

Abstract
Summary: Unsupervised class discovery is a highly useful technique in cancer research, where intrinsic groups sharing biological characteristics may exist but are unknown. The consensus clustering (CC) method provides quantitative and visual stability evidence for estimating the number of unsupervised classes in a dataset. ConsensusClusterPlus implements the CC method in R and extends it with new functionality and visualizations including item tracking, item-consensus and cluster-consensus plots. These new features provide users with detailed information that enable more specific decisions in unsupervised class discovery.
Availability: ConsensusClusterPlus is open source software, written in R, under GPL-2, and available through the Bioconductor project (http://www.bioconductor.org/).
Contact: mwilkers@med.unc.edu
Supplementary Information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Unsupervised class discovery is a data mining technique for the detection of unknown possible groups of items based on intrinsic features and no external information. For this technique, an investigator seeks to answer two questions: how many groups are present in a dataset, and what is the confidence in the number of groups and the group memberships. Consensus clustering (CC) (Monti et al., 2003) is a method for evaluating these questions and is popular in cancer research [e.g. lung adenocarcinoma (Hayes et al., 2006)]. CC provides quantitative and visual ‘stability’ evidence derived from repeated subsampling and clustering. CC reports a consensus of these repetitions, which is robust relative to sampling variability. The CC method is available in the GenePattern software (Reich et al., 2006). ConsensusClusterPlus implements the CC method in the R language (http://www.r-project.org) and adds new functionality and visualizations.
2 SOFTWARE FEATURES
Input to ConsensusClusterPlus is a data matrix and user-specified options. The data matrix represents a collection of features for a set of samples (items); for example, this could be microarray items and gene expression features. Output is stability evidence for a given number of groups (k) and cluster assignments. The output consists of R data objects, text files, graphical plots and a log file.
2.1 Algorithm
ConsensusClusterPlus extends the CC algorithm and is briefly described here. The algorithm begins by subsampling a proportion of items and a proportion of features from a data matrix. Each subsample is then partitioned into up to k groups by a user-specified clustering algorithm: agglomerative hierarchical clustering, k-means or a custom algorithm. This process is repeated for a specified number of repetitions. Pairwise consensus values, defined as ‘the proportion of clustering runs in which two items are [grouped] together’ (Monti et al., 2003), are calculated and stored in a consensus matrix (CM) for each k. Then for each k, a final agglomerative hierarchical consensus clustering using distance of 1−consensus values is completed and pruned to k groups, which are called consensus clusters.
New features of ConsensusClusterPlus algorithm are the 2D feature and item subsampling, which can be performed according to particular distributions such as gene variability, and the option for a custom clustering algorithm. The 2D subsampling provides assessments of clusters' sensitivity to both item and feature sampling variability. Because a custom clustering algorithm can be used to generate consensus, users can utilize the many existing clustering algorithms available in R or can write their own.
2.2 Output and visualizations
ConsensusClusterPlus produces graphical plots extending the CC visualizations. For each k, CM plots depict consensus values on a white to blue colour scale, are ordered by the consensus clustering which is shown as a dendrogram, and have items' consensus clusters marked by coloured rectangles between the dendrogram and consensus values (Fig. 1A). This new feature of ConsensusClusterPlus enables quick and accurate visualization of cluster boundaries, which are not labelled in CC. The purpose of CM plots is to find the ‘cleanest’ cluster partition where items nearly always either cluster together giving a high consensus (dark blue colour) or do not cluster together giving a low consensus (white). Empirical cumulative distribution function (CDF) plots display consensus distributions for each k (Fig. 1C). The purpose of the CDF plot is to find the k at which the distribution reaches an approximate maximum, which indicates a maximum stability and after which divisions are equivalent to random picks rather than true cluster structure.
Fig. 1.
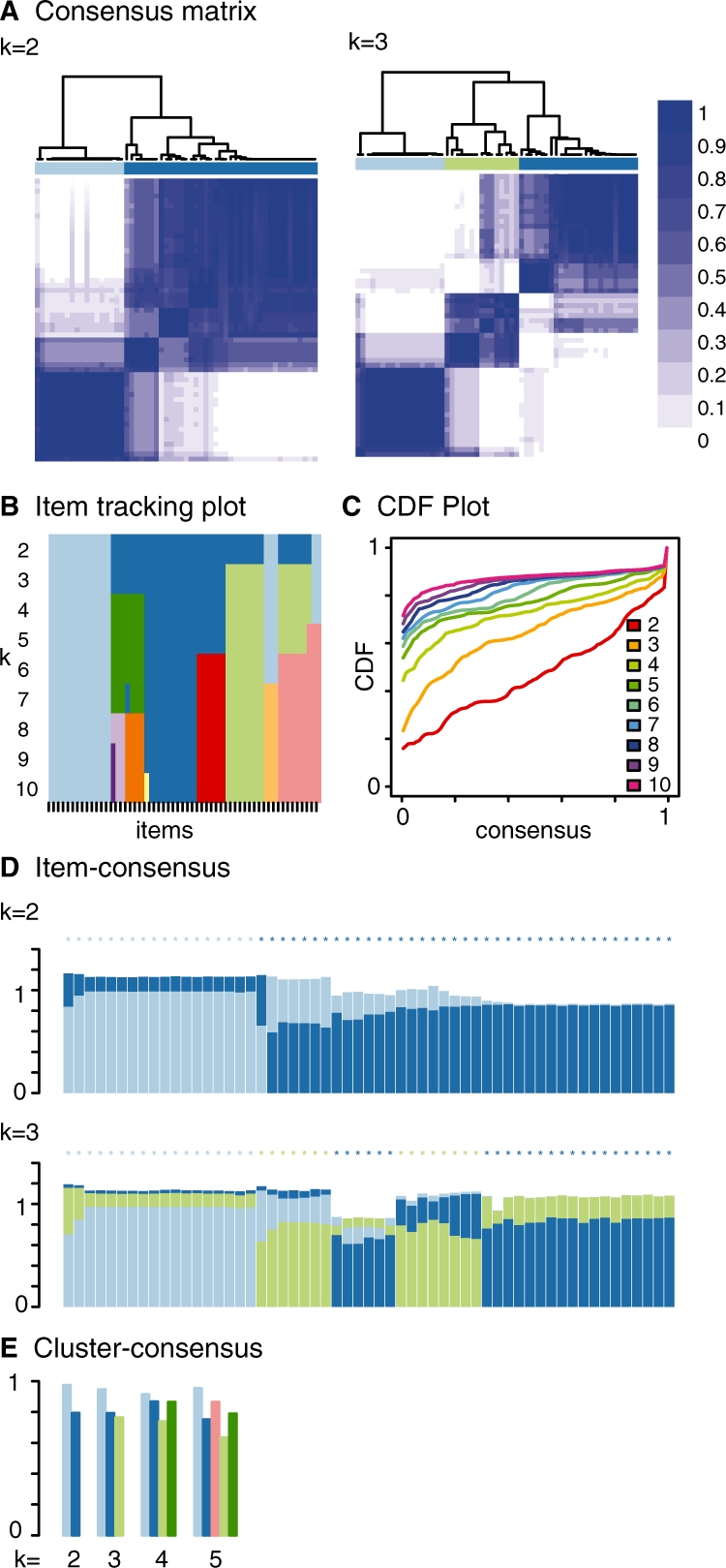
Example application of lung cancer gene expression microarrays. (A) consensus matrix, (B) item tracking plot, (C) CDF plot, (D) item-consensus plot and (E) cluster-consensus plot.
The item tracking plot (Fig. 1B) shows the consensus cluster of items (in columns) at each k (in rows). This allows a user to track an item's cluster assignments across different k, to identify promiscuous items that are suggestive of weak class membership, and to visualize the distribution of cluster sizes across k (Supplementary Fig. 1 for example of promiscuous samples). This plot is similar to colour maps (Hoffmann et al., 2007). Item-consensus (IC) is the average consensus value between an item and members of a consensus cluster, so that there are multiple IC values for an item at a k corresponding to the k clusters. IC plots display items as vertical bars of coloured rectangles whose height corresponds to IC values (Fig. 1D). Consensus clusters of items are marked by coloured asterisks atop the bars. IC plots enable a user to view which samples are highly representative of a cluster and which samples have mixed cluster association and to possibly select cluster-representative samples. Cluster-consensus (CLC) is the average pairwise IC of items in a consensus cluster. The CLC plot displays these values as a bar plot that are grouped at each k (Fig. 1E). The CLC plots enable a user to assess the impact adding a new cluster on the CLC values of existing clusters. The colour schemes between the CM, item tracking, IC and CLC plots are coordinated which enable cross-plot analysis. The colour scheme is defined by the rule that clusters at a k are given the same colour as a cluster at k−1 if the majority of their members are shared. Otherwise, a new colour is assigned.
3 EXAMPLE APPLICATION
For demonstration, we obtained published lung cancer gene expression microarrays (Garber et al., 2001). We selected microarrays of adenocarcinoma, squamous cell carcinoma or normal histologies and sought to rediscover these known classes. We executed ConsensusClusterPlus which resulted in four clusters. These discovered clusters correspond to the pre-selected classes (Supplementary Table 1 and Fig. 2). Two clusters completely contain and segregate squamous cell carcinoma and normal histologies. Adenocarcinoma is spread over the four clusters and is the only histology in two clusters. Adenocarcinoma's expression diversity is consistent with the earlier reports (Garber et al., 2001; Hayes et al., 2006). As an integrity check, we executed GenePattern CC with the same input and found identical cluster assignments.
The item tracking plot showed cluster assignments were stable and that new clusters at k > 4 are small. The IC plot showed that some items with mixed IC (bars with appreciable light blue and dark blue portions) at k = 2 become a new cluster at k = 3 (coloured light green) (Fig. 1D). CLC plots at k = 4 showed reasonably high CLC among the clusters (Fig. 1E). The item tracking, IC and CLC data were useful in deciding cluster number and could be used to select representative samples for further analysis.
4 CONCLUSIONS
ConsensusClusterPlus is open source, Bioconductor-compatible software for unsupervised class discovery. ConsensusClusterPlus extends CC with new, easy-to-use functionality and visualizations that enable detailed analysis.
Funding: National Cancer Institute (NCI) F32CA142039 to M.D.W., Thomas G. Labrecque Foundation through Joan's Legacy Foundation to D.N.H., and National Institutes of Health (NIH) U24CA126554. The content is solely the responsibility of the authors and does not necessarily represent the official views of NCI or NIH.
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Garber ME, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc. Natl Acad. Sci. USA. 2001;98:13784–13789. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes DN, et al. Gene expression profiling reveals reproducible human lung adenocarcinoma subtypes in multiple independent patient cohorts. J. Clin. Oncol. 2006;24:5079–5090. doi: 10.1200/JCO.2005.05.1748. [DOI] [PubMed] [Google Scholar]
- Hoffmann M, et al. Optimized alignment and visualization of clustering results. In: Decker R, Lenz H.-J, editors. Advances in Data Analysis. Berlin Heidelberg: Springer; 2007. pp. 75–82. [Google Scholar]
- Monti S, et al. Consensus clustering: a resampling-based method for class discovery and visualization of gene expression microarray data. Mach. Learn. 2003;52:91–118. [Google Scholar]
- Reich M, et al. GenePattern 2.0. Nat. Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.