

Abstract
The neurodegenerative disorder Alzheimer’s disease (AD) is the 6th leading cause of death in the USA. In addition to neurological and psychiatric symptoms, AD is characterized by deficiencies in S–adenylmethionine (SAM), vitamin B12, and folate. Deficiency in these nutrients has been shown to result in gene promoter methylation with upregulation of AD–associated genes. While some cases of AD are due to specific mutations in genes such as presenilin 1 (PSEN) and the amyloid–β peptide precursor protein (APP), these familial AD (FAD) cases account for a minority of cases. The majority of genetic contribution consists of risk factors with incomplete penetrance. Several environmental risk factors, such as cholesterol and diet, head trauma, and reduced levels of exercise, have also been determined for AD. Nevertheless, the majority of risk for AD appears to be established early in life. We propose to explain this via the LEARn (Latent Early–life Associated Regulation) model. LEARn-AD (LAD) would be a “two-hit” disorder, wherein the first hit would occur due to environmental stress within the regulatory sequences of AD–associated genes, maintained by epigenetic changes such as in DNA methylation. This hit would most likely come in early childhood. The second hit could consist of further stress, such as head trauma, poor mid–life diet, or even general changes in expression of genes that occur later in life independent of any pathogenesis. Given that the primary risk for LAD would be maintained by DNA (hypo)methylation, we propose that long–term nutritional remediation based on the LEARn model, or LEARn–based nutritional gain (LEARnING), beginning early in life, would significantly reduce risk for AD late in life.
Introduction: Hallmarks of and major molecular players in Alzheimer’s disease
Alzheimer’s disease is the most common form of dementia, which is a group of brain disorders that cause memory loss and a decline in mental function, over time. In fact, AD, which is a progressive neurodegenerative disorder, is the 6th leading cause of death in the USA. It has recently been cited as afflicting 5.3 million people, and further affecting at least another 9.9 million unpaid caregivers, according to the Alzheimer’s Association (2009). The disease usually appears late in life and is characterized by extracellular plaque in specific brain regions, composed mainly of β–amyloid peptide (Aβ), intraneuronal tangles composed of microtubule–associated protein tau (τ, MAPT), and progressive loss of synaptic markers and cholinergic neurons, eventually resulting in dementia (Hardy, 2006; Maslow, 2008).
Many factors point to a significant genetic element in the etiology of AD. Significant risk for developing AD is established by early adulthood (Ashford and Mortimer, 2002) or potentially earlier in life (Borenstein et al., 2006). Incidence of early onset familial AD (FAD) led to the discovery of several disease–associated mutations in the presenilin 1 (PSEN1), presenilin 2 (PSEN2), and Aβ precursor protein (APP) genes. In addition, the apolipoprotein E gene (APOE) ε4 genotype has a dose–dependent association as a risk factor for AD (Corder et al., 1993), and this genotype has been recently associated with altered temporal lobe activity in young adults (Dennis et al., 2009). Polymorphisms in the APOE gene promoter have been associated with AD and changes in promoter activity, irrespective of ε4 status (Maloney et al., 2009). Twin studies of AD show 75%–80% concordance in monozygotic and 26%–46% in dizygotic twins (Bergem et al., 1997). Additional genetic associations have been found with the MAPT, insulin degrading enzyme (IDE), α2–macroglobulin, and endothelin converting enzyme 2 (ECE2), among many other genetic associations (Lahiri et al., 2009).
Familial Alzheimer’s disease (FAD) represents minority of cases
On the other hand, evidence suggests that genetics alone cannot explain the incidence of AD. The FAD mutations in PSEN1, PSEN2, and APP only account for no more than 5% of AD in general (Maslow, 2008). The APOEε4 genotype has weak predictive power for AD, and APOEε4 homozygotes do not necessarily develop AD (Myers et al., 1996). Twin studies rest on certain assumptions that may not always be valid (Winerman, 2004). Likewise, significant environmental risk factors have been determined for AD, including, but not limited to cholesterol/diet (Sambamurti et al., 2004) head injury (Mortimer et al., 1991), inflammation (Bales et al., 2000), and reduced midlife physical activity (Friedland et al., 2001). However, the greatest risk factor for AD, as reported by the Alzheimer’s Association (2009), is advancing age.
FAD vs. Sporadic Alzheimer’s disease (SAD)
Any complete model for the etiology of AD, or neurobiological disorders in general, must take into account incomplete penetrance of associated genes and of non–genetic risk factors. A currently popular model posits polygenic and multifactorial effects, with no single factor either necessary or sufficient. Such a hypothesis actually sidesteps the issue of etiology. We propose that AD, and potentially other neurobiological disorders, is a “cluster” of symptomatically similar conditions with distinct etiologies, and we propose three likely parallel explanations (Fig. 1). First, there are “fully” genetic cases, wherein mutation in a critical gene leads to an autosomally inherited disorder, such as FAD. These appear to be a minority of cases. A second category of cases would be “fully environmental”, in terms of AD, they would be precipitated by events such as head trauma, exposure to certain toxicological agents, or acute oxidative stress (Fig. 1). Such sporadic AD (SAD) would rely upon a cascade of events proximally associated with the stress in question. Given the solid evidence in favor of (incomplete) genetic associations for AD, we conclude that SAD would also be a minority of cases. SAD can, in theory, be caused at any time of life and could affect expression of genes at any point, as it is an essentially random process.
Fig. 1. Pathways to AD, FAD, SAD, LAD.
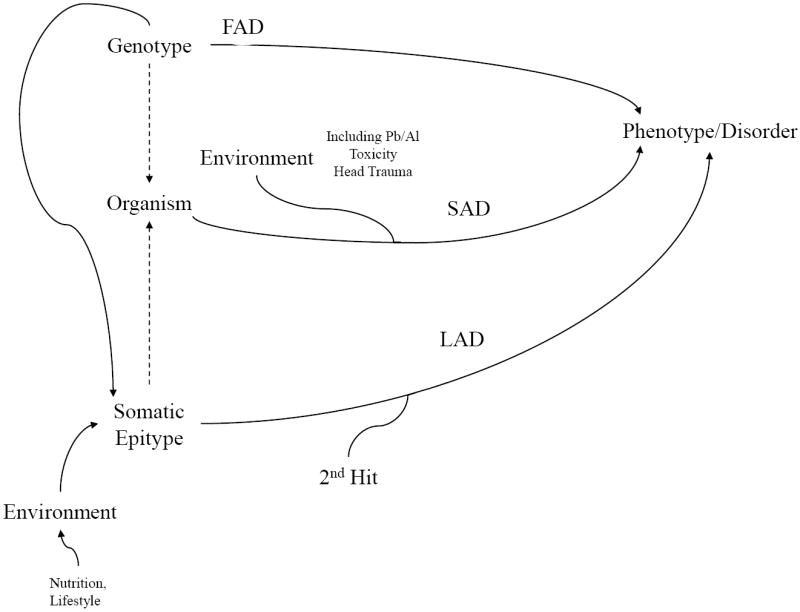
Three pathways are posited that can lead to pathogenic conditions such as AD. In the familial AD (FAD) pathway, genotype results directly in AD. This pathway explains a minority of AD cases. In the sporadic (SAD) pathway, genotype or somatic genotype have minimal input (excluding cases of FAD), and it is direct, non–latent response to environmental insults (such as metal toxicity or head trauma) that lead to AD. In the LEARn AD (LAD) pathway, environment induces epigenetic marker shifts in critical gene sequences, resulting in potentially pathogenic, but latent, somatic epitype(s). These somatic epitypes would undergo “second hit” environmental effects that result in disease.
“Latent Early–life Associated Regulation” or “LEARn” model
We propose that the majority of AD cases would, instead, follow an etiology based on “Latent Early–life Associated Regulation” or “LEARn” (Lahiri et al., 2007; Lahiri et al., 2009; Lahiri et al., 2008). Briefly, LEARn posits that early–life influences, such as exposure to metals, nutritional variation, variation in maternal care, and other stressors modify potential expression levels of disorder–associated genes in a latent fashion. Such latent changes are maintained by epigenetic markers in the promoter sequences of such genes, such as changes in DNA methylation, DNA oxidation, and chromatin organization. Later in life, LEARn–modified genes would require additional triggers or “hits” to express at pathological levels (Fig. 1), similar to the “two–hit” model of oncology (Scrable et al., 1989), also proposed for AD (Zhu et al., 2007). In addition, LEARn proposes two structurally distinct categories of target genes, “LEARned” and “unLEARned”. These structural differences are in densities of CpG and GG dinucleotides, sites of DNA methylation and oxidation, respectively, of the primary DNA sequences of the gene. Specifically, LEARned gene promoters have greater density of CpG and GG dinucleotides than do unLEARned promoters at critical locations proximal to the +1 transcription start site (Lahiri et al., 2009). Early–life exposure to materials such as lead (Pb) may induce a short term upregulation of LEARned AD–associated genes, followed by a long latency period of “normal” levels of gene expression, but latency ends when disease–associated gene expression levels increase later in life (Basha et al., 2005; Wu et al., 2008). Oxidative DNA damage (measured by Oxo8dG), remains low early in life but later increases, potentially due to suppression of repair enzyme expression (Bolin et al., 2006).
LEARn AD (LAD) explains the incomplete penetrance of genetic factors for idiopathic AD using two avenues. First, specific DNA sequences would be more or less vulnerable to LEARn “first hit” modification, according to sequence details. This could include promoter polymorphic sites, especially those that would create or delete a CpG or GG dinucleotide. Second, lack of an appropriate “second hit” would preclude development of LAD, even if a “first hit” had been experienced.
The APP protein, Aβ peptide, and protein are all expressed in healthy individuals. Their simple presence is not a sign of active or incipient AD. A trigger for the disease that is independent of the presence of these proteins is likely to exist. What would trigger APP and Aβ peptides to be overproduced in sporadic cases of AD? The LEARn model proposes that the initial APP triggering mechanism occurs early in life, at developmental stages. Sites of action would be within the promoter of APP and other AD associated genes. The trigger would be maintained through differences in DNA methylation or other epigenetic structures. It is also possible that genes with products protective against AD would have altered methylation patterns due to environmental stress, much as some cancers have been found to associate with specific hypomethylation of oncogenes accompanied with hypermethylation of protective genes (Suzuki et al., 2009). However, a long–latent condition, such as LAD, is likely to function as a “two–hit” disorder, similarly to those found in currently accepted models of cancer etiology (Knudson, 1971), and previously proposed for AD by other workers (Zhu et al., 2007). In the 1980s, Barker and colleagues raised the possibility of developmental origins of late–life disorders (Barker et al., 1989). More recently a detailed “DOHaD” (developmental origins of health and disease) model (Gluckman et al., 2007) has been proposed as has been a toxicological model of Szyf et al (Szyf, 2007). We extend the Barker hypothesis by proposing specific, testable, molecular mechanisms. Likewise, in contrast to Zhu et al, we propose that the first hit in the majority of AD would be during a critical period of early post–natal development, and that it would not be a generalized “oxidative damage” but would, instead, be present in specific vulnerable locations in LEARned gene promoters. Regarding the second hit, it could be a specific environmental influence or even the changes in gene expression, especially upregulation of inflammatory factors, that have been shown to be a function of normal aging (Lu et al., 2004). This is not to say that these environmental insults “intentionally” target AD–related genes in the brain. Instead, certain genes, by juxtaposition of CpG sites with important active/inactive transcription factor sites, would be particularly vulnerable to the effects of environmental stresses that alter CpG methylation patterns.
Genotype vs. Somatic epitype, and the role of Oxidative damage in AD
LEARn introduces an intermediate step between genotype and phenotype, specifically the “somatic epitype”, which is an epigenotype acquired after birth (Lahiri and Maloney, 2006). Evidence for the somatic epitype includes changes in DNA methylation induced by maternal behavior (Weaver et al., 2004). Furthermore, early overfeeding alters methylation and expression levels of specific gene promoters (Plagemann et al., 2009).
Oxidative damage of DNA usually appears as conversion of d–guanosine to oxo8d–guanosine. The 5’ “G” of a “GG” dinucleotide is a hot spot of such oxidation (Hall et al., 1996). This interferes with the binding of methyl CpG–binding protein (MeCP) to methylated cytosine (Valinluck et al., 2004), an “effective demethylation”. Heavy metals such as Pb are known to induce oxidative stress (Fowler et al., 2004), and oxidative stress modulates DNA methylation during malignant transformation (Campos et al., 2007). Methylation in mammalian DNA occurs as the addition of a methyl group to cytosine residues at CpG dinucleotides (Valinluck et al., 2004). Hypomethylation in the promoter region leads to elevated gene expression, whereas hypermethylation results in decreased gene expression. Methylation is catalyzed by DNA methyltransferase (DNMT) enzymes (Bestor, 2000). The activity of DNMT is reduced by heavy metal (cadmium) exposure (Takiguchi et al., 2003). Environmental stressors, including exposure to metals and dietary factors, may interfere with the methylation of CpG clusters, thus altering affinity with potential transcription factors proteins, such as MeCP and specificity protein 1 (SP1). Aluminum has been shown to induce Z–DNA conformation at CCG repeats (Latha et al., 2002).
Role of nutrition in the development of LEARn-AD (LAD)
Nutrition plays a vital role in methylation of DNA, specifically the homocysteine (HCY)/S–adenosylmethionine (SAM) cycle (Fig. 2A). This cycle requires the presence of folate and B12, which facilitate the conversion of HCY to methionine, which is then converted to SAM. SAM then serves as a source of methyl groups for multiple methylation reactions, including methylation of DNA. SAM is thereby converted to S–adenosylhomocysteine, which then converts to HCY in a reversible reaction (Fuso et al., 2005). Deficiency of folate and B12 would reverse the cycle, resulting in increased HCY. HCY can convert to S–adenosylhomocysteine, excess of which inhibits methyl transfer from SAM to various substrates, potentially resulting in DNA hypomethylation (Fig 2B).
Fig. 2. The HCY/SAM cycle.
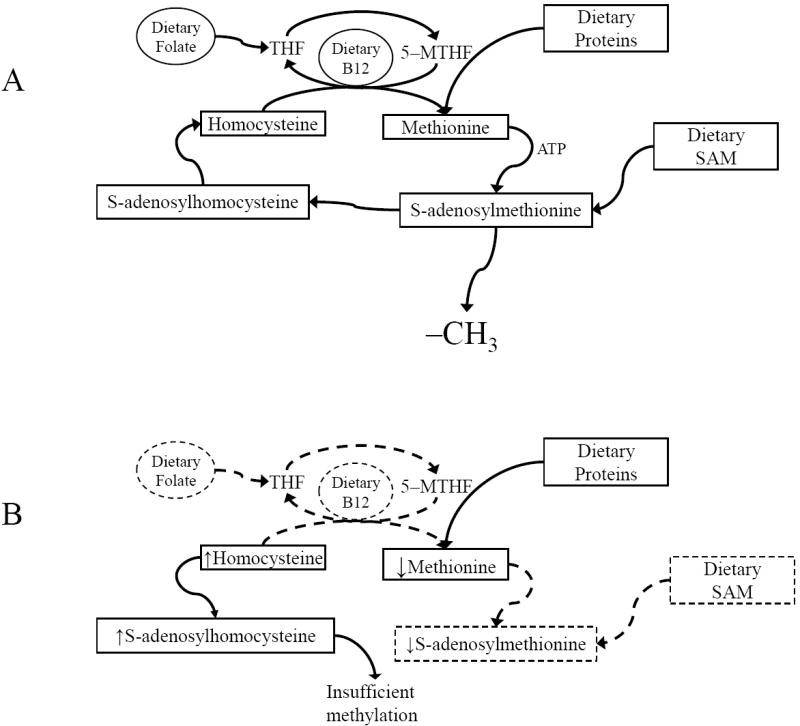
B) Dietary folate and B12 facilitate the conversion of HCY to methionine, which is then converted to SAM. SAM can also be directly supplemented. SAM provides methyl groups for transfer to DNA by DNA methylases and is converted to S–adenosylhomocysteine in the process. Low levels of HCY drive conversion of S–adenosylhomocysteine to HCY, which is then reconverted to methionine. A) Pathogenic (reverse) pathway. Lack of folate and B12 permit accumulation of homocysteine (HCY), which is converted to S–adenosylhomocysteine. High levels of S–adenosylhomocysteine block transfer of methyl groups from S–adenylmethionine (SAM) to DNA or other substrates.
In the context of pathogenic gene expression, induction and remediation of B12/folate deficiency have been shown to alter levels of PSEN1 and BACE1 expression and to alter levels of Aβ in cell culture and in mice (Fuso et al., 2008; Fuso et al., 2009). The structure of the BACE1 promoter would be illustrative of the process. This promoter has multiple CpG sites along a 4 kilobase length. An SP1 transcription factor binding site that contains a CpG dinucleotide has been determined to be active in this promoter (Christensen et al., 2004). B12/folate deficiency would drive the HCY/SAM cycle in the “reverse” direction, inhibiting DNA methylation. An unmethylated SP1 site would be available for transcription factor binding, which would drive elevated BACE1 expression, leading to elevated amyloidogenesis (Fig. 3). On the other hand, supplementation of the deficiency by folate/B12 or direct SAM supplementation would result in driving the cycle “forward”, increasing DNA methylation. Restored methylation would permit binding of methylated DNA binding proteins (MDBP), which would block binding of SP1, reducing BACE1 gene transcription and, ultimately, amyloidogenesis (Fig. 4). It should be noted in a similar context that overfeeding–induced hypermethylation of the pro-opiomelanocortin (POMC) promoter that led to reduced gene expression specifically interfered with SP1–binding sites (Plagemann et al., 2009).
Fig. 3. Deregulation of BACE1 promoter from B12/folate deficiency.
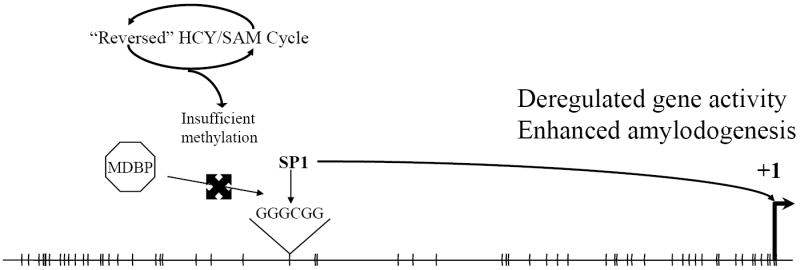
Under nutrient–deficient conditions, the HCY/SAM cycle runs in “reverse”, resulting in reduced DNA methylation efficiency. Lack of methylation of critical CpG dinucleotides prevents binding of methyl–DNA binding proteins (MDBP) and permits binding of transcription factors such as SP1. SP1 binding drives increased gene expression, which ultimately results in elevated amyloidogenesis.
Fig. 4. Regulation of BACE1 promoter following B12/folate or SAM supplementation.
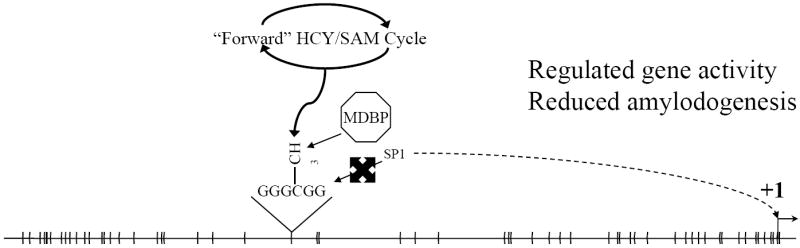
Methylation deficiency can be remediated by dietary supplementation with B12, folate, and/or SAM. This would drive the HCY/SAM cycle “forwards” and provide more abundant methyl groups for transfer to substrates such as CpG dinucleotides. Methylated CpG would permit binding of MDBP, which would inhibit binding of SP1. Inhibition of SP1 binding would regulate gene expression and reduce levels of BACE1 and amyloidogenesis.
Methylation levels for an individual person can change over lifespan
It has recently been shown that methylation levels for an individual person can change over lifespan in a significant portion of a human population (Bjornsson et al., 2008). A differential delayed neurological response has also been demonstrated in rats subjected to postnatal inflammation. The treatment resulted in increased susceptibility to seizure in adulthood (Galic et al., 2008). When considering AD, a pair of identical twins with discordant development of AD had been brought up in the same household. One twin developed AD, while the other did not. It was determined that DNA methylation levels in the temporal neocortex and the superior frontal gyrus, but not the cerebellum were different from each other, with the AD twin having a lower level of methylation than the non-AD twin (Mastroeni et al., 2009). Likewise, decrements in 10 markers of DNA methylation were found in the entorhinal cortex layer II in AD post mortem brains compared to non–AD samples (Mastroeni et al., 2008). Hyperhomocysteinema is well associated with AD (Seshadri, 2006), and this feature has been associated with enhanced levels of gamma–secretase and BACE1 and elevated levels of Aβ peptide in cell culture and animal studies (Fuso et al., 2008; Pacheco-Quinto et al., 2006; Zhang et al., 2009). The alterations in gamma–secretase and BACE1 expression are related to changes in gene methylation (Fuso et al., 2009). Vitamin B deprivation resulted in hyperhomocysteinemia and upregulation of gamma–secretase and BACE1 (Fuso et al., 2008).
These facets of AD vis HCY, folate, and B12 could reasonably suggest an acute relationship among HCY, folate, B12, and AD. High dose supplementation has been shown to reduce plasma HCY levels in AD (Aisen et al., 2003). However, a later controlled study of supplementation with B12 and B6 indicated no change in cognitive decline in mild or moderate AD among patients receiving supplementation vs. those not receiving supplementation (Aisen et al., 2008). If AD were due to an acute nutritional deficiency or imbalance, reversal of this imbalance should at least slow progress of the disorder.
Nutritional remedy: LEARn–induced nutritional gain, or LEARnING
Our LEARn model proposes that connections among AD, HCY/SAM cycle, and DNA methylation are, on the other hand, indicative of a latent condition that may be reversed given sufficiently early intervention. Risk for development and progress of AD is likely to be established by early adulthood (Riley et al., 2005; Snowdon et al., 2000), and a significant body of data points to adopting a “life–course approach” to AD etiology (Whalley et al., 2006). Such a long term of latency suggests the possibility of early–life nutritional remediation. It would be expected that, while vitamin B supplementation would not be efficacious at slowing the progression of AD, higher folate intake has been related to reduced risk of AD in a USA population (Luchsinger et al., 2008). Fruit juices, such as 0.5% apple juice concentrate, have been shown to be a useful source of SAM, reversing DNA hypomethylation in mice (Chan and Shea, 2006). In addition, exercise in rats has been shown to modulate the activity of mucosal betaine–homocysteine methyltransferase 2, potentially reducing aberrant methylation (Buehlmeyer et al., 2008). This suggests the possibility that lifestyle habits suspected to protect against AD, such as physical exercise (Kivipelto and Solomon, 2008), may work through remediation of early–life aberrant DNA methylation. We refer to nutritionally–based early–life therapeutic modification of epigenetic markers of LEARn as LEARn–based induced nutritional gain, or LEARnING.
Summary of three etiological pathways for AD: FAD, SAD and LAD
We have distinguished among three distinct etiological pathways for AD. Each pathway has distinctive actors and suggests unique approaches for treatment or cure (Table 1). When the major factor is the genotype, FAD occurs. In such cases, the cause is mutation in specific genes, such as APP718V/M. Remediation would be limited to symptomatic treatment or extremely difficult techniques such as gene therapy. However, while tragic, FAD is rare. If Environment is the primary operator, SAD would occur. In such a case, reversing the unhealthy environmental factor at an early enough stage should halt or at least slow down the disorder. Work associated with the HCY/SAM cycle indicates that, if SAD occurs, it is sufficiently rare to not strongly influence studies based on nutritional remediation. On the other hand, if somatic epitypes are produced and induced in a LEARn fashion to produce LAD, this suggests the use of early dietary and other lifestyle supplementation to act as a prophylactic measure against AD to be the most desirable course. This suggests directing research toward discovering early–life epigenetic changes that associate with AD, determining methods to efficiently detect these changes and early–life remediation methods to eliminate AD before it occurs. In addition, understanding which specific epigenetic pathways are involved in LAD could lead to more effective later–life prophylactic and remediation that would reduce the need for “catch–up” symptom–based therapies.
Table 1.
From LEARn to LEARnING–paths from cause to remedy
| Major Factor | Major Players | Remedy |
|---|---|---|
| Genotype → G (FAD) | Mutation in particular genes such as APP718 V/M | Difficult/non–existent (future gene therapy technique?) |
| Environment → E (SAD) | Head trauma, nutritional imbalance, pestichemicals, metals | Restore healthy environment |
| Somatic Epitype → GSE (LAD) | Epigenetic markers via LEARned and unLEARned promoter sequences | Proper dietary supplements (e.g., folate) during developmental period (LEARnING) |
Acknowledgments
Sources of Support: This work was supported by grants from the Alzheimer’s association and NIH (AG18379 and AG18884) to DKL. The authors wish to thank Dr. Nasser Zawia for his contribution.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aisen PS, Egelko S, Andrews H, Diaz-Arrastia R, Weiner M, DeCarli C, Jagust W, Miller JW, Green R, Bell K, Sano M. A pilot study of vitamins to lower plasma homocysteine levels in Alzheimer disease. Am J Geriatr Psychiatry. 2003;11:246–9. [PubMed] [Google Scholar]
- Aisen PS, Schneider LS, Sano M, Diaz-Arrastia R, van Dyck CH, Weiner MF, Bottiglieri T, Jin S, Stokes KT, Thomas RG, Thal LJ. High-dose B vitamin supplementation and cognitive decline in Alzheimer disease: a randomized controlled trial. JAMA. 2008;300:1774–83. doi: 10.1001/jama.300.15.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer’s Association. 2009 Alzheimer’s disease facts and figures. Alzheimers Dement. 2009;5:234–70. doi: 10.1016/j.jalz.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Ashford JW, Mortimer JA. Non-familial Alzheimer’s disease is mainly due to genetic factors. J Alzheimers Dis. 2002;4:169–77. doi: 10.3233/jad-2002-4307. [DOI] [PubMed] [Google Scholar]
- Bales KR, Du Y, Holtzman D, Cordell B, Paul SM. Neuroinflammation and Alzheimer’s disease: critical roles for cytokine/Abeta-induced glial activation, NF-kappaB, and apolipoprotein E. Neurobiology of Aging. 2000;21:427–432. doi: 10.1016/s0197-4580(00)00143-3. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;2:577–80. doi: 10.1016/s0140-6736(89)90710-1. [DOI] [PubMed] [Google Scholar]
- Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge YW, Lahiri DK, Zawia NH. The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. Journal of Neuroscience. 2005;25:823–9. doi: 10.1523/JNEUROSCI.4335-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergem AL, Engedal K, Kringlen E. The role of heredity in late-onset Alzheimer disease and vascular dementia. A twin study Arch Gen Psychiatry. 1997;54:264–70. doi: 10.1001/archpsyc.1997.01830150090013. [DOI] [PubMed] [Google Scholar]
- Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:2395–402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, Yu W, Rongione MA, Ekstrom TJ, Harris TB, Launer LJ, Eiriksdottir G, Leppert MF, Sapienza C, Gudnason V, Feinberg AP. Intra-individual change over time in DNA methylation with familial clustering. Journal of the American Medical Association. 2008;299:2877–83. doi: 10.1001/jama.299.24.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolin CM, Basha R, Cox D, Zawia NH, Maloney B, Lahiri DK, Cardozo-Pelaez F. Exposure to lead and the developmental origin of oxidative DNA damage in the aging brain. FASEB Journal. 2006;20:788–90. doi: 10.1096/fj.05-5091fje. [DOI] [PubMed] [Google Scholar]
- Borenstein AR, Copenhaver CI, Mortimer JA. Early-life risk factors for Alzheimer disease. Alzheimer Dis Assoc Disord. 2006;20:63–72. doi: 10.1097/01.wad.0000201854.62116.d7. [DOI] [PubMed] [Google Scholar]
- Buehlmeyer K, Doering F, Daniel H, Kindermann B, Schulz T, Michna H. Alteration of gene expression in rat colon mucosa after exercise. Ann Anat. 2008;190:71–80. doi: 10.1016/j.aanat.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Campos AC, Molognoni F, Melo FH, Galdieri LC, Carneiro CR, D’Almeida V, Correa M, Jasiulionis MG. Oxidative stress modulates DNA methylation during melanocyte anchorage blockade associated with malignant transformation. Neoplasia. 2007;9:1111–21. doi: 10.1593/neo.07712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan A, Shea TB. Supplementation with apple juice attenuates presenilin-1 overexpression during dietary and genetically-induced oxidative stress. Journal of Alzhiemers Disease. 2006;10:353–358. doi: 10.3233/jad-2006-10401. [DOI] [PubMed] [Google Scholar]
- Christensen MA, Zhou W, Qing H, Lehman A, Philipsen S, Song W. Transcriptional regulation of BACE1, the beta-amyloid precursor protein beta-secretase, by Sp1. Molecular and Cellular Biology. 2004;24:865–874. doi: 10.1128/MCB.24.2.865-874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Dennis NA, Browndyke JN, Stokes J, Need A, Burke JR, Welsh-Bohmer KA, Cabeza R. Temporal lobe functional activity and connectivity in young adult APOE epsilon4 carriers. Alzheimers Dement. 2009 doi: 10.1016/j.jalz.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler BA, Whittaker MH, Lipsky M, Wang G, Chen XQ. Oxidative stress induced by lead, cadmium and arsenic mixtures: 30-day, 90-day, and 180-day drinking water studies in rats: an overview. Biometals. 2004;17:567–8. doi: 10.1023/b:biom.0000045740.52182.9d. [DOI] [PubMed] [Google Scholar]
- Friedland RP, Fritsch T, Smyth KA, Koss E, Lerner AJ, Chen CH, Petot GJ, Debanne SM. Patients with Alzheimer’s disease have reduced activities in midlife compared with healthy control-group members. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:3440–5. doi: 10.1073/pnas.061002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuso A, Nicolia V, Cavallaro RA, Ricceri L, D’Anselmi F, Coluccia P, Calamandrei G, Scarpa S. B-vitamin deprivation induces hyperhomocysteinemia and brain S-adenosylhomocysteine, depletes brain S-adenosylmethionine, and enhances PS1 and BACE expression and amyloid-beta deposition in mice. Mol Cell Neurosci. 2008;37:731–46. doi: 10.1016/j.mcn.2007.12.018. [DOI] [PubMed] [Google Scholar]
- Fuso A, Nicolia V, Pasqualato A, Fiorenza MT, Cavallaro RA, Scarpa S. Changes in Presenilin 1 gene methylation pattern in diet-induced B vitamin deficiency. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.02.013. [DOI] [PubMed] [Google Scholar]
- Fuso A, Seminara L, Cavallaro RA, D’Anselmi F, Scarpa S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol Cell Neurosci. 2005;28:195–204. doi: 10.1016/j.mcn.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Galic MA, Riazi K, Heida JG, Mouihate A, Fournier NM, Spencer SJ, Kalynchuk LE, Teskey GC, Pittman QJ. Postnatal inflammation increases seizure susceptibility in adult rats. J Neurosci. 2008;28:6904–13. doi: 10.1523/JNEUROSCI.1901-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Beedle AS. Early life events and their consequences for later disease: a life history and evolutionary perspective. Am J Hum Biol. 2007;19:1–19. doi: 10.1002/ajhb.20590. [DOI] [PubMed] [Google Scholar]
- Hall DB, Holmlin RE, Barton JK. Oxidative DNA damage through long-range electron transfer. Nature. 1996;382:731–5. doi: 10.1038/382731a0. [DOI] [PubMed] [Google Scholar]
- Hardy J. A hundred years of Alzheimer’s disease research. Neuron. 2006;52:3–13. doi: 10.1016/j.neuron.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Kivipelto M, Solomon A. Alzheimer’s disease - the ways of prevention. J Nutr Health Aging. 2008;12:89S–94S. doi: 10.1007/BF02982595. [DOI] [PubMed] [Google Scholar]
- Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proceedings of the National Academy of Sciences of the United States of America. 1971;68:820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri DK, Maloney B. Genes are not our destiny: the somatic epitype bridges between the genotype and the phenotype. Nature Review of Neuroscience. 2006;7 doi: 10.1038/nrn2022-c1. [DOI] [Google Scholar]
- Lahiri DK, Maloney B, Basha MR, Ge YW, Zawia NH. How and when environmental agents and dietary factors affect the course of Alzheimer’s disease: the “LEARn” model (Latent Early Associated Regulation) may explain the triggering of AD. Current Alzheimer Research. 2007;4:219–228. doi: 10.2174/156720507780362164. [DOI] [PubMed] [Google Scholar]
- Lahiri DK, Maloney B, Zawia NH. The LEARn model: an epigenetic explanation for idiopathic neurobiological diseases. Molecular Psychiatry. 2009 doi: 10.1038/mp.2009.82. IN PRESS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri DK, Zawia NH, Greig NH, Sambamurti K, Maloney B. Early-life events may trigger biochemical pathways for Alzheimer’s disease: the “LEARn” model. Biogerontology. 2008;9:375–9. doi: 10.1007/s10522-008-9162-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latha KS, Anitha S, Rao KS, Viswamitra MA. Molecular understanding of aluminum-induced topological changes in (CCG)12 triplet repeats: relevance to neurological disorders. Biochim Biophys Acta. 2002;1588:56–64. doi: 10.1016/s0925-4439(02)00133-3. [DOI] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–91. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Luchsinger JA, Tang MX, Miller J, Green R, Mayeux R. Higher folate intake is related to lower risk of Alzheimer’s disease in the elderly. J Nutr Health Aging. 2008;12:648–50. doi: 10.1007/BF03008276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney B, Ge Y-W, Peterson RC, Hardy J, Rogers JT, Pérez–Tur J, Lahiri DK. Functional characterization of three single–nucleotide polymorphisms present in the human APOE promoter sequence. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2009 doi: 10.1002/ajmg.b.30973. IN PRESS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslow K. 2008 Alzheimer’s disease facts and figures. Alzheimers Dement. 2008;4:110–33. doi: 10.1016/j.jalz.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Mastroeni D, Coleman PD, Grover A, Sue L, McKee A, Rogers J. Differential DNA methylation in neurons of identical twins discordant for Alzheimer’s disease. Alzheimer’s & Dementia. 2009;5:P145. [Google Scholar]
- Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic changes in Alzheimer’s disease: Decrements in DNA methylation. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, Jorm AF, Kokmen E, Kondo K, Rocca WA, et al. Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol. 1991;20(Suppl 2):S28–35. doi: 10.1093/ije/20.supplement_2.s28. [DOI] [PubMed] [Google Scholar]
- Myers RH, Schaefer EJ, Wilson PW, D’Agostino R, Ordovas JM, Espino A, Au R, White RF, Knoefel JE, Cobb JL, McNulty KA, Beiser A, Wolf PA. Apolipoprotein E epsilon4 association with dementia in a population-based study: The Framingham study. Neurology. 1996;46:673–7. doi: 10.1212/wnl.46.3.673. [DOI] [PubMed] [Google Scholar]
- Pacheco-Quinto J, Rodriguez de Turco EB, DeRosa S, Howard A, Cruz-Sanchez F, Sambamurti K, Refolo L, Petanceska S, Pappolla MA. Hyperhomocysteinemic Alzheimer’s mouse model of amyloidosis shows increased brain amyloid beta peptide levels. Neurobiol Dis. 2006;22:651–6. doi: 10.1016/j.nbd.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Plagemann A, Harder T, Brunn M, Harder A, Roepke K, Wittrock-Staar M, Ziska T, Schellong K, Rodekamp E, Melchior K, Dudenhausen JW. Hypothalamic POMC promoter methylation becomes altered by early overfeeding: An epigenetic model of obesity and the metabolic syndrome. J Physiol. 2009 doi: 10.1113/jphysiol.2009.176156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley KP, Snowdon DA, Desrosiers MF, Markesbery WR. Early life linguistic ability, late life cognitive function, and neuropathology: findings from the Nun Study. Neurobiol Aging. 2005;26:341–7. doi: 10.1016/j.neurobiolaging.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Sambamurti K, Granholm AC, Kindy MS, Bhat NR, Greig NH, Lahiri DK, Mintzer JE. Cholesterol and Alzheimer’s disease: clinical and experimental models suggest interactions of different genetic, dietary and environmental risk factors. Current Drug Targets. 2004;5:517–28. doi: 10.2174/1389450043345335. [DOI] [PubMed] [Google Scholar]
- Scrable H, Cavenee W, Ghavimi F, Lovell M, Morgan K, Sapienza C. A model for embryonal rhabdomyosarcoma tumorigenesis that involves genome imprinting. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:7480–4. doi: 10.1073/pnas.86.19.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshadri S. Elevated plasma homocysteine levels: risk factor or risk marker for the development of dementia and Alzheimer’s disease? J Alzheimers Dis. 2006;9:393–8. doi: 10.3233/jad-2006-9404. [DOI] [PubMed] [Google Scholar]
- Snowdon DA, Greiner LH, Markesbery WR. Linguistic ability in early life and the neuropathology of Alzheimer’s disease and cerebrovascular disease. Findings from the Nun Study. Ann N Y Acad Sci. 2000;903:34–8. doi: 10.1111/j.1749-6632.2000.tb06347.x. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Toyota M, Kondo Y, Shinomura Y. Inflammation-related aberrant patterns of DNA methylation: detection and role in epigenetic deregulation of cancer cell transcriptome. Methods Mol Biol. 2009;512:55–69. doi: 10.1007/978-1-60327-530-9_5. [DOI] [PubMed] [Google Scholar]
- Szyf M. The dynamic epigenome and its implications in toxicology. Toxicol Sci. 2007;100:7–23. doi: 10.1093/toxsci/kfm177. [DOI] [PubMed] [Google Scholar]
- Takiguchi M, Achanzar WE, Qu W, Li G, Waalkes MP. Effects of cadmium on DNA-(Cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation. Exp Cell Res. 2003;286:355–65. doi: 10.1016/s0014-4827(03)00062-4. [DOI] [PubMed] [Google Scholar]
- Valinluck V, Tsai HH, Rogstad DK, Burdzy A, Bird A, Sowers LC. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2) Nucleic Acids Res. 2004;32:4100–8. doi: 10.1093/nar/gkh739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–54. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Whalley LJ, Dick FD, McNeill G. A life-course approach to the aetiology of late-onset dementias. Lancet Neurol. 2006;5:87–96. doi: 10.1016/S1474-4422(05)70286-6. [DOI] [PubMed] [Google Scholar]
- Winerman L. A second look at twin studies. Monitor on Psychology. 2004;35:46. [Google Scholar]
- Wu J, Basha MR, Brock B, Cox DP, Cardozo-Pelaez F, McPherson CA, Harry J, Rice DC, Maloney B, Chen D, Lahiri DK, Zawia NH. Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD. Journal of Neuroscience. 2008;28:3–9. doi: 10.1523/JNEUROSCI.4405-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CE, Wei W, Liu YH, Peng JH, Tian Q, Liu GP, Zhang Y, Wang JZ. Hyperhomocysteinemia increases beta-amyloid by enhancing expression of gamma-secretase and phosphorylation of amyloid precursor protein in rat brain. Am J Pathol. 2009;174:1481–91. doi: 10.2353/ajpath.2009.081036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Lee HG, Perry G, Smith MA. Alzheimer disease, the two-hit hypothesis: an update. Biochim Biophys Acta. 2007;1772:494–502. doi: 10.1016/j.bbadis.2006.10.014. [DOI] [PubMed] [Google Scholar]