

Abstract
We have previously reported on the anti-tumoral potential of a chaperone-rich cell lysate (CRCL) vaccine. Immunization with CRCL generated from tumors elicits specific T and NK cell-dependent immune responses leading to protective immunity in numerous mouse tumor models. CRCL provides both a source of tumor antigens and danger signals leading to dendritic cell activation. In humans, tumor-derived CRCL induces dendritic cell activation and CRCL-loaded dendritic cells promote the generation of cytotoxic T lymphocytes in vitro. The current study was designed to identify the signaling events and modifications triggered by CRCL in antigen presenting cells. Our results indicate that tumor-derived CRCL not only promotes the activation of dendritic cells, but also significantly fosters the function of macrophages that thus appear as major targets of this vaccine. Activation of both cell types is associated with the induction of the MAP kinase pathway, the phosphorylation of STAT1, STAT5 and AKT and with transcription factor NF-κB activation in vitro and in vivo. These results thus provide important insights into the mechanisms by which CRCL-based vaccines exert their adjuvant effects on antigen presenting cells.
Keywords: Tumor vaccines, Dendritic cells, Macrophages, Cell signaling
Introduction
Heat shock proteins (HSP), also known as stress proteins or chaperone proteins, are the most abundant intracellular proteins present in all living organisms (Smith et al., 1998). They display a multitude of housekeeping functions, including the folding of nascent polypeptides, refolding of denatured proteins and stabilizing proteins against aggregation and intracellular transport. Chaperone proteins are essential for cellular survival and for protecting cells from a variety of damaging environments. Purified individual tumor-derived chaperone proteins such as HSP 70, HSP 90, GRP94/gp96, and calreticulin (CRT) have also shown promise as vaccines which are capable of generating tumor-specific T cell responses leading to protective anti-cancer immunity (Heike et al., 1999; Li, 1997; Srivastava and Amato, 2001; Srivastava et al., 1998; Wells and Malkovsky, 2000). The underlying principle for the use of chaperone proteins as vaccines is based on their intrinsic function as intracellular carriers of antigenic peptides. The capture of chaperone proteins and their associated antigenic peptides by antigen presenting cells (APC) may foster the delivery of these peptides into the antigen-processing pathways and may lead to their MHC-dependent presentation to T cells (Li et al., 2002; Srivastava, 2002a; Srivastava, 2002b). Some groups have reported that chaperone proteins may also act as natural adjuvants or ‘danger signals’ that are capable of activating antigen-presenting cells (Segal et al., 2006; Srivastava et al., 1998). This effect has however been disputed by other studies suggesting that endotoxin contamination or LPS binding to chaperone proteins purified from tissues may play a major role in APC activation (Gao and Tsan, 2003; Nicchitta, 2003; Radsak et al., 2003; Reed et al., 2003; Wallin et al., 2002).
A cancer cell-based vaccine has been developed in our laboratory which consists of tumor lysates enriched for the major immunogenic chaperone proteins using a free solution-isoelectric focusing technique (FS-IEF) (Graner et al., 2000a; Graner et al., 2004; Zeng et al., 2006). The enriched, tumor-derived chaperone proteins form large, multi-chaperone complexes termed Chaperone Rich Cell Lysate(s) (CRCL) that contain the HSP90 and HSP70 family members, the endoplasmic reticulum chaperones glucose regulated protein (GRP)94/glycoprotein (gp)96, and calreticulin (Graner et al., 2000a; Graner et al., 2000b; Graner et al., 2003; Zeng et al., 2006). CRCL is thus different from conventional individually purified chaperone preparation used as vaccine. In numerous animal models, CRCL demonstrated a more pronounced anti-tumor effect per unit material of protein than the individual purified chaperone protein alone (Graner et al., 2000a; Graner et al., 2000b; Graner et al., 2003; Zeng et al., 2003). CRCL elicited specific T cell responses resulting in tumor regression (Graner et al., 2000a; Graner et al., 2000b; Graner et al., 2003). Previous results have indicated that the efficacy of this vaccine stems in part from its ability to induce dendritic cell (DC) activation through upregulation of CD40 expression and IL-12 production (Zeng et al., 2003).
The objective of this study was to identify the main signaling events and modifications associated with CRCL-induced activation of APC. We herein demonstrate that tumor-derived CRCL, in addition to triggering DC pro-inflammatory cytokine secretion also promotes the upregulation of the recently described co-stimulatory molecule, CD70. In addition, CRCL stimulates the direct tumor killing activity of macrophages, which is associated with inducible nitric oxide synthase (iNOS) expression and nitric oxide (NO) production. The molecular signaling events associated with CRCL-mediated activation of APC involve the induction of the MAP kinase pathway evidenced by increased phosphorylation of ERK1/2 and p38, the activation of the transcription factor NF-κB and the phosphorylation of STAT1, STAT5 and AKT.
Materials and Methods
Mice
Mice were housed under specific pathogen-free conditions and cared for according to the guidelines of the University of Arizona Institutional Animal Care and Use Committee. Female BALB/c (H2d) mice were obtained from the National Cancer Institute (Bethesda, MD) and used at the age of 6-8 weeks.
Cell lines
The murine leukemia cell line 12B1 (given by Dr Wei Chen (Cleveland Clinic, Cleveland, OH) was obtained by retroviral transformation of BALB/c mouse bone marrow cells with the human bcr/abl (b3a2) fusion gene and express the p210 bcr-abl protein (McLaughlin et al., 1987). This is an aggressive leukemia, with the 100% lethal dose (LD100) being 100 cells after tail vein injection (He et al., 2001). The murine macrophage cell line RAW264.7 and the melanoma tumor cell line B16 were purchased from the American Type Culture Collection (Manassas, Virginia). Cells were cultured at 37°C and 10% CO2 in DMEM or RPMI media (Gibco/BRL, Gaithersburg, MD) containing 10% heat-inactivated fetal calf serum (Gemini Bio-products, Woodland, CA) and supplemented with 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin sulfate, 0.025 μg/ml amphotericin B, 0.5× MEM nonessential amino acids and 1 mM sodium pyruvate (complete media, CM). Cells were tested routinely and found to be free of Mycoplasma contamination.
Bone marrow-derived DC and peritoneal macrophages
DC were generated from BALB/c bone marrow cells as previously reported (Larmonier et al., 2008b). Cells were harvested from femurs and tibiae and filtered through a Falcon 100-μm nylon cell strainer (Becton Dickinson Labware, Franklin Lakes, NJ). Red blood cells were lysed in a hypotonic buffer (150 mM NH4Cl, 1 mM KHCO3, 0.1 mM Na2EDTA) and total bone marrow cells were cultured at a density of 5 × 105 cells/ml in a complete RPMI medium. Murine granulocyte-macrophage colony-stimulating factor (GM-CSF; Peprotech, Rocky Hill, NJ) and Interleukin-4 (IL-4; Peprotech) were added at the concentration of 10 ng/ml each. Complete RPMI medium containing GM-CSF and IL-4 was added on day 2. On day 5 non-adherent and loosely adherent cells were collected, washed in complete RPMI and used in further experiments. Flow cytometry analysis indicated that approximately 70% of the cells expressed CD11c. Macrophages were obtained by washing BALB/c naïve mouse peritoneal cavity with 5 ml of cold RPMI medium. Macrophage number was determined by counting adherent cells after a 1-hr attachment period on a hemocytometer glass surface at 37°C in 5% CO2. Cells were seeded and used after a 24 hr-adherent step in a flat bottom 96-well plate following elimination of floating cells by extensive washes.
CRCL preparation
Tumor-derived CRCL (12B1 or B16) was prepared as described previously (Graner et al., 2000a; Graner et al., 2000b; Graner et al., 2003). The endotoxin level contained in the CRCL preparation was determined using the Limulus Amebocyte Lysate (LAL) assay kit (Cambrex Bio Science, Walkersville, MD) according to the manufacturer's instructions. The level of endotoxin in CRCL was lower than that in media control (<0.01 EU/μg).
Detection of cytokine and chemokine production by ELISA
The concentration of TNF-α, IL-12 or RANTES in DC or macrophage culture supernatants was determined using enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer's instructions (eBiosciences, San Diego, CA).
Real Time PCR
Total RNA was extracted using TRIzol reagent (GibcoBRL). Ten micrograms of total RNA was treated with DNase I according to the manufacturer's protocol (Ambion, Austin, TX). The resulting RNA was evaluated by agarose gel electrophoresis and concentrations determined by spectrophotometry on an MBA 2000 spectrophotometer (Perkin Elmer, Boston, MA). 500 ng of DNase I-treated RNA was reverse-transcribed using Bio-Rad iScript (Bio-Rad, Hercules, CA) according to the manufacturer's protocol. Subsequently, 20-μl PCR reactions were set up in 96-well plates containing 10 μl of TaqMan universal PCR master mix, 1 μl of TaqMan primer/probe set, 2 μl of cDNA synthesis reaction (out of a 20-μl total volume), and 7 μl of molecular grade water. Reactions were run and analyzed on a Bio-Rad iCycler iQ Real-Time PCR detection system. Data were analyzed using the comparative Ct method as a means of relative quantization, normalized to an endogenous reference (18S ribosomal RNA) and relative to a calibrator (normalized CT value obtained from medium-alone cells) and expressed as 2–Δ CT according to Applied Biosystems User Bulletin 2: Rev B Relative Quantitation of Gene Expression. The real-time PCR products were separated on a 2% agarose E-Gel (Invitrogen, Carlsbad, CA) and visualized by ethidum bromide.
Flow cytometry analysis and antibodies
Day 5 DC cultured as described above and treated or not for 48 hrs with LPS or tumor-derived CRCL, with or without anti-CD40 Abs were incubated for 10 min with an Fc receptor-blocking antibody (BD Biosciences Pharmingen, San Diego, CA, USA), then for 30 min with saturating amounts of the appropriate primary antibodies in PBS. Cells were then washed three times in PBS and analyzed using a FACScan (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA). A minimum of 10,000 events were recorded for each sample and data were analyzed using a CellQwest software (Becton Dickinson Immunocytometry Systems). The following antibodies were used: PE-anti-CD70 (eBioscience, San Diego, CA, USA) and APC-anti-CD11c (Miltenyi Biotech). Isotype control antibodies were obtained from BD Biosciences Pharmingen.
Detection of NF-κB and iNOS by immunohistochemistry
Mice were injected (s.c) with PBS, LPS (1 μg/ml) or 12B1 tumor-derived CRCL (25 μg/ml). After 24 hrs, draining lymph nodes were removed, embedded in Tissue-Tek (Sakura Finetek USA, Torrance, CA) or snap-frozen in liquid nitrogen. Cryostat tissue sections were prepared and slides were treated as described (Larmonier et al., 2008a). RAW264.7 macrophages were plated on slide chambers, exposed for 24 hrs to tumor-derived CRCL (25 μg/ml) or LPS (1 μg/ml), fixed with methanol and permeabilized with 0.1% saponin. Slides were incubated with a primary antibody against iNOS or phospho-NF-kB p65 (RelA) (Ser276; 1/50 dilution, Cell Signaling Technology, Danvers, MA) in PBS containing 1% BSA, overnight at 4°C. After 3 washes in PBS, slides were incubated with biotinylated secondary antibody and streptavidin/peroxidase complex according to the manufacturer's recommendation (KPL). Slides were then incubated with 3,3′-diaminobenzidine (Vector laboratories, Burlingame, CA) and mounted with Vectamount medium (Vector Laboratories). Slide examination was performed independently by two experienced scientists in a blind manner using a Zeiss Axioplan microscope (Carl Zeiss MicroImaging, Thornwood, NY). Images were captured with Nikon Digital Sight DS-Fi1 camera and NIS-Element software (Nikon Instruments, Melville, NY). Staining was scored by counting the number of positive cells per high-power field (200× magnification). A total of six fields in three randomly chosen sections were analyzed for each group.
Detection of the expression of iNOS and of the phosphorylation of I-κB, STAT1, STAT3, STAT5, ERK1/2, p38 and AKT by western blotting
DC and peritoneal macrophages were cultured as described above. RAW264.7 cells were cultured in 25 cm2 flasks. Cells were lysed in a lysis buffer (1% Nonidet P40, 50 mM Tris HCl, pH 7.4, 2 mM EDTA, 100mM NaCl, 0.2 mg/mL Aprotinin, 0.2 mg/mL Leupeptin, 1 mM PMSF, 10 mM NaF, 30 mM NaPPi, 10 mM Na3VO4). DC or macrophages treated with LPS were used as positive control. Negative controls consisted of DC or macrophages cultured alone. Western blot analysis was then performed as described (Larmonier et al., 2007), using the following antibodies: anti-phospho I-κB, anti I-κB, anti-phospho STAT1, anti-STAT1, anti-phospho STAT3, anti-STAT3, anti-phospho STAT5, anti-STAT5, anti-phospho ERK1/2, anti-ERK1/2, anti-phospho p38, anti-p38, anti-phospho AKT, anti-AKT, anti-phospho IRAK1 or anti-IRAK1 (Cell Signaling, Beverly, MA) or anti-iNOS (Santa Cruz Biotechnology, Inc, Santa Cruz, CA), or anti-β-actin (Sigma, St. Louis, MO).
Determination of Nitrite production
The amount of NO2- released by macrophages was detected in fresh cell-free supernatants by the colorimetric Griess reagent kit as described by the manufacturer's instructions (Molecular Probes, Eugene, OR). The results were expressed in micromoles of NO2- per ml.
Evaluation of macrophage killing activity
Peritoneal macrophages obtained as described above and treated with LPS (1 μg/ml) or CRCL (25 μg/ml) were cultured with tumor cells for 24 hrs. Tumor cell killing was assessed using crystal violet assays as reported (Larmonier et al., 2006). The death of detached tumor cells was confirmed by trypan blue staining and by the inability of these cells to form colonies when sub-cultured in fresh culture medium for 4 days.
Detection of NF-κB activation by DNA-binding transcription factor ELISA assay
Nuclear extracts were harvested from DC (1 × 106 cells) or macrophages (5 × 105 cells) using a nuclear extract kit (Active Motif, Carlsbad, CA). NF-κB P50 DNA-binding activity was then assessed using 15 μg of nuclear extracts according to the manufacturer's recommendations (NF-κB P50 Trans-AM™ kit, Active Motif). DC or macrophages treated with LPS (1 μg/ml) were used as positive control.
NF-κB plasmid transfection and Luciferase assay
Cells were transiently transfected using the TransIT express transfection reagent according to manufacturer's instructions (Mirus Bio, Madison, WI). After cell lysis, reporter gene assay was performed according to manufacturer's instruction (Luciferase Reporter assay system; Promega, Madison, WI). Luciferase activity was measured with a tube luminometer (Femtomaster FB12, Berthold Detection System, Pforzheim, Germany) and expressed as normalized luciferase activity relative to protein concentration (BCA assay; Pierce, Rockford, IL).
Statistical analysis
Unless specified otherwise, all experiments were performed in triplicate and reproduced three times. Statistical analyses comparing values between groups were performed using the Student's t test or by the ANOVA followed by Fisher paired least-significant difference (PLSD) post hoc test with StatView software package v.4.53 (SAS Institute, Cary, NC).
Results
Tumor-derived CRCL induces CD70 expression by DC
CRCL was produced from 12B1 leukemia tumors as previously reported (Graner et al., 2000a). Before analyzing the signaling events and changes induced by CRCL in APC, it was critical to ensure that this particular vaccine preparation was biologically active. Consistent with our previous results (Larmonier et al., 2007; Zeng et al., 2003), tumor-derived CRCL enhanced pro-inflammatory cytokine and chemokine production by DC and primary macrophages (Supplemental figure S1) and by the murine macrophage cell line RAW264.7 (Supplemental figure S2).
We have previously reported that CRCL up-regulates the expression of co-stimulatory molecules CD40 and to a lesser extent CD80 and CD86 in DC (Zeng et al., 2003). CD70 is a recently described co-stimulatory molecule expressed by mature DC. The interaction of CD70 with its ligand CD27, expressed by resting T cells, activated B cells, and NK cells, is critical for the development of potent cell-mediated immunity (Tesselaar et al., 2003). It has been suggested that some TLR ligands such as Pam3Cys-SK4 (TLR1/2 agonist), Poly(I:C) (TLR3 agonist), and MPL (TLR4 agonist) combined with CD40 signaling may promote optimal expression of CD70 (Sanchez et al., 2007). Based on these observations, we reasoned that tumor-derived CRCL may induce up-regulation of DC CD70 expression. To test this hypothesis bone marrow-derived DC were treated with LPS or 12B1 tumor-derived CRCL, in the presence or absence of an anti-CD40 antibody. Expression of CD70 was then determined by flow cytometry. The results depicted in Figure 1A indicate that CRCL was capable of inducing the upregulation of CD70 by DC. Furthermore, CD70 expression was significantly enhanced by the presence of anti-CD40 antibodies (Figure 1B and 1C). These results thus highlight CD70 as an additional co-stimulatory molecule induced by tumor-derived CRCL.
Figure 1. CRCL induces the upregulation of the co-stimulatory molecule CD70 by DC.
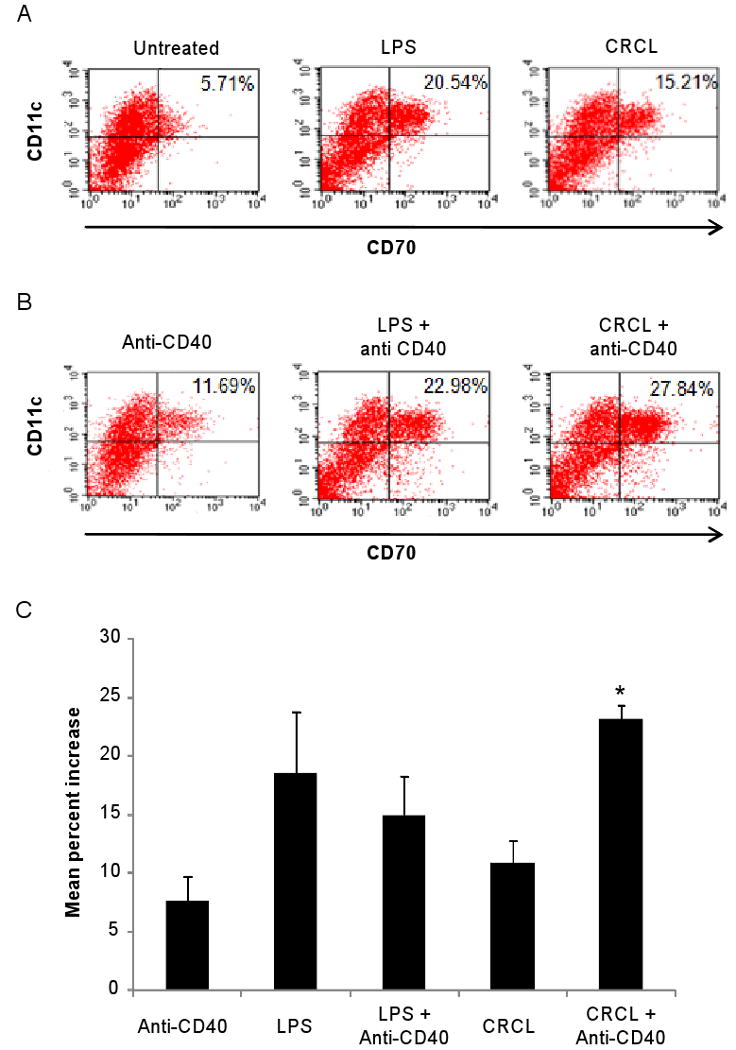
(A) Day 5 bone marrow derived DC were treated with tumor-derived CRCL (25 μg/ml), or LPS (1 μg/ml) as positive control. (B) Similar as (A) but in presence of anti-CD40 antibody (5 μg/ml). After 48 hrs cells were stained with APC-conjugated anti-CD11c mAb and PE-conjugated anti-CD70 mAb and analyzed by flow cytometry. (A, B), Representative dot plots of three independent experiments. (C) Mean percent increase in CD70 expression by DC following the indicated treatments compared to untreated DC. *, a significant difference compared to DC treated with CRCL alone (p<0.05).
CRCL enhances macrophage iNOS expression, NO production and tumoricidal activity
Macrophages represent a population of cells located at the nexus between innate and adaptive immunity. They may act as antigen presenting cells as well as effector killer cells and have been reported to be involved in the regulation of tumor immunity (Bonnotte et al., 2001; Klimp et al., 2002; Kryczek et al., 2006). Based on the central role of these cells in tumor growth control and on our observation that tumor-derived CRCL prominently promote macrophage activation, we sought to further define which aspect(s) of macrophage function(s) may be modulated by the CRCL vaccine. Activated macrophages up-regulate iNOS expression, the inducible form of nitric oxide synthase, which leads to nitric oxide (NO) production (Bogdan, 2001). NO is a major effector molecule involved in macrophage cytotoxic activity (Bogdan, 2001; Klimp et al., 2002). We therefore sought to determine whether CRCL may stimulate iNOS expression and NO secretion. IHC staining of the RAW264.7 macrophage cell line indicates that the number of iNOS-expressing cells was significantly increased following CRCL (9±5 iNOS positive cells/field, p<0.01) or LPS (16±4 iNOS positive cells/fields, p<0.001) treatment compared to untreated cells (0.5±0.8 iNOS positive cells/fields) (Figure 2A). Western blot analysis further demonstrates that CRCL was capable of triggering iNOS expression in RAW264.7 and primary (peritoneal) macrophages (Figure 2B). Furthermore, the concentration of nitrites, the principal metabolites of NO, was increased in the culture supernatants of CRCL-activated peritoneal or RAW264.7 macrophages (Figure 2C). LPS and CRCL induced the production of comparable levels of nitrites, but the expression of iNOS was higher in CRCL-treated RAW264.7 macrophages suggesting a reduced activity of this enzyme. We next evaluated the influence of CRCL on the direct killing activity of macrophages towards cancer cells. The results depicted in figure 3 demonstrate that CRCL significantly promoted the cytotoxic potential of macrophages against tumor cells (Figure 3). No direct effects of LPS or CRCL on cancer cells were observed (not shown). The death of detached tumor cells upon contact with macrophages was confirmed by trypan blue staining (97±5% trypan blue positive dead cancer cells for the CRCL-treated macrophage group and 94±7% for the LPS-treated macrophage group) and the inability of these detached cells to form colonies when sub-cultured in fresh culture medium for 4 days (not shown). Taken together, these findings demonstrate that CRCL stimulates the tumoricidal function of macrophages which is associated with the induction of iNOS expression and NO production and further highlight these cells as major targets of CRCL.
Figure 2. CRCL induces iNOS expression and NO production by macrophages.
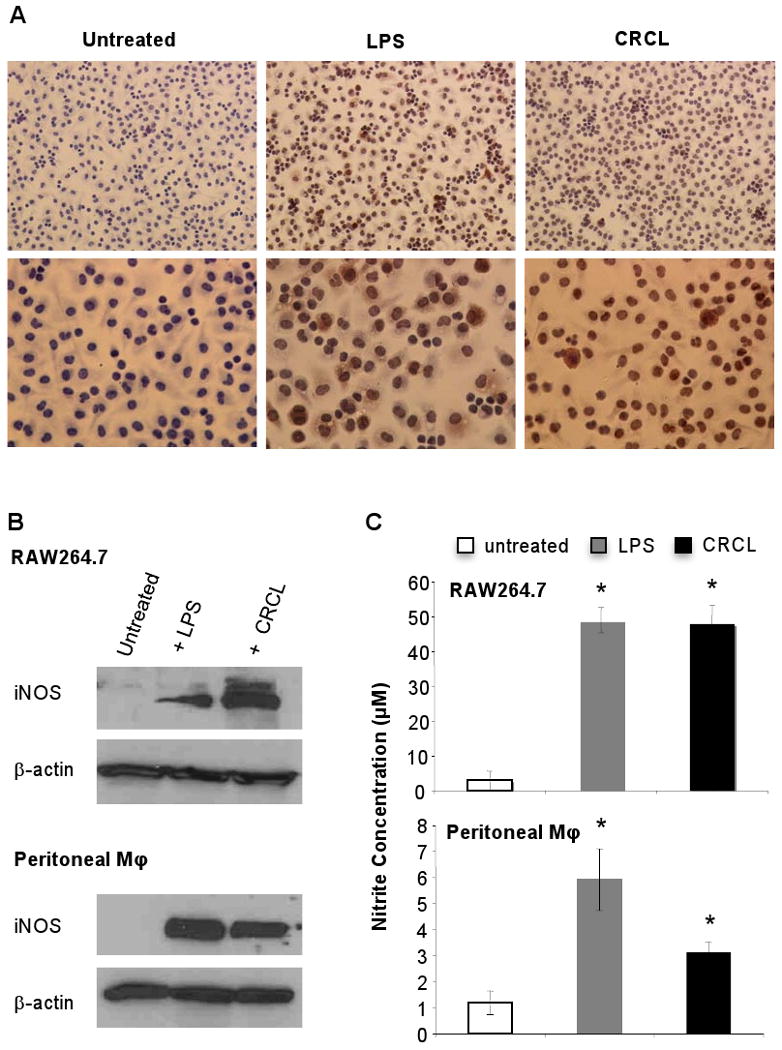
(A) RAW264.7 cells were plated on slide chambers and exposed for 24 hrs to tumor-derived CRCL (25 μg/ml). Cells were then washed and stained for iNOS expression by IHC. Upper panels, 200× magnification, lower panels 400× magnification. (B) Western blot detection of iNOS expression in RAW264.7 or peritoneal macrophage treated with CRCL (25 μg/ml). (C) RAW264.7 cells or peritoneal macrophages were incubated for 12 hrs with CRCL (25 μg/ml) and nitrite concentration was evaluated in the culture supernatants. In all the experiments LPS (1 μg/ml)was used as a positive control. *, a significant difference when compared to untreated control cells (p < 0.05).
Figure 3. CRCL triggers macrophage tumoricidal activity.
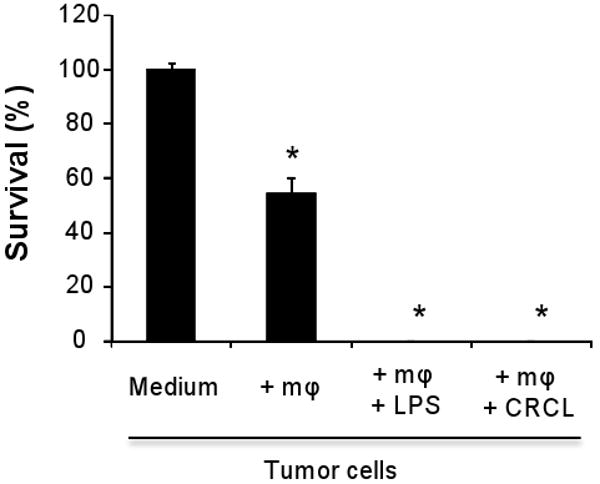
Peritoneal macrophages (50,000 cells per well) were cultured with B16 tumor cells (20,000 cells per well) and treated or not with tumor-derived CRCL (25 μg/ml) or with LPS (1 μg/ml). After 24 hrs cell viability was determined using a crystal violet toxicity assay. B16 cells alone (Medium) and B16 cells cultured with macrophages (+mφ) were used as negative controls. The data are representative of three independent experiments. *, a significant difference when compared to tumor cells culured alone (p < 0.05).
CRCL triggers NF-κB activation in DC and macrophages
The transcription factor NF-κB plays a key role in the activation of DC and macrophages (Kawai and Akira, 2006). Its activation results in the secretion of pro-inflammatory cytokines and chemokines and is associated with NO production (Binder et al., 2004). NF-κB activation depends on the cytoplasmic phosphorylation of its inhibitor I-κB by I-κB kinase (Akira and Takeda, 2004; Kawai and Akira, 2006). Once phosphorylated, I-κB dissociates from NF-κB that translocates into the nucleus where it binds to its specific DNA promoter sequence upstream of target genes. The results depicted in Figure 4A indicate that tumor-derived CRCL is capable of triggering I-κB phosphorylation in both bone marrow derived DC and peritoneal macrophages. Compared to LPS, CRCL induced a higher phosphorylation of I-κB in DC than in macrophages. To confirm these results, we performed DNA binding transcription factor ELISA to detect NF-κB activation in CRCL-treated DC and macrophages. NF-κB p50 binding to its specific oligonucleotide probe was enhanced in the nuclear extracts prepared from CRCL-treated cells (Figure 4B). CRCL-induced NF-κB activation was further confirmed using a NF-κB luciferase reporter gene introduced in the RAW264.7 macrophage cell line. Our results indicate that CRCL treatment results in an increase in NF-κB activation when compared to untreated cells (Figure 4C).
Figure 4. Activation of NF-κB in CRCL treated DC and macrophages.
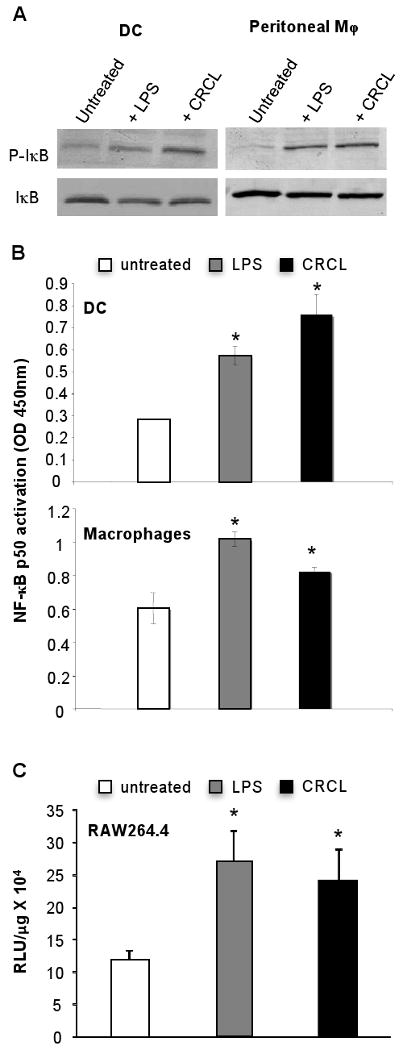
(A) Day 5 bone marrow derived DC or peritoneal macrophages were incubated for 24 hrs with CRCL (25 μg/ml). Total cell extracts were performed and phospho-IκB or IκB expression was analyzed by Western blot. Untreated cells were used as a negative control and cells treated with LPS (1 μg/ml) were used as a positive control. (B) Day 5 bone marrow derived DC or peritoneal macrophages were incubated for 24 hrs with CRCL. Nuclear extracts were performed and the DNA binding activity of NF-κB p50 to a consensus DNA probe was assessed as described in materials and methods. DC or macrophages cultured alone were used as a negative control, and cells treated with LPS were used as a positive control. The data are shown as a mean ± SD of duplicate wells of NF-κB p50 activation determined as the OD value 450nm as indicated by the manufacturer. (C) RAW264.7 cells were transiently transfected with a NF-κB luciferase plasmid. After 36 hrs, cells were treated with LPS or CRCL for 12 hrs as indicated and a luciferase assay was performed as described in material and methods. Data are presented as Relative Luminescent Unit (RLU)/μg of protein. Results are representative of three independent experiments. *, a significant difference when compared to untreated control cells (p < 0.05).
To determine whether our in vitro findings translate in vivo, we investigated the phosphorylation of NF-κB p65 in the draining lymph nodes (DLN) of naïve mice injected with PBS, LPS or CRCL. We detected a significant increase in NF-κB p65 phosphorylation in the DLN of animals administered CRCL vaccine when compared to control (PBS-treated) mice (Figure 5). These results therefore demonstrate that CRCL induces activation of NF-κB both in vitro and in vivo.
Figure 5. CRCL induced phosphorylation of NF-κB p65 in vivo.
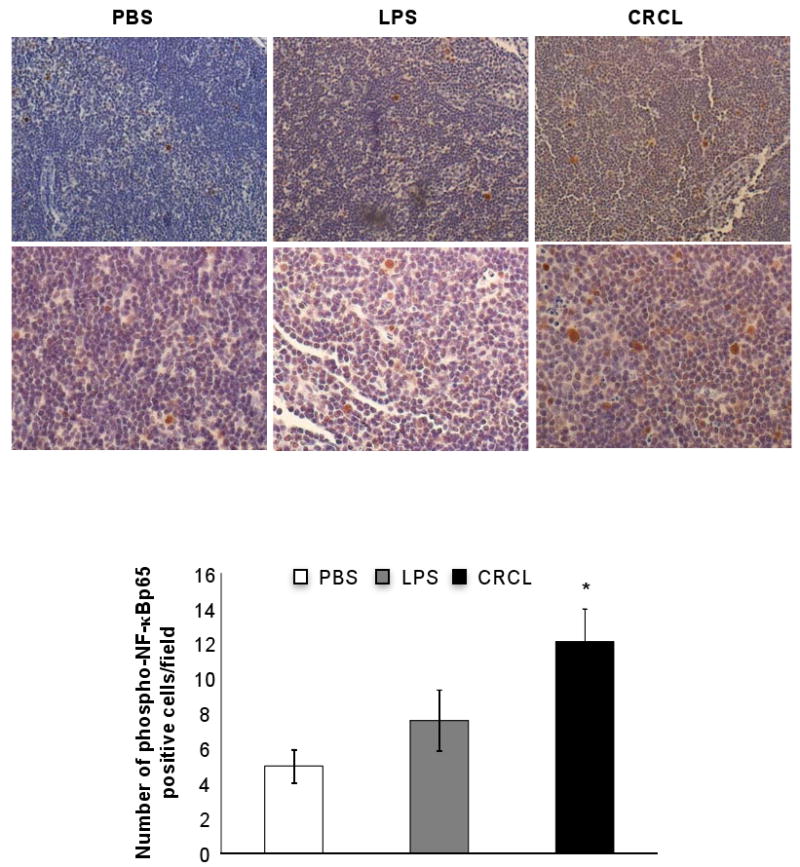
Naïve mice (4 animals per group) were injected (s.c.) with CRCL (100 μl, 25 μg), LPS (100 μl, 1 μg) or PBS as control vehicle. Phospho-NF-κBp65 was detected in the draining lymph nodes by IHC staining. Upper panels, 200× magnification, lower panels 400× magnification. Histogram represents a double blind count of 10 frames of phospho-NF-κB p65 positive cells. *, a significant difference compared to untreated mice (p<0.002).
CRCL induces the activation of STAT1, STAT5, AKT and the MAP kinase pathway in DC and macrophages
To further explore the mechanisms by which CRCL activates DC and macrophages we investigated the role of additional transcription factors and upstream components of the NF-κB signaling cascade.
STAT1, a transcription factor activated following phosphorylation, has been shown to promote DC and macrophage activation (Shuai and Liu, 2003). Compared to untreated cells, an increase of STAT1 Tyr701 phosphorylation was detected in DC and macrophages treated with CRCL (Figure 6 and Supplemental Figure S3). STAT5 is an additional transcription factor activated in response to a variety of cytokines and growth factors and is also involved in the activation of DC and macrophages (Hennighausen and Robinson, 2008). The phosphorylation of STAT5 Tyr694 was enhanced in DC and macrophages treated with CRCL (Figure 6 and Supplemental Figure S3). Conversely, CRCL reduced Tyr694 phosphorylation of STAT3, a transcription factor that dampens DC and macrophage activation (Cheng et al., 2003) (Figure 6).
Figure 6. CRCL activates STAT1, STAT5, AKT and the MAP kinase pathway but impairs the phosphorylation of STAT3 in DC and macrophages.
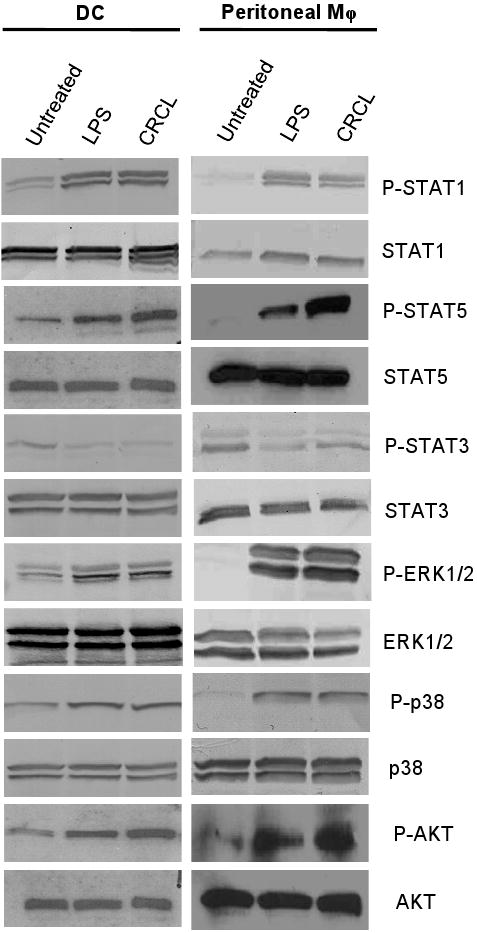
Day 5 bone marrow derived DC or peritoneal macrophages were incubated for 24 hrs with CRCL (25 μg/ml) or LPS (1 μg/ml) as a positive control. (A) Western blot analysis of total cell extracts was then performed probing for phospho STAT1 (P-STAT1), STAT1, phospho STAT5 (P-STAT5), STAT5, phospho ERK1/2 (P-ERK1/2), ERK1/2, phospho p38 (P-p38), p38, phospho AKT (P-AKT), AKT, phospho STAT3 (P-STAT3), or STAT3.
The MAP kinase (MAPK) pathway is activated in response to various cell surface signals that converge to the activation of transcription factors such as NF-κB or STAT-1 and STAT-5 (Akira and Takeda, 2004). To further identify the signaling cascades triggered in response to CRCL in DC and macrophages, we evaluate a possible modifications of MAPK components. Our results indicate that the phosphorylation of ERK1/2 Thr202/Tyr204 and p38 Thr180/Tyr182, critical components of the MAP kinase cascade was increased in CRCL-treated DC and macrophages. The phosphorylation of ERK1/2 induced by LPS or CRCL was higher in macrophages compared to DC (Figure 6 and Supplemental Figure S3).
AKT represents an important signaling protein involved in cellular survival pathways. It regulates apoptotic processes and modulates downstream effector molecules such as NF-κB (Akira and Takeda, 2004). The results depicted in Figure 6 demonstrate that CRCL promotes the phophorylation of AKT Ser473 in DC and to a higher extend in macrophages. These data thus indicates that CRCL is capable of inducing cell survival pathways within antigen presenting cells, particularly in macrophages.
CRCL overcomes TGF-β mediated inhibition of DC
We have previously reported that a subset of immunosuppressive lymphocytes (CD4+CD25+FoxP3+ regulatory T cells, Treg) suppress the induction of anti-tumoral immune responses by impairing the activation of DC. This inhibition partly involves the immunosuppressive cytokine TGF-β (Larmonier et al., 2007). We have also reported that Treg fail to inhibit DC phenotypic maturation or pro-inflammatory cytokine production following exposure to tumor-derived CRCL (Larmonier et al., 2008a). To further evaluate whether CRCL can overcome the inhibitory affects of TGF-β on DC, these cells were treated with LPS or tumor-derived CRCL with or without TGF-β and TNF-α secretion was determined. Our results indicate that DC stimulated with CRCL resist TGF-β-mediated suppression (Figure 7).
Figure 7. CRCL overcomes TGF-β-mediated DC suppression.

Day 5 bone marrow derived DC were incubated for 24 hrs with CRCL (25 μg/ml) or LPS (1 μg/ml), with or without TGF-β (10 ng/ml). The supernatants were harvested after 24 hrs and tested for TNF-α production by ELISA. *, a significant difference when compared to DC treated with LPS without TGF-β (p<0.05). NS, non-significant difference when compated to DC treated with CRCL without TGF-β.
Discussion
Chaperone proteins purified from tumor tissues have been identified as unique mediators of specific anti-cancer immunity. The advantages of using chaperone protein-associated peptides as a source of tumor antigens are multiple: 1) they do not require the identification of tumor specific peptides, 2) they elicit polyclonal T lymphocyte responses following immunization, preventing the outgrowth of immunological escape variants, 3) they consist of both MHC Class I associated peptides and MHC Class II or helper epitopes, which together induce more potent and durable immune responses, and 4) these proteins also act as natural adjuvants or ‘danger signals’ capable of activating antigen presenting cells such as DC and macrophages, enhancing the potential of these cells to process and present antigens and to stimulate T cell responses (Asea et al., 2000; Basu et al., 2001; Binder et al., 2000; Srivastava and Udono, 1994).
Our laboratory has developed an original tumor-derived vaccine, CRCL, capable of eliciting specific T cell responses leading to tumor regression (Graner et al., 2000a; Graner et al., 2000b; Graner et al., 2003). The primary components of CRCL are a combination of chaperones, which include HSP90, HSP70 family members, the endoplasmic reticulum chaperones glucose regulated protein (GRP)94/glycoprotein (gp)96, and calreticulin (Graner et al., 2000a; Graner et al., 2000b; Graner et al., 2003; Zeng et al., 2006). Each of these chaperones has been utilized individually and found to generate specific immunity against their tumor of origin (Graner et al., 2000b; Srivastava, 2002a). All of these chaperones are gathered into a single preparation by the clustering of the proteins that occur during the FS-IEF procedure (Graner et al., 2000a). While there are a number of other unidentified proteins in CRCL, immunodepletion of HSP70, HSP90, GRP94, and calreticulin results in a significantly reduced immune response (Zeng et al., 2003; Zeng et al., 2006), indicating that these chaperones are truly responsible for vaccine efficiency. In numerous animal models, CRCL vaccines have a more pronounced anti-tumor effect per unit material of protein than any of the individual purified chaperone proteins used alone or tumor lysate (Graner et al., 2000a; Graner et al., 2000b; Graner et al., 2003; Zeng et al., 2003). Furthermore, the immunogenic potential of CRCL can be enhanced by loading them into DC. The enhanced immunogenicity of CRCL-pulsed DC may be due to the broad variety of antigenic peptides that are chaperoned by CRCL, but also to the direct activation of DC by virtue of the adjuvant properties of the chaperone proteins within CRCL vaccines (Graner et al., 2003; Zeng et al., 2003; Zeng et al., 2005).
Our previous studies have indicated that tumor-derived CRCL triggers DC expression of CD40 and, to a lesser extent, CD80 and 86, all of which are co-stimulatory molecules that are fundamental for T cell activation. Our current study demonstrates that CRCL additionally promotes the expression of CD70, a newly identified marker of activated DC that binds to CD27 expressed on naïve T cells (Tesselaar et al., 2003). The engagement of CD27 with CD70 leads to sustained potent cellular immunity. The optimal up-regulation of CD70 involves both CD40/CD40L interaction as well as stimulation through TLRs (Sanchez et al., 2007). Importantly, tumor-derived CRCL was as efficient as LPS in inducing CD70 up-regulation in DC, which highlights its effectiveness and further advocates for the advantages of using CRCL as a vaccine with robust adjuvant qualities.
Macrophages represent a subset of immune cells capable of recognizing and killing cancer cells (Bauer et al., 2001; Bogdan, 2001; Nathan, 1992; Pervin et al., 2001; Xie et al., 1996). Our previous studies had primarily focused on the modulation of dendritic cells by CRCL, but the effects of this vaccine on macrophage functions were largely undetermined. We herein demonstrate that macrophages represent prominent targets of CRCL. Indeed, tumor-derived CRCL is not only capable of triggering macrophage pro-inflammatory cytokine production, but also induces their tumoricidal activity. Macrophages can kill tumor cells by direct and/or indirect mechanisms including the production of nitric oxide (Bogdan, 2001). Treatment with tumor-derived CRCL results in the induction of macrophage iNOS expression and is associated with NO production. Furthermore, CRCL triggers secretion of macrophage TNF-α that together with NO may participate in the observed anti-tumoral activity. In addition, CRCL-activated macrophages may indirectly contribute to the induction and maintenance of anti-tumor immune responses by secreting IL-12 and RANTES, which fosters DC activation and T cell recruitment. These results thus demonstrate that by targeting not only DC but also tumor killer macrophages, CRCL is endowed with the potential to activate multiple arms of the anti-tumoral immune responses, thus augmenting the diversity of the killing mechanisms leading to tumor elimination.
The molecular mechanisms underlying CRCL adjuvant effects on DC and macrophages had not been previously defined. To address this question, we evaluated key signaling events that may be modulated by CRCL in these cells. NF-κB is one of the key factors responsible for the transcription of genes involved in DC phenotypic and functional changes during activation (Iwasaki and Medzhitov, 2004). NF-κB controls pro-inflammatory responses by up-regulating MHC Class II and co-stimulatory molecule expression, as well as inducing the secretion of pro-inflammatory cytokines and chemokines by DC and macrophages (Akira and Takeda, 2004; Kawai and Akira, 2006). We here provide evidence that the tumor-derived CRCL vaccine is capable of activating NF-κB, both in vitro in DC and macrophages and in vivo following injection of mice. Further investigation led to the identification of STAT1 and STAT5 as additional transcription factors activated in DC and macrophages in response to CRCL stimulation. Furthermore, CRCL activates the MAP kinase pathway, responsible for cell differentiation, survival, and growth (Dhillon et al., 2007). Additionally, CRCL may foster APC survival by promoting the Akt/PKB pathway. Interestingly CRCL hampers the phosphorylation of STAT3, a transcription factor that dampens the immune response by inhibiting DC and macrophage activation and function (Shuai and Liu, 2003). Together these data demonstrate that tumor-derived CRCL promotes the intracellular signaling cascades leading to DC and macrophage activation while hampering negative signals that may lead to their inhibition.
We have shown that regulatory T cells, a critical population of lymphocytes that contribute to tumor-induced immunosuppression, inhibit DC activation though a mechanism involving TGF-β (Larmonier et al., 2007). However, this suppression of DC and macrophages by regulatory T cells may be overcome by the tumor-derived CRCL vaccine (Larmonier et al., 2008a). Consistent with these data, we have now demonstrated that CRCL overrides TGF-β negative signaling induced in bone marrow-derived DC. These results therefore suggest that the positive cell signaling cascades induced by CRCL in DC may overcome the negative signals triggered by tumor-induced suppressive factors or cells. Understanding the mechanisms of action of this vaccine including its adjuvant effects is critical in optimizing its use in future clinical trials.
Supplementary Material
Day 5 bone marrow derived DC or peritoneal macrophages were treated with CRCL (25 μg/ml) or LPS (1 μg/ml) as a positive control. The culture supernatants were collected after 24 hrs and analyzed by ELISA for TNF-α, IL-12 or RANTES secretion. *, a significant difference when compared to untreated control cells (p < 0.05).
(A) Effects of CRCL on TNF-α and IL-12 production by RAW264.7 cells treated with CRCL (25 μg/ml) or LPS (1 μg/ml). The supernatants were harvested after 12 and 24 hrs and tested for TNF-α or IL-12 production by ELISA. (B, top panel) Real-time PCR analysis of TNF-α mRNA levels in RAW264.7 cells treated with CRCL or LPS. Each sample was normalized to its corresponding ribosomal RNA 18S signal. (B, bottom panel) IL12p40 gene expression is upregulated by treatment of RAW264.7 cells with CRCL (25 μg/ml). RNA was isolated after treatment of the cells with CRCL or LPS for 12 h at 37°C. The presented data are representative of three experiments. *, a significant difference when compared to untreated control cells (p < 0.05).
The histograms were obtained by densitometric analysis of western blot data from 3 independent experiments. *, a significant difference compared to untreated control cells (p<0.05). (A) Peritoneal macrophages, (B) Dendritic cells.
Acknowledgments
This work was supported in part by the NIH grant R01 CA104926 (EK), the Leukemia and Lymphoma Society Fellow Award 5188-07 (NL), the Tee Up for Tots and Raise a Racquet for Kids Funds (EK, NL).
Abbreviations
- CRCL
Chaperone Rich Cell Lysates
- DC
Dendritic cells
- NF-κB
Nuclear Factor Kappa B
- NK
Natural Killer cells
- MAP
Mitogen-Activated Protein
- MHC
Major Histocompatibility Complex
- STAT
Signal Transduction and Activator of Transcription
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 2000;6:435–442. doi: 10.1038/74697. [DOI] [PubMed] [Google Scholar]
- Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14:303–313. doi: 10.1016/s1074-7613(01)00111-x. [DOI] [PubMed] [Google Scholar]
- Bauer PM, Buga GM, Fukuto JM, Pegg AE, Ignarro LJ. Nitric oxide inhibits ornithine decarboxylase via S-nitrosylation of cysteine 360 in the active site of the enzyme. J Biol Chem. 2001;276:34458–34464. doi: 10.1074/jbc.M105219200. [DOI] [PubMed] [Google Scholar]
- Binder RJ, Han DK, Srivastava PK. CD91: a receptor for heat shock protein gp96. Nat Immunol. 2000;1:151–155. doi: 10.1038/77835. [DOI] [PubMed] [Google Scholar]
- Binder RJ, Vatner R, Srivastava P. The heat-shock protein receptors: some answers and more questions. Tissue Antigens. 2004;64:442–451. doi: 10.1111/j.1399-0039.2004.00299.x. [DOI] [PubMed] [Google Scholar]
- Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- Bonnotte B, Larmonier N, Favre N, Fromentin A, Moutet M, Martin M, Gurbuxani S, Solary E, Chauffert B, Martin F. Identification of tumor-infiltrating macrophages as the killers of tumor cells after immunization in a rat model system. J Immunol. 2001;167:5077–5083. doi: 10.4049/jimmunol.167.9.5077. [DOI] [PubMed] [Google Scholar]
- Cheng F, Wang HW, Cuenca A, Huang M, Ghansah T, Brayer J, Kerr WG, Takeda K, Akira S, Schoenberger SP, Yu H, Jove R, Sotomayor EM. A critical role for Stat3 signaling in immune tolerance. Immunity. 2003;19:425–436. doi: 10.1016/s1074-7613(03)00232-2. [DOI] [PubMed] [Google Scholar]
- Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- Gao B, Tsan MF. Endotoxin contamination in recombinant human heat shock protein 70 (Hsp70) preparation is responsible for the induction of tumor necrosis factor alpha release by murine macrophages. J Biol Chem. 2003;278:174–179. doi: 10.1074/jbc.M208742200. [DOI] [PubMed] [Google Scholar]
- Graner M, Raymond A, Akporiaye E, Katsanis E. Tumor-derived multiple chaperone enrichment by free-solution isoelectric focusing yields potent antitumor vaccines. Cancer Immunol Immunother. 2000a;49:476–484. doi: 10.1007/s002620000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graner M, Raymond A, Romney D, He L, Whitesell L, Katsanis E. Immunoprotective activities of multiple chaperone proteins isolated from murine B-cell leukemia/lymphoma. Clin Cancer Res. 2000b;6:909–915. [PubMed] [Google Scholar]
- Graner MW, Likhacheva A, Davis J, Raymond A, Brandenberger J, Romanoski A, Thompson S, Akporiaye E, Katsanis E. Cargo from tumor-expressed albumin inhibits T-cell activation and responses. Cancer Res. 2004;64:8085–8092. doi: 10.1158/0008-5472.CAN-04-1871. [DOI] [PubMed] [Google Scholar]
- Graner MW, Zeng Y, Feng H, Katsanis E. Tumor-derived chaperone-rich cell lysates are effective therapeutic vaccines against a variety of cancers. Cancer Immunol Immunother. 2003;52:226–234. doi: 10.1007/s00262-002-0359-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Feng H, Raymond A, Kreeger M, Zeng Y, Graner M, Whitesell L, Katsanis E. Dendritic-cell-peptide immunization provides immunoprotection against bcr-abl-positive leukemia in mice. Cancer Immunol Immunother. 2001;50:31–40. doi: 10.1007/PL00006680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heike M, Weinmann A, Bethke K, Galle PR. Stress protein/peptide complexes derived from autologous tumor tissue as tumor vaccines. Biochemical Pharmacology. 1999;58:1381–1387. doi: 10.1016/s0006-2952(99)00178-1. [DOI] [PubMed] [Google Scholar]
- Hennighausen L, Robinson GW. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev. 2008;22:711–721. doi: 10.1101/gad.1643908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006 doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- Klimp AH, de Vries EG, Scherphof GL, Daemen T. A potential role of macrophage activation in the treatment of cancer. Crit Rev Oncol Hematol. 2002;44:143–161. doi: 10.1016/s1040-8428(01)00203-7. [DOI] [PubMed] [Google Scholar]
- Kryczek I, Zou L, Rodriguez P, Zhu G, Wei S, Mottram P, Brumlik M, Cheng P, Curiel T, Myers L, Lackner A, Alvarez X, Ochoa A, Chen L, Zou W. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. 2006;203:871–881. doi: 10.1084/jem.20050930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larmonier N, Cantrell J, Lacasse C, Li G, Janikashvili N, Situ E, Sepassi M, Andreansky S, Katsanis E. Chaperone-rich tumor cell lysate-mediated activation of antigen-presenting cells resists regulatory T cell suppression. J Leukoc Biol. 2008a;83:1049–1059. doi: 10.1189/jlb.0907635. [DOI] [PubMed] [Google Scholar]
- Larmonier N, Janikashvili N, LaCasse CJ, Larmonier CB, Cantrell J, Situ E, Lundeen T, Bonnotte B, Katsanis E. Imatinib mesylate inhibits CD4+ CD25+ regulatory T cell activity and enhances active immunotherapy against BCR-ABL- tumors. J Immunol. 2008b;181:6955–6963. doi: 10.4049/jimmunol.181.10.6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larmonier N, Marron M, Zeng Y, Cantrell J, Romanoski A, Sepassi M, Thompson S, Chen X, Andreansky S, Katsanis E. Tumor-derived CD4(+)CD25(+) regulatory T cell suppression of dendritic cell function involves TGF-beta and IL-10. Cancer Immunol Immunother. 2007;56:48–59. doi: 10.1007/s00262-006-0160-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larmonier N, Merino D, Nicolas A, Cathelin D, Besson A, Bateman A, Solary E, Martin F, Katsanis E, Bonnotte B. Apoptotic, necrotic, or fused tumor cells: an equivalent source of antigen for dendritic cell loading. Apoptosis. 2006;11:1513–1524. doi: 10.1007/s10495-006-8765-0. [DOI] [PubMed] [Google Scholar]
- Li Z. Priming of T cells by heat shock protein-peptide complexes as the basis of tumor vaccines. Seminars in Immunology. 1997;9:315–322. doi: 10.1006/smim.1997.0087. [DOI] [PubMed] [Google Scholar]
- Li Z, Menoret A, Srivastava P. Roles of heat-shock proteins in antigen presentation and cross-presentation. Current Opinion in Immunology. 2002;14:45–51. doi: 10.1016/s0952-7915(01)00297-7. [DOI] [PubMed] [Google Scholar]
- McLaughlin J, Chianese E, Witte ON. In vitro transformation of immature hematopoietic cells by the P210 BCR/ABL oncogene product of the Philadelphia chromosome. Proc Natl Acad Sci U S A. 1987;84:6558–6562. doi: 10.1073/pnas.84.18.6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C. Nitric oxide as a secretory product of mammalian cells. Faseb J. 1992;6:3051–3064. [PubMed] [Google Scholar]
- Nicchitta CV. Re-evaluating the role of heat-shock protein-peptide interactions in tumour immunity. Nat Rev Immunol. 2003;3:427–432. doi: 10.1038/nri1089. [DOI] [PubMed] [Google Scholar]
- Pervin S, Singh R, Chaudhuri G. Nitric oxide-induced cytostasis and cell cycle arrest of a human breast cancer cell line (MDA-MB-231): potential role of cyclin D1. Proc Natl Acad Sci U S A. 2001;98:3583–3588. doi: 10.1073/pnas.041603998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radsak MP, Hilf N, Singh-Jasuja H, Braedel S, Brossart P, Rammensee HG, Schild H. The heat shock protein Gp96 binds to human neutrophils and monocytes and stimulates effector functions. Blood. 2003;101:2810–2815. doi: 10.1182/blood-2002-07-2261. [DOI] [PubMed] [Google Scholar]
- Reed RC, Berwin B, Baker JP, Nicchitta CV. GRP94/gp96 elicits ERK activation in murine macrophages. A role for endotoxin contamination in NF-kappa B activation and nitric oxide production. J Biol Chem. 2003;278:31853–31860. doi: 10.1074/jbc.M305480200. [DOI] [PubMed] [Google Scholar]
- Sanchez PJ, McWilliams JA, Haluszczak C, Yagita H, Kedl RM. Combined TLR/CD40 stimulation mediates potent cellular immunity by regulating dendritic cell expression of CD70 in vivo. J Immunol. 2007;178:1564–1572. doi: 10.4049/jimmunol.178.3.1564. [DOI] [PubMed] [Google Scholar]
- Segal BH, Wang XY, Dennis CG, Youn R, Repasky EA, Manjili MH, Subjeck JR. Heat shock proteins as vaccine adjuvants in infections and cancer. Drug Discov Today. 2006;11:534–540. doi: 10.1016/j.drudis.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol. 2003;3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- Smith DF, Whitesell L, Katsanis E. Molecular chaperones: biology and prospects for pharmacological intervention. Pharmacol Rev. 1998;50:493–514. [PubMed] [Google Scholar]
- Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annual Review of Immunology. 2002a;20:395–425. doi: 10.1146/annurev.immunol.20.100301.064801. [DOI] [PubMed] [Google Scholar]
- Srivastava P. Roles of heat-shock proteins in innate and adaptive immunity. Nature Reviews. Immunology. 2002b;2:185–194. doi: 10.1038/nri749. [DOI] [PubMed] [Google Scholar]
- Srivastava PK, Amato RJ. Heat shock proteins: the ‘Swiss Army Knife’ vaccines against cancers and infectious agents. Vaccine. 2001;19:2590–2597. doi: 10.1016/s0264-410x(00)00492-8. [DOI] [PubMed] [Google Scholar]
- Srivastava PK, Menoret A, Basu S, Binder RJ, McQuade KL. Heat shock proteins come of age: primitive functions acquire new roles in an adaptive world. Immunity. 1998;8:657–665. doi: 10.1016/s1074-7613(00)80570-1. [DOI] [PubMed] [Google Scholar]
- Srivastava PK, Udono H. Heat shock protein-peptide complexes in cancer immunotherapy. Curr Opin Immunol. 1994;6:728–732. doi: 10.1016/0952-7915(94)90076-0. [DOI] [PubMed] [Google Scholar]
- Tesselaar K, Xiao Y, Arens R, van Schijndel GM, Schuurhuis DH, Mebius RE, Borst J, van Lier RA. Expression of the murine CD27 ligand CD70 in vitro and in vivo. J Immunol. 2003;170:33–40. doi: 10.4049/jimmunol.170.1.33. [DOI] [PubMed] [Google Scholar]
- Wallin RP, Lundqvist A, More SH, von Bonin A, Kiessling R, Ljunggren HG. Heat-shock proteins as activators of the innate immune system. Trends Immunol. 2002;23:130–135. doi: 10.1016/s1471-4906(01)02168-8. [DOI] [PubMed] [Google Scholar]
- Wells AD, Malkovsky M. Heat shock proteins, tumor immunogenicity and antigen presentation: an integrated view. Immunology Today. 2000;21:129–132. doi: 10.1016/s0167-5699(99)01558-3. [DOI] [PubMed] [Google Scholar]
- Xie K, Dong Z, Fidler IJ. Activation of nitric oxide synthase gene for inhibition of cancer metastasis. J Leukoc Biol. 1996;59:797–803. doi: 10.1002/jlb.59.6.797. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Feng H, Graner MW, Katsanis E. Tumor-derived, chaperone-rich cell lysate activates dendritic cells and elicits potent antitumor immunity. Blood. 2003;101:4485–4491. doi: 10.1182/blood-2002-10-3108. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Graner MW, Katsanis E. Chaperone-rich cell lysates, immune activation and tumor vaccination. Cancer Immunol Immunother. 2006;55:329–338. doi: 10.1007/s00262-005-0694-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Graner MW, Thompson S, Marron M, Katsanis E. Induction of BCR-ABL-specific immunity following vaccination with chaperone-rich cell lysates derived from BCR-ABL+ tumor cells. Blood. 2005;105:2016–2022. doi: 10.1182/blood-2004-05-1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Day 5 bone marrow derived DC or peritoneal macrophages were treated with CRCL (25 μg/ml) or LPS (1 μg/ml) as a positive control. The culture supernatants were collected after 24 hrs and analyzed by ELISA for TNF-α, IL-12 or RANTES secretion. *, a significant difference when compared to untreated control cells (p < 0.05).
(A) Effects of CRCL on TNF-α and IL-12 production by RAW264.7 cells treated with CRCL (25 μg/ml) or LPS (1 μg/ml). The supernatants were harvested after 12 and 24 hrs and tested for TNF-α or IL-12 production by ELISA. (B, top panel) Real-time PCR analysis of TNF-α mRNA levels in RAW264.7 cells treated with CRCL or LPS. Each sample was normalized to its corresponding ribosomal RNA 18S signal. (B, bottom panel) IL12p40 gene expression is upregulated by treatment of RAW264.7 cells with CRCL (25 μg/ml). RNA was isolated after treatment of the cells with CRCL or LPS for 12 h at 37°C. The presented data are representative of three experiments. *, a significant difference when compared to untreated control cells (p < 0.05).
The histograms were obtained by densitometric analysis of western blot data from 3 independent experiments. *, a significant difference compared to untreated control cells (p<0.05). (A) Peritoneal macrophages, (B) Dendritic cells.