of the markers; and e is a (T × 1) vector of environmental effects. In the Bayesian analysis the prior distribution for Ij is Ij = 1 with prior probability 30/m for the 30-QTL+/− data or 3/m for the 3 QTL+/−, and for Ij = 0 the prior probability is 1 − 30/m and 1 − 3/m, respectively. For bj the t-distribution was used as a prior with 4.1 d.f. The t-distribution with 4.1 d.f. approximates the Laplace distribution used for simulation but has tails that are not as fat as the Laplace distribution. The t-distribution was chosen because it is more convenient for analysis (Meuwissen et al. 2001). Four and one-tenth is probably a conservative number of degrees of freedom, since the LaPlace distribution is more fat tailed than the t-distribution with 4.1 d.f. However, a t-distribution with ≤4 d.f. results in the sampling from a posterior inverse-chi-square distribution with ≤4 d.f., which has an infinite variance. The latter results in an instable MCMC sampling process, and thus the number of degrees of freedom was set slightly >4. The software is available under an open source license at genomicselection.net (see www.genomicselection.net). For the 500 individuals in the TEST1 and TEST2 data sets the genetic value was predicted as 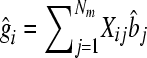 , where
, where 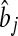 is the estimate of marker effect, and Nm is the number of SNPs. The accuracy of the predictions,
is the estimate of marker effect, and Nm is the number of SNPs. The accuracy of the predictions, 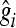 , is calculated as the correlation between gi and
, is calculated as the correlation between gi and 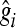 .
.
RESULTS
Ten thousand generations of computer simulation of the whole-genome sequence of one chromosome of 1 M resulted on average in 33,066 SNPs. Either 3 or 30 of the SNPs were designed to be causative, i.e., QTL. Table 1 shows the accuracy of the predicted genetic values when T = 200 individuals were used to estimate SNP effects (training data) and the test individuals come from the same generation as the training individuals. Accuracies of the predictions were high, but the data sets with 3 QTL had higher accuracy than those with 30 QTL. Presumably with 30 QTL per chromosome their effects are smaller and therefore harder to estimate accurately and it is harder to detect the QTL (which should be the only SNPs in the model). In the 3-QTL and 30-QTL data, the inclusion of the causative mutations increased the accuracy by 3.7 and 2.5%, respectively. So, even at this high density of ∼33,000 SNPs per morgan there is a benefit of including the causative mutations, but the effect of having few vs. many QTL is substantially larger.
TABLE 1.
The accuracy of the predictions of total genetic value (±SE), when T = 200 and L = 1 or T = 400 and L = 2, and using test data from the same generation (TEST1) or 10 generations later (TEST2)
| Data set |
|||
|---|---|---|---|
| TEST1: | TEST2: | TEST1: | |
| T = 200, L = 1: | T = 200, L = 1: | T = 400, L = 2a | |
| Causal SNPs | 3 QTL | 3 QTL | 6 QTL |
| Excluded | 0.938 ± 0.013 | 0.943 ± 0.012 | 0.963 ± 0.007 |
| Included | 0.973 ± 0.004 | 0.974 ± 0.004 | 0.979 ± 0.008 |
| Data set |
|||
| TEST1: | TEST2: | TEST1: | |
| T = 200, L = 1: | T = 200, L = 1: | T = 400, L = 2a | |
| Causal SNPs |
30 QTL |
30 QTL |
60 QTL |
| Excluded | 0.806 ± 0.023 | 0.806 ± 0.022 | 0.799 ± 0.020 |
| Included | 0.826 ± 0.019 | 0.824 ± 0.019 | 0.853 ± 0.028 |
Situations with L = 2 were replicated 10 times instead of 20.
When the test and training individuals are separated by 10 generations (TEST2 data), accuracies are very similar to those where test and training data come from the same generation (Table 1). This implies that the SNPs for which effects were estimated are at or so close to the causative SNPs that a genetic distance of 10 generations hardly affected the SNP effects.
Theory predicts that, if λ = Th2/ML is constant, accuracy is constant. This theory is tested by comparing twice the genome size and twice the number of records to the previous results (Table 1; L = 2 vs. L = 1). The marker and QTL density is not changed, and thus there are twice as many markers and QTL when genome size increases to L = 2 M. Since heritability is kept constant at 0.5, this implies that the fraction of the variance explained by each QTL is halved. Differences were small between L = 1 with T = 200 vs. L = 2 with T = 400 records. Across the four comparisons in Table 1 the accuracy increases on average by 1.3% when L = 2 but this is not a consistent trend, so we conclude that the theoretical prediction holds approximately.
Table 2 tests the same theory in that if heritability is halved, the training data set, T, needs to be doubled to maintain the accuracy, which would again maintain the same λ. Accuracy was approximately maintained by doubling the number of training records, although there seemed a general tendency for a slightly reduced accuracy of ∼3% due to a fourfold reduction in heritability.
TABLE 2.
The accuracy of the predictions of total genetic value (±SE) in the TEST1 data set when T = 200 and h2 = 0.5, T = 400 and h2 = 0.25, and T = 800 and h2 = 0.125
| Causative SNPs | T = 200, h2 = 0.5 | T = 400, h2 = 0.25 | T = 800, h2 = 0.125 |
|---|---|---|---|
| 3 QTL | |||
| Excluded | 0.938 ± 0.013 | 0.909 ± 0.016 | 0.912 ± 0.011 |
| Included | 0.973 ± 0.004 | 0.936 ± 0.011 | 0.935 ± 0.011 |
| 30 QTL | |||
| Excluded | 0.806 ± 0.023 | 0.711 ± 0.032 | 0.733 ± 0.030 |
| Included | 0.826 ± 0.019 | 0.796 ± 0.019 | 0.780 ± 0.019 |
Table 3 shows the accuracies of selection when GWBLUP is used to estimate the SNP effects. With this method of analysis, the accuracy is greatly reduced and the inclusion of the causative SNPs has hardly any effect on the accuracy of selection. This may be expected, since the GWBLUP is equivalent to estimating a relationship matrix between the individuals on the basis of the marker data and using this to estimate the genetic values of the animals (Goddard 2009). The relatively few causative SNPs hardly affect the estimated relationship matrix. Also, the accuracy of selection is hardly affected by the genetic model (3 QTL vs. 30 QTL), probably because the genetic relationship matrix is equally appropriate for both genetic models. This suggests that the accuracies obtained by GWBLUP are those that could be obtained when all SNPs had a very small effect, and thus the infinitesimal genetic model (Falconer and Mackay 1996) applies.
TABLE 3.
The accuracy of the predictions of total genetic value (±SE) in the TEST1 data set when the training data contained T = 200 individuals and GWBLUP or BayesB is used to estimate the marker effects
| Causative SNPs |
||||
|---|---|---|---|---|
| GWBLUP |
BayesB |
|||
| Data | Excluded | Included | Excluded | Included |
| 3 QTL | 0.503 ± 0.011 | 0.508 ± 0.011 | 0.938 ± 0.013 | 0.973 ± 0.004 |
| 30 QTL | 0.491 ± 0.016 | 0.493 ± 0.010 | 0.806 ± 0.023 | 0.826 ± 0.019 |
Figure 1 compares the accuracy at reduced marker densities with that of the whole-genome sequence. Absolute differences in accuracy are similar with many or few QTL, but proportional changes are greater when the accuracy is low, i.e., with 30 QTL. It may also be noted that, over a 33-fold increase in density, the accuracy increases approximately linearly with the log of the density, where density is expressed as the number of SNPs per morgan.
Figure 1.—

The accuracy of the predictions of total genetic value in the TEST1 data set as a function of the marker density (d) in SNPs per morgan. The densities evaluated were d = 1000, 11,000, and 33,000, and the training data contained T = 200 individuals.
Figure 2 shows how accurately genomic selection actually detects the causative SNPs by plotting the posterior probability of fitting a causative SNP against the variance explained by the SNP for the first three replicates of the 3-QTL+ data. Figure 2 shows that small QTL may go undetected or not accurately detected and that some large QTL are picked up by a SNP close to the QTL position (∼20 SNPs away). In the latter cases the R2 between the QTL and the detected SNP was 0.99 (third QTL of replicate 1), 1.0 (second QTL of replicate 2), and 1.0 (second QTL of replicate 3); i.e., the detected SNPs were indistinguishable from the causative SNP. Thus, the largest QTL seem to be detected by the analysis and predicted either by the QTL itself or by a SNP that is in very high LD with the QTL. Since there are ∼33,000 SNPs per morgan in these data, a distance of 20 SNPs between the QTL and the SNP fitted by the model is equivalent to 60 kb. The SNPs with posterior probabilities >10% always showed some LD with a QTL and thus helped to improve accuracy of prediction.
Figure 2.—

The posterior probability of a SNP being fitted in the model and the simulated QTL variance plotted against the position along the chromosome for three randomly picked 3-QTL+ data sets (plotted are the 100 SNPs that surround the 3 causative SNPs, resulting in three segments in each graph (separated by  ).
).
Figure 3 shows how much of the variance caused by the QTL is picked up by SNPs that show R2 > 0.95 with the QTL for the 3-QTL+ and 30-QTL+ data. In the 3-QTL+ data, only 2 of 27 SNPs that explained >20% of the genetic variance within their replicate were not detected in the sense that their posterior probability of being causative was <50%. For the 30-QTL+ data these numbers were 4 of 29 QTL. In the 3-QTL+ data, 22 of these 27 causative SNPs were fitted with a posterior probability >0.9, and in the 30-QTL+ data these numbers were 19 of 29. Although these numbers seemed similar for the 3-QTL+ and 30-QTL+ data, it may be noted that there were 10 times as many QTL in the 30-QTL+ data, and thus it was much less likely that a QTL explained >20% of the genetic variance. These high posterior probabilities are achieved despite the fact that the prior probability of a causative SNP is very low, namely on average 9 × 10−5 (= 3/33,066) and 9 × 10−4 for the 3-QTL+ and 30-QTL+ data, respectively. As the number of training records, T, increases, the posterior probabilities are expected to further increase.
Figure 3.—

The posterior probability of fitting a causative SNP vs. the genetic variance explained by this SNP. (a and b) The 3-QTL+ and 30-QTL+ data, with T = 200 records; (c) the 6-QTL+ data with 400 records.
Figure 3C investigates whether the accuracy of detecting the QTL is maintained if the genome size is doubled and therewith the number of QTL. Since each individual QTL is expected to explain only half as much of the total genetic variance, the number of records was doubled as well (T = 400). In this situation, there were 27 QTL (across 10 replicates) that each explained >10% of the genetic variance, of which only 4 had a posterior probability of being causative <50%. Twenty-one of these 27 had a posterior probability >0.9. Thus, with a larger number of records it is possible to detect QTL explaining a smaller proportion of the total variance.
DISCUSSION
The effect of affordable whole-genome sequencing technology on the accuracy of genomic selection was that (1) the accuracy of selection was substantially higher than that achieved with much smaller-sized SNP chips and increased approximately linearly with the log of the number of SNPs (see Figure 1), (2) the accuracy increased a further 2.5–3.7% when the causative mutations were included even if the marker data were already very dense, (3) the estimates of the high-density SNP effects yielded accurate estimated breeding values (EBVs) even when the training and evaluation data were 10 generations apart, and (4) the GWBLUP estimation method does not take full advantage of the high-density marker data and therefore a method such as the Bayesian method used here (BayesB; Meuwissen et al. 2001) gives a much higher accuracy than the BLUP method (Table 3).
The explanation for point 4 probably is that, as marker density increases, the variance due to each marker decreases in BLUP, whereas BayesB is increasingly able to detect the causative SNPs and estimate their effects. The latter, however, assumes that there are a limited number of causative SNPs, which was assumed to be 3 or 30 per morgan here. If in reality the number of causative SNPs is very large (say hundreds or thousands per morgan), the BLUP method may yield as accurate predictions as other methods, because the assumption that every SNP has an effect is then close to reality. Even under an infinitesimal model, the effective number of QTL approaches a limit of M, equal approximately to 2NeL, due to linkage (Daetwyler et al. 2008; Goddard 2009). In our simulation, Ne = 1000 and L = 1, so M = 2000 effective loci. Since we simulated only 3 or 30 QTL per morgan, BayesB is expected to have a large advantage over GWBLUP, as observed. As the number of QTL approaches M, the advantage of BayesB over GWBLUP will vanish. If in livestock breeds, such as Holstein cattle, where Ne ∼ ≤100 (Sorensen et al. 2005), the real number of QTL approaches 200 per morgan, this could explain why GWBLUP yields as high accuracy as BayesB in many cases. Alternatively, if the SNP density is not high enough to generate a single SNP in high LD with each QTL, the effect of each QTL may be spread over many SNPs each with small effects, causing the SNP effects to approach an infinitesimal model more closely than do the QTL effects. In this case, increasing the SNP density should cause BayesB to have higher accuracy than GWBLUP because again it is able to fit fewer larger effects.
In this study gene effects were sampled from a double-exponential distribution, which is a thick-tailed distribution and fits quite well with the assumption of a thick-tailed t prior distribution in BayesB. In practical applications, the distribution of the gene effects will not be known, but most studies that investigated the distribution of allelic effects found an exponential or other thick-tailed distribution (for reviews see Erickson et al. 2004 and Rocha et al. 2004). Habier et al. (2007) and Meuwissen (2009) compared the accuracy of allelic effects simulated from a thick-tailed and a normal distribution and found little difference in accuracy. In the case of normally distributed allelic effects, the accuracy of BayesB estimates of EBV was reduced by 1.5%. The reduced accuracy for the normally distributed effects is expected since there will be fewer big genes; i.e., effectively the number of QTL increases.
Gene effects were assumed to be additive in both the simulated data and the data analysis. Hill et al. (2008) showed that genetic variance is mainly additive in nature, but some effects will be nonadditive. The simplest genetic interaction is the within-locus interaction, which causes dominance variance. We therefore also simulated a scheme where 20% of the genetic variance was dominance variance, which is an upper limit for this variance in the models of Hill et al. (2008). The degree of dominance, d/a, was adjusted for every QTL such that 20% of its total genetic variance was dominance variance, and the error variance was reduced accordingly such that the narrow sense heritability remained 0.5, where a (d) is the additive (dominance) effect (Falconer and Mackay 1996). Table 4 shows that there was almost no reduction in accuracy when there were 30 QTL whereas there was a marked reduction of ∼13% when there were 3 QTL. An explanation for this is that in the case of 30 QTL, dominance deviations of individual QTL are small and taken together act approximately like an independent error component, which is accounted for in the analysis. However, when there are only 3 QTL, dominance deviations of individual QTL are large and not so well modeled as part of the error variance and consequently the accuracy with which the additive effects are estimated is reduced. In conclusion, dominance variance may reduce the accuracy of genomic selection noticeably, if few very large QTL show a lot of dominance, but it does not eliminate its accuracy; and if many QTL are responsible for the dominance variance, there is hardly any reduction in accuracy. This conclusion may also hold for more complicated epistatic interactions; i.e., if few very large genes show strong interactions, accuracy will be reduced, but if the epistatic variance is due to many interacting genes, the reduction in accuracy is expected to be small.
TABLE 4.
The accuracy of the predictions of total genetic value (±SE) in the TEST1 data set when the training data contained T = 200 individuals and 0 or 20% of the total genetic variance was due to dominance variance
| Dominance variance |
||
|---|---|---|
| Data set | 0% | 20% |
| 3 QTL | 0.973 ± 0.004 | 0.841 ± 0.015 |
| 30 QTL | 0.826 ± 0.019 | 0.827 ± 0.009 |
Whole genome-sequence data were used; i.e., the causative SNPs were included in the analysis.
Using the current findings, accuracy can be predicted for other values of the input parameters as long as λ = Th2/ML remains constant. This scaling of the results was investigated in Tables 1 and 2 for increased genome sizes and reduced heritabilities, respectively, and was found to be reasonably accurate. For instance, since the effective number of segments per morgan is approximately linear with Ne (Goddard 2009), a realistic situation in humans with Ne ∼ 8000, L ∼ 37, Nqtl = 1100, h2 ∼ 0.5, and T = 60,000 is expected to yield approximately the same accuracy as Ne = 1000, L = 1, Nqtl = 30, h2 = 0.5, and T = 200 (0.826 from Table 1). Similarly, if a population with many breeds of cattle has Ne ∼ 1000, L ∼ 30, Nqtl = 900, h2 ∼ 0.25, and T = 12,000, this is also expected to yield the same accuracy (0.826). The large effective size of the human population makes the human genome in effect larger than its 37 M: it reduces the size of the LD blocks, i.e., more blocks per morgan, and the total number of SNPs is increased (severalfold larger than in our simulation), since heterozygosity increases with effective size.
The simulations assumed no selection to improve the interpretability of the results, since selection can complicate the genomic architecture markedly and thus needs further investigations. Selection could have a variety of effects on the accuracy of genomic selection. For instance, artificial selection that increased the frequency of an initially rare allele would increase the surrounding LD and probably increase the accuracy of genomic selection. On the other hand, natural selection against mutations that increase disease risk will lead to segregation of alleles that are relatively young, are at low frequency, and have low LD with markers, and hence the accuracy of genomic selection will be reduced.
We expected that with the use of whole-genome sequence data, since it includes the causative SNPs, the BayesB-type of estimation procedure would be able to pinpoint the position of the QTL. Figures 2 and 3 show that this is the case for the biggest QTL, although sometimes SNPs that are in very high LD with the QTL are used to explain the QTL. This high precision in pinpointing the QTL or a SNP that is in very high LD with the QTL explains why the accuracy persisted across generations (Table 2). This is contrary to the results of, e.g., Habier et al. (2007), who used a much lower marker density and found that accuracy decreased substantially over time. However, another important difference with the study of Habier et al. is that both the historical and the recent population sizes were much bigger in our simulations (factors of 10 and 100, respectively), which means that the training data set is much less likely to contain closely related individuals, and thus the SNP effects are not picking up family effects (as reported by Habier et al.). The latter implies that the training data need to contain individuals with as little as possible relationship with each other and SNP density needs to be high for the accuracy to persist across generations.
The population history was chosen such that the training (TRAIN) and test populations were quite unrelated due to the large effective size during the last 10 generations. This large recent effective size does not change the LD patterns much, since genetic drift is low, but reduces the probability of close relationships. In real livestock populations, effective population sizes were often very large in prehistoric times, decreased markedly during the more recent past, and show often a strong family structure, i.e., few dominating families, during the last few generations. This creates an LD structure with relatively high LD over long distances, but the LD does not increase much as distance between the SNPs decreases (Goddard and Hayes 2009). This long-distance LD should make genomic selection relatively accurate even with sparse markers, but the accuracy will not improve much as marker density increases. Thus, the increase in accuracy as density increases will be much less dramatic than in Figure 1. Also, the long-distance LD may make GWBLUP more accurate relative to BayesB since many markers can be used to pick up a single QTL effect and this more closely fits the assumption of GWBLUP that all markers have effects.
For the human population of European descent, the LD structure is quite different from that of cattle (Goddard and Hayes 2009): there is a lot of LD between very closely linked SNPs, due to the relatively small prehistoric effective population size, but very little LD between the more distant SNPs due the recent large increase in effective size. This may increase the steepness of the curves in Figure 1, since accuracies at lower SNP densities will be markedly reduced. Thus for humans, whole-genome sequence data may increase the accuracy of predicting breeding value more than predicted from our simulations.
Even at a cost of $1000 per genome sequence, the sequencing of 12,000–60,000 genomes, as in the above examples of cattle and humans, respectively, will cost $12–60 million. However, these costs may be reduced substantially by the whole-genome sequencing of a limited subset of the individuals and using a SNP chip for the genotyping of the majority of the individuals, followed by the imputation of their missing genotypes (Scheet and Stephens 2006). In this way, the high accuracy from whole-genome sequencing can be combined with the lower costs of a SNP chip (Goddard 2009).
The results imply that accurate prediction of genetic value for complex traits in livestock and humans is within reach. This assumes that the number of QTL per trait is hundreds to thousands and that tens of thousands of individuals can be used to derive the prediction equation. Although the latter assumption implies a high cost, there are already tens of thousands of people included in genome-wide association studies for many traits. Similar numbers of dairy cattle have already been genotyped for the Illumina 50K SNP chip. Compared to our simulation, these data sets have orders of magnitudes larger numbers both of SNPs and of genotyped individuals, and the algorithms used here will not be able to perform the required data analyses. Hence, analyzing these data will require new, faster, and less memory-intensive algorithms. Accurate predictions of genetic value will revolutionize genomic selection in livestock, genetic risk prediction in humans, and personalized medicine. To date genetic predictions of individual disease risk have not been highly accurate, causing some to question their value (Janssens and VanDuijn 2008), but these results suggest that with a combination of genome sequence data, large sample sizes, and a statistical method that detects the polymorphisms that are informative (such as BayesB used here), high accuracy is attainable.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.110.116590/DC1.
References
- Barrett, J. C., S. Hansoul, D. L. Nicolae, J. H. Cho, R. H. Duerr et al., 2008. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat. Genet. 40 955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daetwyler, H. D., B. Villanueva and J. A. Woolliams, 2008. Accuracy of predicting the genetic risk of disease using a genome-wide approach. PLoS ONE 3 e3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson, D. L., C. B. Fenster, H. K. Stenoien and D. Price, 2004. Quantitative trait locus analyses and the study of evolutionary process. Mol. Ecol. 13 2505–2522. [DOI] [PubMed] [Google Scholar]
- Falconer, D., and T. Mackay, 1996. Introduction to Quantitative Genetics. Longman, London. [DOI] [PMC free article] [PubMed]
- Goddard, M. E., 2009. Genomic selection: prediction of accuracy and maximisation of long term response. Genetica 136 245–257. [DOI] [PubMed] [Google Scholar]
- Goddard, M. E., and B. J. Hayes, 2009. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 10 381–391. [DOI] [PubMed] [Google Scholar]
- Habier, D., R. L. Fernando and J. C. Dekkers, 2007. The impact of genetic relationship information on genome-assisted breeding values. Genetics 177 2389–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes, B. J., and M. E. Goddard, 2001. The distribution of the effects of genes affecting quantitative traits in livestock. Genet. Sel. Evol. 33 209–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W. G., M. E. Goddard and P. M. Visscher, 2008. Data and theory point to mainly additive genetic variance for complex traits. PLoS Genet. 4 e1000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggart, C. J., M. Chadeau-Hyam, T. G. Clark, R. Lampariello, J. C. Whittaker et al., 2007. Sequence-level population simulations over large genomic regions. Genetics. 177 1725–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 2002. Generating samples under a Wright-Fischer neutral model of genetic variation. Bioinformatics 18 337–338. [DOI] [PubMed] [Google Scholar]
- Janssens, A. C., and C. M. VanDuijn, 2008. Genome-based prediction of common diseases: advances and prospects. Hum. Mol. Genet. 17 R166–R173. [DOI] [PubMed] [Google Scholar]
- Kimura, M., 1969. The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics 61 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingman, J. F. C., 1982. On the genealogy of large populations. J. Appl. Probab. 19A 27–43. [Google Scholar]
- Mardis, E. R., 2008. Next-generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet. 9 387–402. [DOI] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., 2009. Accuracy of breeding values of ‘unrelated’ individuals predicted by dense SNP genotyping. Genet. Sel. Evol. 41 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., B. J. Hayes and M. E. Goddard, 2001. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha, J. L., E. J. Eisen, L. D. VanVleck and D. Pomp, 2004. A large-sample QTL study in mice: I. Growth. Mamm. Genome 15 83–99. [DOI] [PubMed] [Google Scholar]
- Scheet, P., and M. Stephens, 2006. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 78 629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure, J., and H. Ji, 2008. Next-generation DNA sequencing. Nat. Biotechnol. 26 1135–1145. [DOI] [PubMed] [Google Scholar]
- Sorensen, A. C., M. K. Sorensen and P. Berg, 2005. Inbreeding in Danish dairy cattle breeds. J. Dairy Sci. 88 1865–1872. [DOI] [PubMed] [Google Scholar]
- TenBosch, J. R., and W. W. Grody, 2008. Keeping up with the next generation: massively parallel sequencing in clinical diagnostics. J. Mol. Diagn. 10 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRaden, P. M., C. P. VanTassell, G. R. Wiggans, T. S. Sonstegard, R. D. Schnabel et al., 2009. Invited review: reliability of genomic predictions for North American Holstein bulls. J. Dairy Sci. 92 16–24. [DOI] [PubMed] [Google Scholar]