

Abstract
Synaptic plasticity in the mesolimbic dopamine (DA) system is critically involved in reward-based conditioning and the development of drug addiction. Ca2+ signals triggered by postsynaptic action potentials (APs) drive the induction of synaptic plasticity in the CNS. However, it is not clear how AP-evoked Ca2+ signals and the resulting synaptic plasticity are altered during in vivo exposure to drugs of abuse. We have recently described long-term potentiation (LTP) of NMDA receptor (NMDAR)-mediated transmission onto DA neurons that is induced in a manner dependent on bursts of APs. LTP induction requires amplification of burst-evoked Ca2+ signals by preceding activation of metabotropic glutamate receptors (mGluRs) generating inositol 1,4,5-trisphosphate (IP3). In this study, using brain slices prepared from male rats, we show that repeated in vivo exposure to the psychostimulant amphetamine (5 mg/kg, i.p., 3–7 d) upregulates mGluR-dependent facilitation of burst-evoked Ca2+ signals in DA neurons of the ventral tegmental area (VTA). Protein kinase A (PKA)-induced sensitization of IP3 receptors mediates this upregulation of mGluR action. As a consequence, NMDAR-mediated transmission becomes more susceptible to LTP induction after repeated amphetamine exposure. We have also found that the magnitude of amphetamine-conditioned place preference (CPP) in behaving rats correlates with the magnitude of mGluR-dependent Ca2+ signal facilitation measured in VTA slices prepared from these rats. Furthermore, the development of amphetamine CPP is significantly attenuated by intra-VTA infusion of the PKA inhibitor H89. We propose that enhancement of mGluR-dependent NMDAR plasticity in the VTA may promote the learning of environmental stimuli repeatedly associated with amphetamine experience.
Introduction
Dopamine (DA) neurons, located in the ventral tegmental area (VTA) and substantia nigra pars compacta (SNc), are critically involved in processing and learning of reward-related information (Schultz, 1998; Wise, 2004). In awake animals, action potential (AP) firing of DA neurons switches from tonic single-spike activity to phasic bursts (2–10 APs at 10–50 Hz) in response to the unexpected presentation of primary rewards. After conditioning with repeated pairing of environmental cues and rewards, the phasic burst response transfers from rewards to reward-predicting cues (Schultz, 1998; Pan et al., 2005). It is generally believed that these DA neuron bursts, producing phasic DA release in projection areas, mediate the learning of cue–reward associations and also provide an incentive motivational signal for goal-directed behavior (Berridge, 2007).
Recent evidence indicates important roles for synaptic plasticity at glutamatergic inputs onto DA neurons in reward-based conditioning and the development of drug addiction (Wolf et al., 2004; Hyman et al., 2006; Stuber et al., 2008). Previous studies have mostly focused on the plasticity of AMPA receptor (AMPAR)-mediated transmission. Indeed, it has been repeatedly shown that in vivo exposure to psychostimulants and other drugs of abuse produces persistent potentiation of AMPAR-mediated excitation of DA neurons (Giorgetti et al., 2001; Saal et al., 2003; Bellone and Luscher, 2006). However, glutamatergic inputs activating NMDA receptors (NMDARs) appear to play a predominant role in the generation of DA neuron bursts (Johnson et al., 1992; Overton and Clark, 1992; Deister et al., 2009; Zweifel et al., 2009). We have recently described long-term potentiation (LTP) of NMDAR-mediated transmission that is induced by pairing sustained synaptic stimulation (∼1 s) of glutamatergic inputs with postsynaptic burst firing in DA neurons (Harnett et al., 2009). This LTP induction protocol may resemble the activity pattern experienced during cue–reward conditioning, in that cue presentation may give rise to persistent glutamatergic input while the reward would elicit DA neuron burst firing (Brown et al., 1999). Therefore, this form of NMDAR plasticity might contribute to the acquisition of conditioned DA neuron burst responses to reward-predicting cues.
Mechanistically, the induction of NMDAR LTP requires burst-evoked Ca2+ signals amplified by preceding synaptic activation of metabotropic glutamate receptors (mGluRs) coupled to the generation of inositol 1,4,5-trisphosphate (IP3) (Cui et al., 2007; Harnett et al., 2009). Here, IP3, resulting from mGluR activation, and Ca2+, provided by burst-induced Ca2+ influx, synergistically coactivate IP3 receptors (IP3Rs) to cause Ca2+ release from intracellular stores (Taylor and Laude, 2002). Thus, IP3Rs act as a coincidence detector for presynaptic glutamatergic input activity and postsynaptic burst firing to mediate the induction of NMDAR plasticity.
Acute exposure to the psychostimulant amphetamine has been shown to block the induction of long-term depression of AMPAR-mediated transmission by suppressing Ca2+ influx in DA neurons (Jones et al., 2000). Here, we show that repeated in vivo exposure to amphetamine causes sensitization of IP3Rs in VTA DA neurons, resulting in an enhancement of mGluR-dependent NMDAR LTP. We also provide evidence suggesting that enhanced NMDAR plasticity in the VTA may promote the learning of amphetamine-associated environmental stimuli.
Materials and Methods
Subjects.
Male Sprague Dawley rats [4–6 weeks old; ∼8 weeks old (∼250 g) for the experiments involving intra-VTA injections] were obtained from Harlan. Rats were housed in groups of two to three per cage under a 12 h light/dark cycle (lights on at 7:00 A.M.). Food and water were available ad libitum. All animal procedures were approved by the Universtiy of Texas Institutional Animal Care and Use Committee.
Slices and solutions.
Rats were decapitated under halothane or isoflurane anesthesia, and horizontal midbrain slices (200–220 μm) containing the VTA and SNc were prepared. Slices were cut using a Vibratome (VT1000S; Leica Microsystems) in an ice-cold cutting solution containing (in mm) 205 sucrose, 2.5 KCl, 1.25 NaH2PO4, 7.5 MgCl2, 0.5 CaCl2, 10 glucose, and 25 NaHCO3, saturated with 95% O2 and 5% CO2 (∼305 mOsm/kg), and incubated at 35°C for >1 h in physiological saline containing (in mm) 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 11 glucose, and 21.4 NaHCO3, saturated with 95% O2 and 5% CO2 (pH 7.4, ∼295 mOsm/kg). Recordings were made at 34–35°C in the same physiological saline perfused at 2–3 ml/min. The pipette solution contained (in mm) 115 K-gluconate or K-methylsulfate, 20 KCl, 1.5 MgCl2, 10 HEPES, 0.025 EGTA, 2 Mg-ATP, 0.2 Na2-GTP, and 10 Na2-phosphocreatine (pH 7.25, ∼280 mOsm/kg).
Electrophysiological recording.
Neurons were visualized using a 40× or 60× objective on an upright microscope (BX51WI; Olympus America) with infrared/differential interference contrast or oblique illumination optics. Putative DA neurons were identified by spontaneous pacemaker firing (1–5 Hz) with broad APs (>1.2 ms) in a cell-attached configuration before break-in and by the presence of large whole-cell Ih currents (>200 pA in response to a 1.5 s voltage step from −62 to −112 mV) after break-in. Recordings in the VTA were restricted to those neurons located within ∼150 μm from the border of the medial terminal nucleus of the accessory optic tract in horizontal slices. The criteria for DA neuron identification described above are generally accepted in this lateral part of the VTA as well as in the SNc (Ford et al., 2006; Riegel and Williams, 2008; Wanat et al., 2008). Whole-cell voltage-clamp recordings were routinely made at a holding potential of −62 mV, corrected for a liquid junction potential of −7 mV. Pipettes had an open-tip resistance of 1.7–2.5 MΩ when filled with the pipette solution. A Multiclamp 700A or 700B amplifier (Molecular Devices) was used to record the data, which were filtered at 1–5 kHz, digitized at 2–10 kHz, and collected using AxoGraph X (AxoGraph Scientific).
A 2 ms depolarizing pulse from −62 to −7 mV was used to elicit an unclamped AP. The time integral of the outward tail current [i.e., IK(Ca)] was calculated between 20 and 400–600 ms after the depolarizing pulse. We have shown previously that IK(Ca) thus measured is completely eliminated by TTX and also by apamin, a selective blocker of small-conductance Ca2+-sensitive K+ (SK) channels, and hence can be used as a readout of AP-induced Ca2+ transients (Cui et al., 2007). For a burst (a train of five APs at 20 Hz), the integral of IK(Ca) was calculated after removing a 20 ms window after each depolarizing pulse. The obtained values were normalized by the membrane capacitance and thus expressed in picocoulomb/picofarad.
Ca2+ imaging studies using fluorescent Ca2+ indicator dyes have shown that major Ca2+ influx via L-type Ca2+ channels occurs during ramp depolarization preceding APs in DA neurons firing repetitively (Wilson and Callaway, 2000; Guzman et al., 2009). We found that brief (3–5 min) application of low concentrations of TTX (20–100 nm) to spontaneously firing DA neurons, recorded with a perforated-patch configuration using gramicidin (50–250 μg/ml), revealed occasional APs triggering large afterhyperpolarizations (AHPs) interspersed in the background of small membrane potential oscillations driven by L-type Ca2+ and SK conductances (Wilson and Callaway, 2000) (n = 5 from four rats) (Fig. 1). Therefore, assuming that the size of AHPs is largely determined by Ca2+-sensitive SK conductance (Wolfart et al., 2001), APs themselves appear to provide significant Ca2+ influx in spontaneously firing DA neurons, as reported previously in acutely dissociated DA neurons (Puopolo et al., 2007).
Figure 1.

APs are responsible for the generation of large AHPs. Representative traces of spontaneous DA neuron firing, recorded with a perforated-patch configuration before and during bath application of TTX (50 nm), are shown. APs can be occasionally observed after brief TTX application (3 min). Note that the amplitude of AHPs after APs is significantly larger than the amplitude of hyperpolarizations during subthreshold membrane potential oscillations. APs were completely eliminated after prolonged perfusion of TTX (9 min).
Synaptic stimuli were applied using bipolar tungsten electrodes (∼100 μm tip separation) placed 50–150 μm rostral to the recorded neuron. To isolate NMDAR EPSCs, recordings were performed in the presence of DNQX (10 μm), picrotoxin (100 μm), and eticlopride (100 nm) to block AMPA, GABAA, and DA D2 receptors, respectively, and in low Mg2+ (0.1 mm) to remove blockade of NMDARs. EPSCs thus evoked are completely blocked by the NMDA antagonist AP-5 (Harnett et al., 2009).
Ca2+ imaging.
For Ca2+ imaging experiments, EGTA was replaced with Fluo-5F (25 μm; Invitrogen) in the pipette solution. Images were captured at 8.5–15 Hz with a 60× objective using a spinning disk confocal imaging system (Olympus). Burst-evoked Ca2+ signals from selected regions of interest (ROIs) were expressed as ΔF/F = (F − Fbaseline)/(Fbaseline − Fbackground), which was integrated over a 2 s period after the burst onset.
Flash photolysis.
Caged IP3 (25 or 100 μm; Invitrogen) was loaded into the cytosol through the whole-cell pipette. A 1 ms UV pulse was applied using a xenon arc lamp (Cairn Research) to rapidly release IP3. The UV pulse was focused through a 60× objective onto a ∼350 μm diameter area centered at recorded neurons. The amount of photolysis is known to be proportional to the UV pulse intensity (McCray et al., 1980), which is proportional to the capacitance of the capacitor feeding current to the flash lamp. This capacitance was varied (50–4050 μF) to adjust the UV pulse intensity.
Immunohistochemistry.
Horizontal midbrain slices (200 μm) were fixed with 4% paraformaldehyde in PBS for 30 min and washed three times with PBS. To neutralize free aldehyde groups, slices were treated with 1% Na-borohydrate in PBS for 10 min and rinsed three times in PBS and once in TBS. Subsequently, slices were incubated for 30 min in a blocking solution containing 10% horse serum, 1% BSA, and 0.5% Triton X-100 in PBS. After a brief wash with PBS, slices were incubated overnight at 4°C in primary antibodies against IP3R1 [rat, 1:1000; mAb18A10 from Dr. K. Mikoshiba (University of Tokyo, Tokyo, Japan) (Nakade et al., 1991; Nakanishi et al., 1996)] and tyrosine hydroxylase (TH; rabbit, 1:1000; Calbiochem), an enzymatic marker for DA neurons in the VTA and SNc. Afterward, slices were washed three times in PBS and incubated in Alexa 488-conjugated chicken anti-rat IgG (1:500; Invitrogen) and Alexa 546-conjugated goat anti-rabbit IgG (1:500; Invitrogen) for 90 min at room temperature. After the PBS wash, slices were mounted in Vectashield mounting medium (Vector Laboratories) and visualized with a 100× oil-immersion objective using a spinning disk confocal imaging system (Olympus).
Repeated amphetamine treatment.
Rats received once-daily intraperitoneal injections of saline or d-amphetamine sulfate (5 mg/kg) for 1, 3, or 7 d. Injections were performed in a chamber different from the home cage, and rats were kept in the injection chamber for ∼20 min before being returned to the home cage. Midbrain slices were prepared 1 or 10 d after the final injection.
Conditioned place preference.
Conditioning was performed in a three-chamber apparatus, which consisted of two large compartments (28 × 21 cm) separated by a middle gray chamber (12 × 21 cm) (Med Associates). One compartment had a grid floor with black walls, whereas the other had a mesh floor with black-and-white checkered walls. On the pretest day, rats were allowed to explore the entire apparatus for 20 min, and the percentage of time spent in each compartment was calculated after excluding the time spent in the middle chamber. Any rats that spent >60% of time in one compartment were not used for conditioning. There was no overall initial preference for the two compartments in 11 rats tested, which spent 50.8 ± 1.6% of time on the grid floor side in the pretest. Thus, the conditioned place preference (CPP) apparatus was unbiased. During the next 7 d, rats were given injections of saline and confined to one compartment for 30 min in the morning and given injections of amphetamine (5 mg/kg, i.p.) and confined to the other compartment for 30 min in the afternoon. The initially less preferred compartment in each rat (i.e., the compartment that the rat spent 40–50% of time in the pretest) was assigned as the amphetamine-paired side for conditioning. A 20 min posttest was performed 1 d after the last conditioning session. Both the pretest and the posttest took place in the afternoon. The preference for the amphetamine-paired side (expressed in seconds) was defined as the time spent in the amphetamine-paired compartment minus the time spent in the saline-paired compartment. The CPP score was calculated in each rat by subtracting the preference for the amphetamine-paired side in the pretest from that in the posttest. Immediately after the posttest, rats were killed for brain slice electrophysiology to determine the effect of (S)-3,5-dihydroxyphenylglycine (DHPG) on IK(Ca). Data obtained from one to three neurons were averaged in each rat.
More robust CPP is generally observed when drug administration is paired with the initially nonpreferred compartment than with the preferred one (Tzschentke, 1998). In the present study, it was important that the magnitude of CPP was not affected by the initial preference/compartment assignment, since the CPP score was compared to the DHPG effect measured in vitro after the posttest in individual rats. Thus, we always paired amphetamine injection with the initially nonpreferred side in each rat (biased assignment). In this way, the CPP score was independent of the initial preference for the amphetamine-paired compartment in the 11 rats tested. Although the subject assignment procedure (biased vs unbiased) generally affects the magnitude of CPP, it has been shown not to influence the drug's ability to induce CPP as long as the apparatus is unbiased (Cunningham et al., 2003).
Intra-VTA injections and CPP.
Rats were anesthetized with a mixture of ketamine and xylazine (90 and 10 mg/kg, i.p.) and implanted with bilateral chronic guide cannulae (22 gauge; Plastics One), with dummy cannulae (32 gauge) inside, aimed at 1.5 mm above the VTA (anteroposterior, −5.3; mediolateral, +2.2; dorsoventral, −7.5; 10° angle) (Paxinos and Watson, 1998). The guide cannulae were fixed to the skull with stainless steal screws and dental cement. The rats were housed individually after surgery and allowed to recover for at least 10 d.
For intra-VTA microinjections, we used a syringe pump (Harvard Apparatus) connected to 1 μl Hamilton syringes that were attached to the inner injection cannulae (28 gauge; Plastics One). The tip of the injection cannulae extended 1.5 mm beyond the tip of the guide cannulae. To test the effect of the protein kinase A (PKA) inhibitor H89 on the acquisition of amphetamine CPP, rats received bilateral microinjections (0.3 μl/side) of either saline or H89 (1.2 nmol) into the VTA 30 min before each amphetamine conditioning session. In these experiments, the CPP training was reduced to 3 d to minimize the damage produced by repeated intra-VTA microinjections. As described above, amphetamine injections (5 mg/kg, i.p.) were paired with the initially nonpreferred compartment, and saline sessions and amphetamine sessions were performed in the morning and afternoon, respectively. In a separate series of experiments, to test the effect of H89 microinjection alone, systemic injections of saline were paired with both compartments, and intra-VTA microinjections of H89 were made 30 min before the afternoon sessions in which saline was paired with the initially nonpreferred side.
Rats were killed after the CPP posttest, and their brains were removed and stored in PBS containing 10% paraformaldehyde and 10% sucrose. Coronal sections (50 μm) were cut using a vibratome and stained with cresyl violet for histological verification of the injection sites. One rat had injection sites outside the VTA and was excluded from the analysis.
Data analysis.
Data are expressed as mean ± SEM. Statistical significance was determined by Student's t test or ANOVA, followed by the Bonferroni post hoc test. The difference was considered significant at p < 0.05.
Results
In vivo amphetamine exposure increases mGluR-dependent facilitation of AP-evoked Ca2+ signals in VTA DA neurons
Activation of mGluRs and other IP3-coupled neurotransmitter receptors, such as α1 adrenergic receptors and muscarinic acetylcholine receptors, causes facilitation of AP-evoked Ca2+ signals in DA neurons (Cui et al., 2007). To test whether in vivo amphetamine exposure alters mGluR-mediated regulation of AP-induced Ca2+ signals, we performed whole-cell voltage-clamp recordings in DA neurons from naive rats and rats given injections of saline or amphetamine (5 mg/kg, i.p.) for 7 d. Recordings were made in midbrain slices prepared 1 d after the final injection. AP-evoked Ca2+ signals were assessed by measuring Ca2+-sensitive SK currents (IK(Ca)) after unclamped APs elicited by 2 ms depolarizing pulses (see Materials and Methods). A single unclamped AP and a burst of APs (i.e., a train of five APs at 20 Hz) were evoked every 60 s, and the group I mGluR agonist DHPG (1 μm) was superfused for 5 min. In the VTA, DHPG (1 μm) produced small increases in single AP- and burst-induced IK(Ca) in saline-treated rats that were comparable to the DHPG effects observed in naive animals (Fig. 2A,B). In contrast, DHPG produced significantly larger IK(Ca) facilitation in amphetamine-treated rats compared with naive rats and saline-treated rats (single AP: F(2,34) = 7.90, p < 0.01; burst: F(2,34) = 12.5, p < 0.001; one-way ANOVA). The size of baseline IK(Ca) was not affected by in vivo amphetamine treatment (single AP: F(2,34) = 0.16, p = 0.85; burst: F(2,34) = 0.41, p = 0.66; one-way ANOVA) (Fig. 2C). DHPG caused larger facilitation of IK(Ca) in SNc neurons than in VTA neurons for both naive and saline-treated rats (Fig. 2B). Repeated amphetamine exposure failed to further increase the magnitude of DHPG effect in SNc neurons (single AP: F(2,21) = 0.029, p = 0.97; burst: F(2,21) = 0.017, p = 0.98; one-way ANOVA). We next monitored cytosolic Ca2+ levels using Fluo-5F (25 μm) as a Ca2+ indicator in VTA DA neurons (Fig. 2D). DHPG produced significantly larger enhancement of burst-evoked fluorescence changes in amphetamine-treated rats (40 ± 16%; n = 4 from two rats) compared with saline-treated rats (5 ± 4%; n = 5 from two rats; t(7) = 2.39; p < 0.05, unpaired t test). In these experiments, fluorescence changes were measured in proximal dendrites, where DHPG effects can be more readily observed compared to the soma (Cui et al., 2007). These results demonstrate that repeated amphetamine exposure augments mGluR-mediated facilitation of AP-evoked Ca2+ signals in the VTA but not in the SNc.
Figure 2.

mGluR-dependent facilitation of AP-evoked Ca2+ signals is augmented after withdrawal from repeated, but not single, amphetamine exposure. A, Example traces of IK(Ca) (top) and summary time graphs (bottom) illustrating the effects of DHPG (1 μm) on single AP- and burst-evoked IK(Ca) in VTA DA neurons from saline- and amphetamine-treated rats. B, Summary bar graphs demonstrating that in vivo amphetamine exposure augmented DHPG-induced facilitation of IK(Ca) in the VTA (naïve, n = 5 from 3 rats; saline, n = 14 from 12 rats; amphetamine, n = 18 from 17 rats) but not in the SNc (naïve, n = 5 from 4 rats; saline, n = 9 from 7 rats; amphetamine, n = 10 from 8 rats). *p < 0.05; **p < 0.01; ***p < 0.001. C, A summary bar graph showing that the size of baseline IK(Ca) was not affected by in vivo amphetamine treatment in VTA DA neurons. The time integral of IK(Ca) (expressed in picocoulombs) was normalized to the membrane capacitance in each neuron to evaluate the size of baseline IK(Ca). The numbers of neurons per rats are the same as those for VTA data in B. D, Left, Confocal fluorescence image of a VTA DA neuron filled with Fluo-5F (25 μm) from an amphetamine-treated rat. Fluorescence changes were measured at the ROI covering an area in the proximal dendrite that extended ∼10 μm from the soma. Scale bar, 20 μm. Right, Representative traces illustrating the effect of DHPG on burst-induced Ca2+ signals in saline- and amphetamine-treated rats. Bursts of five APs were elicited at the times indicated. Traces from an amphetamine-treated rat were obtained from the neuron shown on the left. E, Summary graph showing the magnitude of DHPG-induced facilitation of IK(Ca) (single AP) after various amphetamine treatment regimens as indicated (saline, n = 14 from 12 rats; 1 d amphetamine, n = 11 from 7 rats; 3 d amphetamine, n = 5 from 4 rats; 7 d amphetamine, n = 18 from 17 rats; 7 d amphetamine plus 10 d withdrawal, n = 9 from 8 rats). F, Repeated amphetamine exposure did not affect DHPG-induced inward currents. The numbers of neurons per rats are the same as those for VTA data in B. Error bars indicate SEM.
Although the magnitude of DHPG-induced IK(Ca) facilitation in the VTA was not changed after single (1 d) amphetamine exposure, it was increased after 3 d of amphetamine exposure to a level comparable with that observed after 7 d treatment (Fig. 2E). Furthermore, the augmentation of DHPG effect persisted for at least 10 d after withdrawal from 7 d amphetamine exposure. Subsequent experiments in the present study were conducted in slices prepared 1 d after 7 d saline/amphetamine exposure.
Superfusion of DHPG also produced a small, sustained inward current (10–40 pA). This mGluR-induced inward current is known to be independent of intracellular Ca2+ signaling in DA neurons (Guatteo et al., 1999). In vivo amphetamine treatment failed to affect the amplitude of DHPG-induced inward current in VTA neurons (F(2,34) = 0.51; p = 0.60, one-way ANOVA) (Fig. 2F), suggesting that the increase in DHPG effect on IK(Ca) is attributable to changes in the signaling pathway downstream of mGluRs. In line with this, the α1 adrenergic receptor agonist phenylephrine (10 μm) also produced significantly larger facilitation of IK(Ca) in amphetamine-treated rats (77 ± 17%; n = 9 from eight rats) compared with saline-treated rats (19 ± 10%; n = 9 from six rats; t(16) = 3.02; p < 0.01, unpaired t test).
PKA-mediated sensitization of IP3Rs underlies the increase in mGluR-induced Ca2+ signal facilitation
mGluR-dependent facilitation of AP-evoked Ca2+ signals is mediated by an increase in cytosolic IP3 levels, leading to enhanced Ca2+-induced Ca2+ release via IP3Rs (Taylor and Laude, 2002; Cui et al., 2007). To examine the effect of in vivo amphetamine exposure on IP3 sensitivity of IP3Rs, we performed flash photolysis of caged IP3 (100 μm) and measured the evoked SK-mediated outward current (IIP3) (Morikawa et al., 2000). The concentration of IP3 released was varied by applying different UV pulse intensities (expressed in microfarads; see Materials and Methods) (Fig. 3A). Concentration–response curves thus constructed revealed a leftward shift after repeated amphetamine exposure in VTA neurons [group (saline/amphetamine):F(1,68) = 5.62, p < 0.05; UV intensity: F(4,68) = 530, p < 0.001; group × UV intensity: F(4,68) = 3.25, p < 0.05; mixed two-way ANOVA] (Fig. 3B). Accordingly, amphetamine treatment produced a significant decrease in the EC50 value (i.e., the UV pulse intensity causing a half-maximal response) in the VTA (saline group: 325 ± 42 μF, n = 9 from six rats; amphetamine group: 220 ± 21 μF, n = 10 from eight rats; t(17) = 2.30; p < 0.05, unpaired t test) but not in the SNc (saline group: 133 ± 22 μF, n = 5 from five rats; amphetamine group: 168 ± 36 μF, n = 4 from three rats; t(7) = 0.86; p = 0.42, unpaired t test). SNc neurons had lower EC50 values than VTA neurons in saline-treated rats, consistent with the larger DHPG effect on IK(Ca) in the SNc (Fig. 2B). The maximal IIP3 amplitude was not affected by amphetamine treatment in VTA neurons (saline group, 5.8 ± 0.7 pA/pF; amphetamine group, 6.5 ± 0.7 pA/pF; t(17) = 0.68; p = 0.50, unpaired t test), suggesting that IP3R expression levels were not altered. Therefore, repeated amphetamine exposure increases IP3R sensitivity in VTA DA neurons.
Figure 3.

IP3R sensitivity is increased after repeated amphetamine exposure. A, Traces of IIP3 evoked with different UV pulse intensities (50, 150, 450, 1350, and 4050 μF) in a VTA DA neuron from an amphetamine-treated rat. The cytosol was loaded with caged IP3 (100 μm). B, Averaged concentration (UV pulse intensity)–response (IIP3) curves in VTA neurons from saline- and amphetamine-treated rats (saline, n = 9 from 6 rats; amphetamine, n = 10 from 8 rats). Data are fitted to a logistic equation. The IIP3 amplitude is normalized to the maximal value. *p < 0.05; **p < 0.01 versus saline group. Error bars indicate SEM.
Exposure to psychostimulants and other drugs of abuse leads to upregulation of the cAMP–PKA pathway in many brain areas, including the VTA (Nestler and Aghajanian, 1997; Tolliver et al., 1999). It is well known that PKA-mediated phosphorylation increases IP3 sensitivity of the neuronal type 1 IP3R (IP3R1) (Tang et al., 2003; Wagner et al., 2008). To examine the expression of IP3R1 in DA neurons, we performed double-labeling immunohistochemistry for IP3R1 and TH, an enzyme responsible for DA synthesis, in midbrain slices prepared from two rats (Fig. 4A). Colocalization of IP3R1 and TH immunoreactivity was found in 124 cells of 126 TH-positive cells and 150 IP3R1-positive cells in the VTA. A similar degree of colocalization was also found in the SNc (93 cells of 93 TH-positive cells and 106 IP3R1-positive cells). Thus, IP3R1 appears to be expressed in virtually all DA neurons and also in certain non-DA neurons in the VTA/SNc. Next, we tested the effect of the PKA inhibitor H89 (10 μm; >1 h preincubation plus intracellular dialysis via the whole-cell pipette) to determine the involvement of PKA in the enhancement of IP3-induced facilitation of IK(Ca) after in vivo amphetamine exposure. In these experiments, we used a low concentration of caged IP3 (25 μm) photolyzed with a low-intensity UV pulse (100 μF), which produced no measurable IIP3 by itself. This subthreshold IP3 photolysis caused facilitation of IK(Ca) when delivered 50 ms before the unclamped AP (Fig. 4B), as was reported previously (Cui et al., 2007). The magnitude of IP3-induced IK(Ca) facilitation thus measured in VTA neurons was significantly increased in amphetamine-treated rats (Fig. 4B,C). H89 largely suppressed this increase in IP3-induced IK(Ca) facilitation caused by in vivo amphetamine exposure [group (saline/amphetamine): F(1,28) = 5.41, p < 0.05; recording condition (control/H89): F(1,28) = 12.5, p < 0.01; group × recording condition: F(1,28) = 6.07, p < 0.05; two-way ANOVA]. Increasing the UV pulse intensity (200–500 μF), which releases a larger concentration of IP3, was able to produce IP3-induced IK(Ca) facilitation in the presence of H89 (saline group: 113 ± 12%, n = 4 from four rats; amphetamine group: 128 ± 41%, n = 5 from four rats; t(7) = 0.31; p = 0.76, unpaired t test) (Fig. 4D), consistent with the idea that H89 reduced the IP3 sensitivity of IP3Rs. The augmentation of DHPG-induced IK(Ca) facilitation observed in amphetamine-treated animals was also suppressed by H89 (group: F(1,22) = 11.4, p < 0.01; recording condition: F(1,22) = 16.0, p < 0.001; group × recording condition: F(1,22) = 13.7, p < 0.01; two-way ANOVA) (Fig. 4E). Furthermore, PKA blockade with H89 eliminated the difference in IP3- and DHPG-induced facilitation of IK(Ca) between saline- and amphetamine-treated animals. Together, these results suggest that repeated amphetamine exposure results in PKA-mediated sensitization of IP3Rs to augment mGluR-dependent facilitation of AP-evoked Ca2+ signals.
Figure 4.
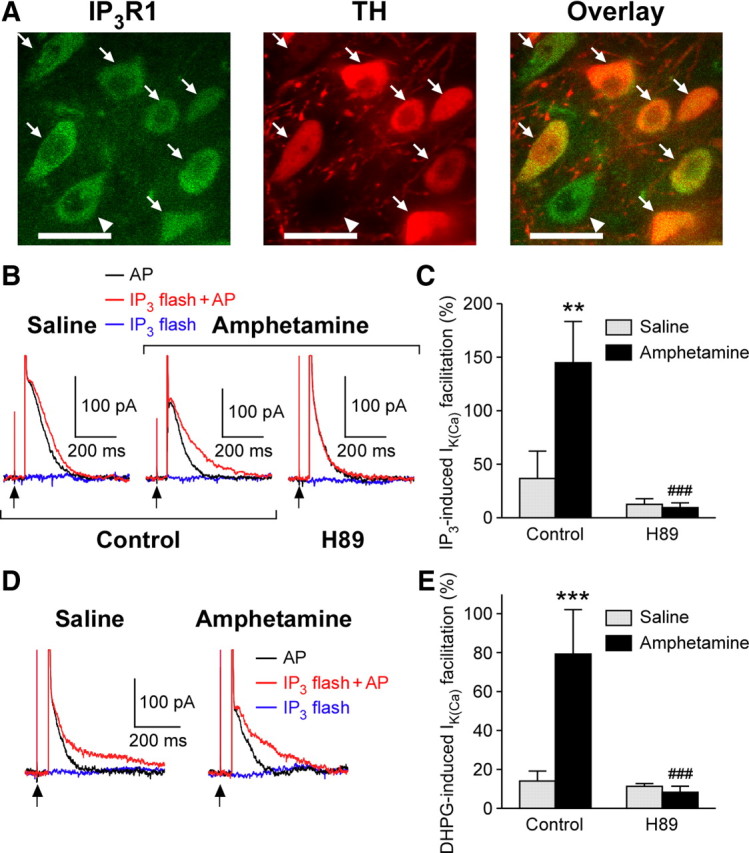
Involvement of PKA in IP3R sensitization after in vivo amphetamine exposure. A, Confocal photomicrographs showing coexpression of IP3R1 (green; left) and TH (red; middle) in VTA neurons. Both images are superimposed to better illustrate cellular colocalization of IP3R1 and TH immunoreactivities (right). Arrows indicate cells coexpressing IP3R1 and TH, whereas the cell with an arrowhead expressed IP3R1 but not TH. Scale bars, 20 μm. B, Representative traces of IK(Ca) (single AP) with and without flash photolysis of caged IP3 (25 μm) in VTA neurons from saline- and amphetamine-treated rats. A low-intensity UV pulse (100 μF) was applied 50 ms before the 2 ms depolarization. Current traces observed with the UV pulse alone without the following depolarization are also shown (blue). Note that IP3-induced facilitation of IK(Ca) observed in amphetamine-treated rats is blocked by H89 (10 μm; right traces). C, Summary bar graph showing the magnitude of IP3-induced facilitation of IK(Ca) under control recording conditions (saline, n = 7 from 3 rats; amphetamine, n = 8 from 6 rats) and in H89 (saline, n = 9 from 4 rats; amphetamine, n = 8 from 4 rats). **p < 0.01 versus saline group; ###p < 0.001 versus control condition. D, Representative traces illustrating that IP3-induced facilitation of IK(Ca) can be observed in the presence of H89 when stronger UV pulses are used (200 μF in the experiments shown). E, Summary bar graph demonstrating the DHPG effect on IK(Ca) under control recording conditions (saline, n = 6 from 5 rats; amphetamine, n = 5 from 5 rats) and in H89 (saline, n = 9 from 7 rats; amphetamine, n = 6 from 4 rats). ***p < 0.001 versus saline group; ###p < 0.001 versus control condition. Error bars indicate SEM.
Increased susceptibility to NMDAR LTP in amphetamine-treated rats
We have recently reported that synaptic activation of mGluRs drives the induction of LTP of NMDAR EPSCs via facilitation of burst-evoked Ca2+ signals in DA neurons (Harnett et al., 2009). We thus asked whether NMDAR LTP induction is enhanced by in vivo amphetamine exposure. In these experiments, pharmacologically isolated NMDAR EPSCs were recorded at −62 mV in VTA neurons (see Materials and Methods). The intensity of synaptic stimulation was adjusted so that it evoked NMDAR EPSCs of ∼40 pA (saline group: 38 ± 1 pA, n = 7 from three rats; amphetamine group: 39 ± 1 pA, n = 5 from four rats). It should be noted that in vivo exposure to psychostimulants has been shown to produce no global change in NMDAR-mediated transmission (Ungless et al., 2001; Borgland et al., 2004). First, synaptic facilitation of IK(Ca) was assessed using trains of synaptic stimulation (50 Hz) having different durations (0.25, 0.5, 1.0, and 1.5 s). A single AP or a burst of APs was evoked in isolation or 100 ms after the synaptic stimulation train. We found that in vivo amphetamine treatment significantly augmented synaptic facilitation of IK(Ca) at all durations tested for both single APs (group: F(1,30) = 17.5, p < 0.01; synaptic stimulation duration: F(3,30) = 26.2, p < 0.001; group × synaptic stimulation duration: F(3,30) = 10.0, p < 0.001; mixed two-way ANOVA) and bursts (group: F(1,21) = 19.3, p < 0.01; synaptic stimulation duration: F(3,21) = 8.89, p < 0.001; group × synaptic stimulation duration: F(3,21) = 2.50, p = 0.087; mixed two-way ANOVA) (Fig. 5). The magnitude of IK(Ca) facilitation became larger with prolongation of the synaptic stimulation train, most likely reflecting gradual accumulation of cytosolic IP3 during sustained mGluR activation (Cui et al., 2007; Harnett et al., 2009).
Figure 5.

Synaptic facilitation of IK(Ca) is enhanced after repeated amphetamine exposure. A, Representative traces illustrating the difference in synaptic facilitation of IK(Ca) between saline- and amphetamine-treated rats using synaptic stimulation trains of various durations (0.25, 0.5, 1.0, and 1.5 s). A single AP was evoked 100 ms after the offset of each synaptic stimulation train. Traces of IK(Ca) after synaptic stimulation are shown after subtracting the trace elicited by synaptic stimulation alone. All traces obtained with different synaptic stimulation durations are overlaid. B, C, Summary graphs plotting the magnitude of synaptic facilitation of IK(Ca) versus stimulation duration for single APs (saline, n = 7 from 3 rats; amphetamine, n = 5 from 4 rats) (B) and bursts (saline, n = 6 from 3 rats; amphetamine, n = 3 from 3 rats) (C). *p < 0.05; **p < 0.01; ***p < 0.001 versus saline group. Stim, stimulation. Error bars indicate SEM.
We next examined NMDAR LTP in VTA neurons from saline- and amphetamine-treated rats. Again, the baseline NMDAR EPSC amplitude was set at ∼40 pA to control for the synaptic stimulation intensity (saline group: 41 ± 2 pA, n = 6 from four rats; amphetamine group: 41 ± 2 pA, n = 5 from four rats). The LTP induction protocol consisted of a 1.4 s synaptic stimulation train paired with a burst, in which the burst onset was delayed by 1 s from the onset of synaptic stimulation (Fig. 6A). Under these conditions, no LTP was observed in saline-treated rats (−1 ± 4%), whereas the same induction protocol resulted in significant LTP in amphetamine-treated rats (24 ± 5%; t(9) = 3.90; p < 0.01 vs saline group, unpaired t test) (Fig. 6A,B). LTP observed in amphetamine-treated rats was not accompanied by changes in the paired-pulse ratio (EPSC2/EPSC1, 50 ms interstimulus interval; 0.87 ± 0.06 before LTP, 0.90 ± 0.06 after LTP; t(4) = 1.36; p = 0.25, paired t test), suggesting a postsynaptic locus of LTP expression, as reported previously for SNc DA neurons in naive rats (Harnett et al., 2009). The magnitude of synaptic facilitation of IK(Ca), assessed immediately before LTP induction in each neuron using a 1 s synaptic stimulation train preceding a single AP, averaged 7 ± 5% and 37 ± 6% in saline- and amphetamine-treated rats, respectively (t(9) = 3.88; p < 0.01, unpaired t test) (Fig. 6C). Pretreatment with H89 (10 μm) suppressed synaptic facilitation of IK(Ca) (6 ± 2%) as well as NMDAR LTP (−1 ± 3%; baseline EPSC amplitude, 40 ± 2 pA) in amphetamine-treated rats (Fig. 6B,C). Furthermore, the magnitude of NMDAR LTP displayed a positive correlation with that of IK(Ca) facilitation when analyzed in all neurons from both saline- and amphetamine-treated rats (r = 0.87) (Fig. 6C). These data suggest that PKA-mediated increase in mGluR-dependent facilitation of AP-induced Ca2+ signals drives enhanced NMDAR plasticity in VTA neurons from amphetamine-treated animals.
Figure 6.

NMDAR-mediated transmission onto VTA DA neurons is more susceptible to LTP induction by synaptic stimulation–burst pairing in amphetamine-treated rats. A, Example experiments to induce NMDAR LTP in saline- and amphetamine-treated rats. Time graphs of NMDAR EPSC amplitude are shown on the left. The LTP induction protocol, which consisted of repetitive (10 times every 20 s) synaptic stimulation–burst pairing (top right), was delivered at the time indicated by the arrow. Traces of NMDAR EPSCs at times indicated by numbers in the time graphs are also shown. B, Summary time graph of NMDAR LTP experiments performed under control recording conditions from saline- and amphetamine-treated rats and in H89 from amphetamine-treated rats (saline, n = 6 from 4 rats; amphetamine, n = 5 from 4 rats; amphetamine–H89, n = 5 from 5 rats). Each symbol represents mean normalized EPSC amplitude from a 2 min window. Error bars indicate SEM. C, The magnitude of NMDAR LTP is plotted versus the magnitude of synaptic facilitation of IK(Ca) in neurons examined for NMDAR LTP in B. The solid line is a linear fit to all data points.
Upregulation of mGluR-dependent Ca2+ signal facilitation may promote amphetamine place conditioning
NMDAR plasticity in DA neurons may contribute to cue–reward learning (Harnett et al., 2009; Zweifel et al., 2009). To assess the learning of environmental cues associated with repeated amphetamine exposure in behaving animals, we performed CPP experiments, in which rats were given injections of saline and amphetamine (5 mg/kg, i.p.) in two distinct compartments for 7 d. In the CPP posttest performed 1 d after 7 d conditioning, all 11 rats exhibited an increase in the time spent on the amphetamine-paired side (t(10) = 11.2, p < 0.001, paired t test) (Fig. 7A). Immediately after the CPP posttest, midbrain slices were prepared from these rats, and IK(Ca) facilitation produced by DHPG (1 μm) was measured in VTA DA neurons. IK(Ca) facilitation averaged 73 ± 14% and 39 ± 7% for single APs and bursts, respectively. These values were comparable with those shown for VTA neurons from amphetamine-treated rats (Fig. 2B). Furthermore, we found that the CPP score was positively correlated with the magnitude of IK(Ca) facilitation for both single APs (r = 0.81) and bursts (r = 0.90) (Fig. 7B).
Figure 7.

Amphetamine-induced CPP correlates with DHPG-induced facilitation of IK(Ca) measured in brain slices. A, The preference for the amphetamine-paired side during the pretest and the posttest are plotted in 11 rats. ***p < 0.001. B, The magnitude of CPP is plotted versus the magnitude of DHPG-induced facilitation of IK(Ca) for both single APs and bursts. Solid lines represent linear fit to the data.
We next tested whether H89 administered directly into the VTA affects the acquisition of amphetamine CPP (Fig. 8). In these experiments, we performed 3 d amphetamine conditioning. It should be noted that upregulation of the mGluR-induced IK(Ca) facilitation is already observed after 3 d of amphetamine exposure (Fig. 2E). We first confirmed that intra-VTA injection of H89 by itself did not affect the preference for the two compartments (Fig. 8B). However, the development of CPP was significantly attenuated when H89 was injected into the VTA before each amphetamine conditioning session (intra-VTA saline: 391 ± 83 s, n = 6; intra-VTA H89: 116 ± 30 s, n = 6; t(10) = 3.13; p < 0.05, unpaired t test) (Fig. 8C). These observations suggest that PKA-mediated upregulation of mGluR-induced Ca2+ signal facilitation in the VTA, which would augment NMDAR plasticity, promotes the learning of environmental cues repeatedly paired with amphetamine exposure.
Figure 8.

PKA blockade in the VTA attenuates the acquisition of amphetamine CPP. A, Left, Representative photomicrograph of a cresyl violet-stained section illustrating bilateral cannulae placements. This section was obtained from a rat that was injected with H89 during amphetamine conditioning. Arrowheads indicate the tips of injection cannulae. Right, Schematic diagram depicting the approximate locations of cannulae tips in 18 rats from which the data in B and C were obtained. The number on each panel represents the distance (in millimeters) from bregma as indicated by Paxinos and Watson (1998). B, A graph demonstrating that intra-VTA injection of H89 does not affect side preference. Both compartments were paired with intraperitoneal injection of saline in these six rats. C, Changes in the preference for the amphetamine-paired side are shown for rats that received intra-VTA injection of saline (left; 6 rats) or H89 (right; 6 rats) before each amphetamine conditioning session. *p < 0.05; **p < 0.01.
Discussion
Previous life experiences, including exposure to drugs of abuse, can induce alterations in the capacity of synapses to exhibit activity-dependent plasticity in the CNS (Abraham, 2008; Mockett and Hulme, 2008). This “plasticity of synaptic plasticity,” termed metaplasticity, may affect the future learning ability of animals. Metaplasticity of glutamatergic transmission most commonly involves changes in NMDAR function/expression that lead to altered capacity of AMPAR-mediated transmission to undergo NMDAR-dependent plasticity (Philpot et al., 2007). Here, we have described a form of metaplasticity of NMDAR-mediated transmission as a consequence of alterations in IP3Rs after in vivo amphetamine exposure. The main finding is that repeated, but not single, exposure to amphetamine markedly enhances mGluR- and IP3-induced facilitation of AP-evoked Ca2+ signals in VTA DA neurons. This leads to increased susceptibility to the induction of NMDAR LTP that is dependent on burst-evoked Ca2+ signals amplified by preceding activation of mGluRs (Fig. 9).
Figure 9.

Schematic diagram illustrating the NMDAR plasticity mechanism and the effect of repeated amphetamine exposure. Sustained stimulation of glutamatergic inputs produces a gradual increase in cytosolic IP3 levels via activation of mGluRs. Burst-evoked Ca2+ signals, triggered by Ca2+ influx through voltage-gated Ca2+ channels (VGCCs), are amplified if the burst occurs when IP3 levels are elevated. These amplified burst-evoked Ca2+ signals drive the induction of NMDAR LTP. After repeated amphetamine exposure, PKA-mediated phosphorylation of IP3Rs will be upregulated, causing an increase in IP3 sensitivity of IP3Rs. This will promote the induction of NMDAR LTP in amphetamine-treated rats.
PKA mediates IP3R sensitization
IP3 facilitates Ca2+-induced Ca2+ release by shifting the Ca2+ sensitivity of IP3Rs (Taylor and Laude, 2002). Thus, mGluR/IP3-mediated facilitation of small, single AP-induced IK(Ca) was greater in magnitude (approximately twofold) compared with facilitation of large, burst-induced IK(Ca) in both control and amphetamine-treated rats.
Our data with the PKA inhibitor H89 demonstrate the involvement of PKA in the augmentation of mGluR/IP3-dependent Ca2+ signal facilitation after amphetamine exposure. This is consistent with previous studies reporting PKA regulation of IP3R-mediated Ca2+ release in DA neurons (Riegel and Williams, 2008; Harnett et al., 2009). PKA phosphorylation of IP3R1 increases its IP3 sensitivity by ∼4- to 15-fold (Tang et al., 2003; Wagner et al., 2008). We observed an ∼1.5-fold increase in the potency of IP3 to evoke SK-mediated outward currents in amphetamine-treated rats, suggesting that the increase in the fraction of PKA phosphorylated IP3R1 may be small. However, this relatively small shift in the sensitivity of IP3Rs resulted in dramatic increases (approximately fourfold to fivefold) in the magnitude of Ca2+ signal facilitation produced by subthreshold levels of IP3, a low concentration of the mGluR agonist DHPG, or low-intensity synaptic stimulation of mGluRs.
It is well known that chronic stimulation of Gi-coupled receptors, including DA D2 receptors, leads to upregulation of the cAMP/PKA pathway (Nevo et al., 1998). Thus, repetitive stimulation of D2 autoreceptors, resulting from amphetamine-induced somatodendritic DA release (Mercuri et al., 1989), may be a potential mechanism causing PKA-mediated IP3R sensitization in DA neurons. In vivo exposure to opiates and alcohol has been shown to increase GABA release via upregulation of the cAMP/PKA pathway at GABAergic terminals onto VTA DA neurons (Bonci and Williams, 1997; Melis et al., 2002). Our data have identified IP3Rs in DA neurons as another target enhanced by PKA upregulation in the VTA. It remains to be determined whether similar sensitization of IP3Rs can be induced with exposure to drugs of abuse other than amphetamine, which would also increase DA levels in the VTA (Yoshida et al., 1993; Campbell et al., 1996).
It is unlikely that voltage-gated Ca2+ channels and/or SK channels are significantly affected by in vivo amphetamine exposure, since the basal size of IK(Ca) was not altered in amphetamine-treated rats. Hence, the influence of repeated amphetamine exposure on AP-induced Ca2+ signals can be detected only when cytosolic IP3 levels are elevated via activation of mGluRs and other IP3-coupled neurotransmitter receptors.
NMDAR metaplasticity: a potential role in “stamping in” the memory of drug-associated stimuli
The magnitude of NMDAR LTP was positively correlated with that of synaptic facilitation of IK(Ca) in our previous study, in which variable synaptic stimulation intensity was used (Harnett et al., 2009). These two parameters displayed a similar positive correlation in VTA DA neurons when the data were pooled from both saline- and amphetamine-treated rats in the present study. Furthermore, the magnitude of synaptic facilitation of IK(Ca) was significantly larger in amphetamine-treated rats when the synaptic stimulation intensity was controlled using the NMDAR EPSC amplitude. Therefore, larger Ca2+ signals during the synaptic stimulation–burst pairing protocol most likely account for the enhancement of NMDAR LTP in amphetamine-treated rats.
The upregulation of mGluR-induced facilitation of AP-evoked Ca2+ signals after amphetamine exposure was observed selectively in the VTA but not in the SNc. It is generally believed that the VTA→NAc pathway plays a predominant role in the initial learning of reward-related cues (Everitt and Robbins, 2005; Beeler et al., 2009). In the present study, we observed a positive correlation between amphetamine CPP measured in behaving rats and mGluR-induced IK(Ca) facilitation in the VTA measured in brain slices prepared from those rats. Furthermore, PKA blockade in the VTA attenuated the development of amphetamine CPP. Therefore, PKA-mediated enhancement of NMDAR plasticity might strengthen the learning of environmental cues experienced during repeated amphetamine exposure, although it is unlikely to be an obligatory requirement for amphetamine CPP, which can be induced even after a single amphetamine conditioning session (Capriles and Cancela, 1999). Interestingly, intra-VTA injection of a cAMP antagonist has been shown to suppress morphine CPP (Harris et al., 2004), suggesting a general role of the cAMP/PKA cascade in the VTA in drug-induced conditioning.
Curiously, SNc neurons exhibited a significantly more potent mGluR-induced Ca2+ signal facilitation and higher IP3R sensitivity compared with VTA neurons in control animals. This implies that SNc DA neurons are more susceptible to NMDAR LTP induction. Indeed, robust NMDAR LTP was observed in SNc DA neurons from naive rats in our previous study (Harnett et al., 2009), whereas no significant LTP was induced in VTA DA neurons from saline-treated rats in the present study using the range of synaptic stimulation intensity that was included in the previous study. The functional significance of this difference between the VTA and the SNc in behaving animals is not clear. Conditioned DA neuron burst responses to reward-predicting cues are acquired rather homogeneously among DA neurons in these two areas (Schultz, 1998; Pan et al., 2005). In this regard, however, it is interesting to note a recent report demonstrating that DA neurons exhibiting burst responses to cues predicting aversive outcomes are mostly located in the SNc but not in the VTA (Matsumoto and Hikosaka, 2009), suggesting that SNc DA neurons are more susceptible to conditioning under certain training conditions.
Accumulating evidence indicates important roles for both AMPAR- and NMDAR-mediated glutamatergic transmissions in the VTA in reward- and drug-induced conditioning (Harris et al., 2004; Engblom et al., 2008; Stuber et al., 2008; Zweifel et al., 2009). Notably, in vivo psychostimulant experience has been shown to produce global potentiation of AMPAR-mediated transmission onto VTA DA neurons, which occludes further LTP while facilitating LTD induction (Ungless et al., 2001; Faleiro et al., 2004; Bellone and Luscher, 2006; Chen et al., 2008) (but see Pu et al., 2006). This generalized increase in AMPAR-mediated excitation of VTA neurons is thought to drive persistent synaptic plasticity in the NAc, probably via enhanced DA release (Wolf et al., 2004; Mameli et al., 2009). In contrast, no global NMDAR potentiation has been detected in VTA DA neurons after in vivo psychostimulant exposure, despite the known regulation of NMDAR function/expression by PKA (Chen and Roche, 2007). We hypothesize that NMDAR potentiation will take place in an input-specific fashion, so that only NMDARs at those inputs activated during amphetamine conditioning would be potentiated. NMDAR LTP induced by synaptic stimulation–burst pairing is input specific, in that only those inputs activated during induction undergo LTP (Harnett et al., 2009). The input specificity of potentiation may mediate the learning of specific environmental stimuli associated with drug experience and with rewards in general. In this scenario, the resulting enhanced NMDAR-dependent DA neuron bursting and phasic DA release in target structures would signal the incentive/motivational salience of those environmental stimuli (Berridge, 2007; Zweifel et al., 2009).
Maladaptive learning of the environments and behaviors associated with drug experience is one of the key pathophysiologies underlying drug addiction (Hyman et al., 2006). A number of studies have demonstrated that previous exposure to drugs of abuse, including psychostimulants, opiates, and nicotine, promotes subsequent cue learning driven by the same drug or different drugs and also by natural rewards (Lett, 1989; Shippenberg et al., 1996; Harmer and Phillips, 1998; Kim et al., 2004; Klein et al., 2007). The NMDAR metaplasticity described in this study may contribute to this generalized “sensitization” of reward-based learning after previous drug experience. In support of this idea, it has been shown that the enhancement of morphine CPP after cocaine exposure is blocked by injection of an NMDAR antagonist into the VTA (Kim et al., 2004). An increased sensitivity to reward-based conditioning has been observed when the training session starts after a period of abstinence, ranging from 1 to 21 d, after previous drug exposure. In our study, the increase in mGluR-dependent facilitation of AP-evoked Ca2+ signals was observed 1 d after repeated amphetamine exposure and lasted for at least 10 d. The increased “conditionability” of DA neurons may act to stamp in the memory of environmental events encountered during initial days of experience with drugs of abuse and may also play a role in the intensification of responsiveness to drug-related cues that develops over the course of abstinence (Grimm et al., 2001).
Footnotes
This work was supported by National Institutes of Health Grant DA015687. B.E.B. was supported by a National Research Service Award. We thank Dr. Katsuhiko Mikoshiba for the gift of the monoclonal anti-IP3R1 antibody (mAb18A10).
References
- Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci. 2008;9:387. doi: 10.1038/nrn2356. [DOI] [PubMed] [Google Scholar]
- Beeler JA, Cao ZF, Kheirbek MA, Zhuang X. Loss of cocaine locomotor response in Pitx3-deficient mice lacking a nigrostriatal pathway. Neuropsychopharmacology. 2009;34:1149–1161. doi: 10.1038/npp.2008.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone C, Luscher C. Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci. 2006;9:636–641. doi: 10.1038/nn1682. [DOI] [PubMed] [Google Scholar]
- Berridge KC. The debate over dopamine's role in reward: the case for incentive salience. Psychopharmacology (Berl) 2007;191:391–431. doi: 10.1007/s00213-006-0578-x. [DOI] [PubMed] [Google Scholar]
- Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. J Neurosci. 1997;17:796–803. doi: 10.1523/JNEUROSCI.17-02-00796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgland SL, Malenka RC, Bonci A. Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J Neurosci. 2004;24:7482–7490. doi: 10.1523/JNEUROSCI.1312-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J, Bullock D, Grossberg S. How the basal ganglia use parallel excitatory and inhibitory learning pathways to selectively respond to unexpected rewarding cues. J Neurosci. 1999;19:10502–10511. doi: 10.1523/JNEUROSCI.19-23-10502.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell AD, Kohl RR, McBride WJ. Serotonin-3 receptor and ethanol-stimulated somatodendritic dopamine release. Alcohol. 1996;13:569–574. doi: 10.1016/s0741-8329(96)00069-9. [DOI] [PubMed] [Google Scholar]
- Capriles N, Cancela LM. Effect of acute and chronic stress restraint on amphetamine-associated place preference: involvement of dopamine D(1) and D(2) receptors. Eur J Pharmacol. 1999;386:127–134. doi: 10.1016/s0014-2999(99)00746-3. [DOI] [PubMed] [Google Scholar]
- Chen BS, Roche KW. Regulation of NMDA receptors by phosphorylation. Neuropharmacology. 2007;53:362–368. doi: 10.1016/j.neuropharm.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM, Chou JK, Bonci A. Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron. 2008;59:288–297. doi: 10.1016/j.neuron.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G, Bernier BE, Harnett MT, Morikawa H. Differential regulation of action potential- and metabotropic glutamate receptor-induced Ca2+ signals by inositol 1,4,5-trisphosphate in dopaminergic neurons. J Neurosci. 2007;27:4776–4785. doi: 10.1523/JNEUROSCI.0139-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CL, Ferree NK, Howard MA. Apparatus bias and place conditioning with ethanol in mice. Psychopharmacology (Berl) 2003;170:409–422. doi: 10.1007/s00213-003-1559-y. [DOI] [PubMed] [Google Scholar]
- Deister CA, Teagarden MA, Wilson CJ, Paladini CA. An intrinsic neuronal oscillator underlies dopaminergic neuron bursting. J Neurosci. 2009;29:15888–15897. doi: 10.1523/JNEUROSCI.4053-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engblom D, Bilbao A, Sanchis-Segura C, Dahan L, Perreau-Lenz S, Balland B, Parkitna JR, Lujan R, Halbout B, Mameli M, Parlato R, Sprengel R, Luscher C, Schutz G, Spanagel R. Glutamate receptors on dopamine neurons control the persistence of cocaine seeking. Neuron. 2008;59:497–508. doi: 10.1016/j.neuron.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8:1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- Faleiro LJ, Jones S, Kauer JA. Rapid synaptic plasticity of glutamatergic synapses on dopamine neurons in the ventral tegmental area in response to acute amphetamine injection. Neuropsychopharmacology. 2004;29:2115–2125. doi: 10.1038/sj.npp.1300495. [DOI] [PubMed] [Google Scholar]
- Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J Neurosci. 2006;26:2788–2797. doi: 10.1523/JNEUROSCI.4331-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgetti M, Hotsenpiller G, Ward P, Teppen T, Wolf ME. Amphetamine-induced plasticity of AMPA receptors in the ventral tegmental area: effects on extracellular levels of dopamine and glutamate in freely moving rats. J Neurosci. 2001;21:6362–6369. doi: 10.1523/JNEUROSCI.21-16-06362.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JW, Hope BT, Wise RA, Shaham Y. Neuroadaptation. Incubation of cocaine craving after withdrawal. Nature. 2001;412:141–142. doi: 10.1038/35084134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guatteo E, Mercuri NB, Bernardi G, Knopfel T. Group I metabotropic glutamate receptors mediate an inward current in rat substantia nigra dopamine neurons that is independent from calcium mobilization. J Neurophysiol. 1999;82:1974–1981. doi: 10.1152/jn.1999.82.4.1974. [DOI] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci. 2009;29:11011–11019. doi: 10.1523/JNEUROSCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmer CJ, Phillips GD. Enhanced appetitive conditioning following repeated pretreatment with D-amphetamine. Behav Pharmacol. 1998;9:299–308. [PubMed] [Google Scholar]
- Harnett MT, Bernier BE, Ahn KC, Morikawa H. Burst-timing-dependent plasticity of NMDA receptor-mediated transmission in midbrain dopamine neurons. Neuron. 2009;62:826–838. doi: 10.1016/j.neuron.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris GC, Wimmer M, Byrne R, Aston-Jones G. Glutamate-associated plasticity in the ventral tegmental area is necessary for conditioning environmental stimuli with morphine. Neuroscience. 2004;129:841–847. doi: 10.1016/j.neuroscience.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Johnson SW, Seutin V, North RA. Burst firing in dopamine neurons induced by N-methyl-D-aspartate: role of electrogenic sodium pump. Science. 1992;258:665–667. doi: 10.1126/science.1329209. [DOI] [PubMed] [Google Scholar]
- Jones S, Kornblum JL, Kauer JA. Amphetamine blocks long-term synaptic depression in the ventral tegmental area. J Neurosci. 2000;20:5575–5580. doi: 10.1523/JNEUROSCI.20-15-05575.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JA, Pollak KA, Hjelmstad GO, Fields HL. A single cocaine exposure enhances both opioid reward and aversion through a ventral tegmental area-dependent mechanism. Proc Natl Acad Sci U S A. 2004;101:5664–5669. doi: 10.1073/pnas.0401373101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein ED, Gehrke BJ, Green TA, Zentall TR, Bardo MT. Repeated cocaine experience facilitates sucrose-reinforced operant responding in enriched and isolated rats. Learn Motiv. 2007;38:44–55. doi: 10.1016/j.lmot.2006.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lett BT. Repeated exposures intensify rather than diminish the rewarding effects of amphetamine, morphine, and cocaine. Psychopharmacology (Berl) 1989;98:357–362. doi: 10.1007/BF00451687. [DOI] [PubMed] [Google Scholar]
- Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR, Spanagel R, Luscher C. Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci. 2009;12:1036–1041. doi: 10.1038/nn.2367. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Hikosaka O. Two types of dopamine neuron distinctly convey positive and negative motivational signals. Nature. 2009;459:837–841. doi: 10.1038/nature08028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCray JA, Herbette L, Kihara T, Trentham DR. A new approach to time-resolved studies of ATP-requiring biological systems; laser flash photolysis of caged ATP. Proc Natl Acad Sci U S A. 1980;77:7237–7241. doi: 10.1073/pnas.77.12.7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Camarini R, Ungless MA, Bonci A. Long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J Neurosci. 2002;22:2074–2082. doi: 10.1523/JNEUROSCI.22-06-02074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri NB, Calabresi P, Bernardi G. The mechanism of amphetamine-induced inhibition of rat substantia nigra compacta neurones investigated with intracellular recording in vitro. Br J Pharmacol. 1989;98:127–134. doi: 10.1111/j.1476-5381.1989.tb16872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockett BG, Hulme SR. Metaplasticity: new insights through electrophysiological investigations. J Integr Neurosci. 2008;7:315–336. doi: 10.1142/s0219635208001782. [DOI] [PubMed] [Google Scholar]
- Morikawa H, Imani F, Khodakhah K, Williams JT. Inositol 1,4,5-triphosphate-evoked responses in midbrain dopamine neurons. J Neurosci. 2000;20:RC103. doi: 10.1523/JNEUROSCI.20-20-j0003.2000. (1–5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakade S, Maeda N, Mikoshiba K. Involvement of the C-terminus of the inositol 1,4,5-trisphosphate receptor in Ca2+ release analysed using region-specific monoclonal antibodies. Biochem J. 1991;277:125–131. doi: 10.1042/bj2770125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi S, Fujii A, Nakade S, Mikoshiba K. Immunohistochemical localization of inositol 1,4,5-trisphosphate receptors in non-neural tissues, with special reference to epithelia, the reproductive system, and muscular tissues. Cell Tissue Res. 1996;285:235–251. doi: 10.1007/s004410050641. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- Nevo I, Avidor-Reiss T, Levy R, Bayewitch M, Heldman E, Vogel Z. Regulation of adenylyl cyclase isozymes on acute and chronic activation of inhibitory receptors. Mol Pharmacol. 1998;54:419–426. doi: 10.1124/mol.54.2.419. [DOI] [PubMed] [Google Scholar]
- Overton P, Clark D. Iontophoretically administered drugs acting at the N-methyl-D-aspartate receptor modulate burst firing in A9 dopamine neurons in the rat. Synapse. 1992;10:131–140. doi: 10.1002/syn.890100208. [DOI] [PubMed] [Google Scholar]
- Pan WX, Schmidt R, Wickens JR, Hyland BI. Dopamine cells respond to predicted events during classical conditioning: evidence for eligibility traces in the reward–learning network. J Neurosci. 2005;25:6235–6242. doi: 10.1523/JNEUROSCI.1478-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 4. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Philpot BD, Cho KK, Bear MF. Obligatory role of NR2A for metaplasticity in visual cortex. Neuron. 2007;53:495–502. doi: 10.1016/j.neuron.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu L, Liu QS, Poo MM. BDNF-dependent synaptic sensitization in midbrain dopamine neurons after cocaine withdrawal. Nat Neurosci. 2006;9:605–607. doi: 10.1038/nn1687. [DOI] [PubMed] [Google Scholar]
- Puopolo M, Raviola E, Bean BP. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci. 2007;27:645–656. doi: 10.1523/JNEUROSCI.4341-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riegel AC, Williams JT. CRF facilitates calcium release from intracellular stores in midbrain dopamine neurons. Neuron. 2008;57:559–570. doi: 10.1016/j.neuron.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Schultz W. Predictive reward signal of dopamine neurons. J Neurophysiol. 1998;80:1–27. doi: 10.1152/jn.1998.80.1.1. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS, Heidbreder C, Lefevour A. Sensitization to the conditioned rewarding effects of morphine: pharmacology and temporal characteristics. Eur J Pharmacol. 1996;299:33–39. doi: 10.1016/0014-2999(95)00852-7. [DOI] [PubMed] [Google Scholar]
- Stuber GD, Klanker M, de Ridder B, Bowers MS, Joosten RN, Feenstra MG, Bonci A. Reward-predictive cues enhance excitatory synaptic strength onto midbrain dopamine neurons. Science. 2008;321:1690–1692. doi: 10.1126/science.1160873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TS, Tu H, Wang Z, Bezprozvanny I. Modulation of type 1 inositol (1,4,5)-trisphosphate receptor function by protein kinase a and protein phosphatase 1α. J Neurosci. 2003;23:403–415. doi: 10.1523/JNEUROSCI.23-02-00403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CW, Laude AJ. IP3 receptors and their regulation by calmodulin and cytosolic Ca2+ Cell Calcium. 2002;32:321–334. doi: 10.1016/s0143416002001859. [DOI] [PubMed] [Google Scholar]
- Tolliver BK, Ho LB, Fox LM, Berger SP. Necessary role for ventral tegmental area adenylate cyclase and protein kinase A in induction of behavioral sensitization to intraventral tegmental area amphetamine. J Pharmacol Exp Ther. 1999;289:38–47. [PubMed] [Google Scholar]
- Tzschentke TM. Measuring reward with the conditioned place preference paradigm: a comprehensive review of drug effects, recent progress and new issues. Prog Neurobiol. 1998;56:613–672. doi: 10.1016/s0301-0082(98)00060-4. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Wagner LE, 2nd, Joseph SK, Yule DI. Regulation of single inositol 1,4,5-trisphosphate receptor channel activity by protein kinase A phosphorylation. J Physiol. 2008;586:3577–3596. doi: 10.1113/jphysiol.2008.152314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanat MJ, Hopf FW, Stuber GD, Phillips PE, Bonci A. Corticotropin-releasing factor increases mouse ventral tegmental area dopamine neuron firing through a protein kinase C-dependent enhancement of Ih. J Physiol. 2008;586:2157–2170. doi: 10.1113/jphysiol.2007.150078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CJ, Callaway JC. Coupled oscillator model of the dopaminergic neuron of the substantia nigra. J Neurophysiol. 2000;83:3084–3100. doi: 10.1152/jn.2000.83.5.3084. [DOI] [PubMed] [Google Scholar]
- Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5:483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- Wolf ME, Sun X, Mangiavacchi S, Chao SZ. Psychomotor stimulants and neuronal plasticity. Neuropharmacology. 2004;47(Suppl 1):S61–S79. doi: 10.1016/j.neuropharm.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Wolfart J, Neuhoff H, Franz O, Roeper J. Differential expression of the small-conductance, calcium-activated potassium channel SK3 is critical for pacemaker control in dopaminergic midbrain neurons. J Neurosci. 2001;21:3443–3456. doi: 10.1523/JNEUROSCI.21-10-03443.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Yokoo H, Tanaka T, Mizoguchi K, Emoto H, Ishii H, Tanaka M. Facilitatory modulation of mesolimbic dopamine neuronal activity by a mu-opioid agonist and nicotine as examined with in vivo microdialysis. Brain Res. 1993;624:277–280. doi: 10.1016/0006-8993(93)90087-4. [DOI] [PubMed] [Google Scholar]
- Zweifel LS, Parker JG, Lobb CJ, Rainwater A, Wall VZ, Fadok JP, Darvas M, Kim MJ, Mizumori SJ, Paladini CA, Phillips PE, Palmiter RD. Disruption of NMDAR-dependent burst firing by dopamine neurons provides selective assessment of phasic dopamine-dependent behavior. Proc Natl Acad Sci U S A. 2009;106:7281–7288. doi: 10.1073/pnas.0813415106. [DOI] [PMC free article] [PubMed] [Google Scholar]