

Abstract
Motivation: In silico prediction of drug–target interactions from heterogeneous biological data is critical in the search for drugs and therapeutic targets for known diseases such as cancers. There is therefore a strong incentive to develop new methods capable of detecting these potential drug–target interactions efficiently.
Results: In this article, we investigate the relationship between the chemical space, the pharmacological space and the topology of drug–target interaction networks, and show that drug–target interactions are more correlated with pharmacological effect similarity than with chemical structure similarity. We then develop a new method to predict unknown drug–target interactions from chemical, genomic and pharmacological data on a large scale. The proposed method consists of two steps: (i) prediction of pharmacological effects from chemical structures of given compounds and (ii) inference of unknown drug–target interactions based on the pharmacological effect similarity in the framework of supervised bipartite graph inference. The originality of the proposed method lies in the prediction of potential pharmacological similarity for any drug candidate compounds and in the integration of chemical, genomic and pharmacological data in a unified framework. In the results, we make predictions for four classes of important drug–target interactions involving enzymes, ion channels, GPCRs and nuclear receptors. Our comprehensively predicted drug–target interaction networks enable us to suggest many potential drug–target interactions and to increase research productivity toward genomic drug discovery.
Supplementary information: Datasets and all prediction results are available at http://cbio.ensmp.fr/~yyamanishi/pharmaco/.
Availability: Softwares are available upon request.
Contact: yoshihiro.yamanishi@ensmp.fr
1 INTRODUCTION
The identification of drug–target interactions (interactions between drugs and target proteins) is a key area in genomic drug discovery. Interactions with ligands can modulate the function of many classes of pharmaceutically useful protein targets including enzymes, ion channels, G protein-coupled receptors (GPCRs) and nuclear receptors. Owing to the completion of the human genome sequencing and the development of various biotechnologies, we are beginning to analyze the ‘genomic space’ populated by these protein classes. At the same time, the high-throughput screening (HTS) of large-scale chemical libraries is enabling us to explore the entire ‘chemical space’ of possible compounds. However, our knowledge about the relationship between the chemical space and the genomic space is very limited.
In recent years, the importance of chemical genomics is growing fast to relate the chemical space with the genomic space (Dobson et al., 2004; Kanehisa et al., 2006; Stockwell, 2000). The genome-wide detection of compound–protein interactions is a key issue in chemical genomics research, which can lead to identification of new drug leads and therapeutic targets for known diseases such as cancers. Although various biological assays are becoming available, experimental determination of compound–protein interactions remains challenging and very expensive even nowadays. There is therefore a strong incentive to develop new in silico methods capable of detecting these potential compound–protein interactions efficiently.
Traditional computational approaches are categorized into ligand-based approach and docking approach. Ligand-based approach like QSAR (Quantitative Structure Activity Relationship) compares a candidate ligand with the known ligands of a target protein to predict its binding using machine learning methods (Butina et al., 2002; Byvatov et al., 2003). However, the performance of the ligand-based approach is poor when the number of known ligands for a target protein of interest decreases. The docking is a powerful approach, but the docking cannot be applied to proteins whose 3D structures are unknown (Rarey et al., 1996). This limitation is serious for membrane proteins. For example, there are only two GPCRs with 3D structure information as of writing. Therefore it is difficult to use the docking on a genome-wide scale.
Recently, a variety of statistical methods have been developed to predict compound–protein interactions on a genome-wide scale, following the spirit of chemical genomics. The underlying idea is that similar ligands are likely to interact with similar proteins, and the prediction is performed based on compound chemical structures, protein sequences and the currently known compound–protein interactions. A straightforward statistical approach is to use binary classification methods where they take compound–protein pairs as an input for machine learning classifiers such as neural network and support vector machine (SVM) (Bleakley and Yamanishi, 2009; Bock and Gough, 2005; Erhan et al., 2006; Faulon et al., 2008; Jacob and Vert, 2008; Nagamine and Sakakibara, 2007). The other statistical approach is the distance learning in the framework of supervised bipartite graph inference (Yamanishi, 2009; Yamanishi et al., 2008).
Another promising approach is to use pharmacological information. The use of side-effect similarity has been recently proposed, which is based on the assumption that drugs with similar side-effects are likely to interact with similar target proteins (Campillos et al., 2008). However, the method requires drug package inserts that describe the detailed side-effect information, so it is applicable only to marketed drugs for which side-effect information is available. Therefore, it is not possible to infer potential interactions between new drug candidate compounds and target proteins.
In this article, we investigate the relationship between the chemical space, the pharmacological space and the topology of drug–target interactions networks. We then develop a new method to predict unknown drug–target interactions from chemical structure information, genomic sequence information and pharmacological effect information on a large scale. The proposed method consists of two steps: (i) prediction of pharmacological effects from chemical structures of given compounds and (ii) inference of unknown drug–target interactions based on the pharmacological effect similarity in the framework of supervised bipartite graph inference. The algorithm proposed in the first step enables us to obtain pharmacological information about not only marketed drugs but also any compounds, based on the correlation between chemical structures and pharmacological/adverse effects (Scheiber et al., 2009), which makes it possible to perform screening of any drug candidate compounds against many target candidate proteins. To our knowledge, there are no methods which predict drug–target interactions based on chemical, genomic and pharmacological data simultaneously. In the results, we make predictions for four classes of important drug–target interactions involving enzymes, ion channels, GPCRs and nuclear receptors. A comprehensive prediction of drug–target interaction networks enables us to suggest new potential drug–target interactions.
2 MATERIALS
In this study, we focus on drugs targeting four pharmaceutically useful target classes: enzymes, ion channels, GPCRs and nuclear receptors.
2.1 Chemical data
Chemical structures of drugs and other compounds were obtained from the KEGG DRUG and KEGG LIGAND databases (Kanehisa et al., 2008). We computed the chemical structure similarities between compounds using SIMCOMP (Hattori et al., 2003), a program that finds the common substructures between two compounds and outputs the global similarity score based on a graph alignment algorithm. The similarity between two compound structures x and x′ is evaluated by Tanimoto coefficient defined as schem(x, x′)=|x∩x′|/|x∪x′|. The similarity score is referred to as ‘chemical structure similarity’ in this study. Applying this operation to all compound pairs, we construct a similarity matrix denoted as C. The similarity matrix C is considered to represent ‘chemical space’.
2.2 Pharmacological data
Pharmacological effect keywords for drugs (pharmaceutical molecules) were obtained from the JAPIC (Japan Pharmaceutical Information Center) database (http://www.japic.or.jp/). JAPIC manages all package insert information of pharmaceutical products in Japan, under the approval of Health and Welfare Minister of Japan. We used the JAPIC entries (package inserts) of ethical drugs described in natural Japanese language, which were morphologically analyzed to obtain the nouns or phrases using the MeCab program (http://mecab.sourceforge.net/). The resulted set of keywords were translated into English followed by the unification of synonymous words, using life science dictionary (http://lsd.pharm.kyoto-u.ac.jp/en/index.html). Since a pharmaceutical molecule is usually involved in various commercial products, each KEGG DRUG entry of a drug molecule is represented as a logical sum of the presence/absence (1 or 0, respectively) of the unified keywords found in the corresponding JAPIC entries. We obtained 18 653 keywords in total, representing the pharmaceutical effects, adverse effects, caution, usage, properties, etc.
We also performed a simple investigation of the context of the keywords. JAPIC entries are described in an XML format, where the sentences are tagged by the category words such as ‘effect’, ‘side-effect’, ‘caution’ and ‘warning’. Unnecessary information in terms of analyzing pharmacological effects, such as manufacturers, are removed using the corresponding XML tag. Various types of profiles can be generated for a drug using different set of the XML tags. We tested using every tag independently to generate a profile, although we found it ineffective. Then we tested grouping the similar XML tags to form five tag groups: ‘caution’ (unwanted characteristics of the drug, such as adverse event, caution for application or handling, overdose and warning), ‘interaction’ (the combined use of drugs), ‘patient’ (the types of patients, such as children, pregnant, elder people, or the people having chronic diseases), ‘pharmaceutical effect’ (efficacy, usage and pharmacology) and ‘property’ (such as partition coefficient, pharmacokinetics, melting point and solubility). The number of keywords with the ‘caution’, ‘interaction’, ‘patient’, ‘pharmaceutical effect’ and ‘property’ tags are 16 849, 14 223, 16 362, 17 109, and 17 142, respectively. All the keywords for each tag are put on the supplementary website. In this study, we used keywords with the ‘pharmaceutical effect’ tag as pharmacological keywords.
Each drug is represented by a profile (binary vector) y=(y1, y2,…, yK)⊤ in which a pharmacological keyword is coded 1 or 0, respectively, across the 17 109 keywords. The similarity between two drugs y and y′ is evaluated by the weighted cosine correlation coefficient between the above profiles as follows:
| (1) |
where wk is the weight function for the k-th keyword defined as
where dk is the frequency of the k-th keyword in the data, and K is the total number of keywords in the data, σ is the SD of {dk}k=1K, and h is a parameter (set to 0.1 in this study). The weight function is introduced to put more emphasis on infrequent keywords rather than frequent keywords across different drug package inserts, because rare keywords (e.g. ‘cytopenia’, ‘pancytopenia’, ‘photosensitivity’, ‘teratogenic’) are more informative than common keywords (e.g. ‘disease’, ‘receptor’, ‘stability’, ‘biological’) in terms of characteristics of drugs.
The similarity score is referred to as ‘pharmacological effect similarity’ or ‘pharmacological similarity’ in this study. Applying this operation to all drug pairs, we construct a similarity matrix denoted as P. The similarity matrix P is considered to represent ‘pharmacological space’.
2.3 Genomic data
Amino acid sequences of proteins coded in the human genome were obtained from the KEGG GENES database (Kanehisa et al., 2008). We computed the sequence similarities between two proteins z and z′ using a normalized version of Smith–Waterman scores (Smith and Waterman, 1981). The similarity score is denoted as sgeno(z, z′) and referred to as ‘genomic sequence similarity’ in this study. Applying this operation to all protein pairs, we construct a similarity matrix denoted as G. In this study the similarity matrix G is considered to represent ‘genomic space’.
2.4 Drug–target interaction data
The information about the interactions between drugs and target proteins was obtained from the KEGG BRITE (Kanehisa et al., 2008), BRENDA (Schomburg et al., 2004), SuperTarget (Gunther et al., 2008) and DrugBank (Wishart et al., 2008) databases. According to our survey, the numbers of known drugs with pharmacological information in JAPIC are 212, 99, 105 and 27, for their targets enzymes, ion channels, GPCRs and nuclear receptors, respectively. The numbers of the corresponding target proteins in these classes are 664, 204, 95 and 26, respectively. The numbers of the corresponding interactions are 1515, 776, 314 and 44, respectively.
The set of known drug–target interactions is regarded as the ‘gold standard’ data in this study, and is used for evaluating the performance of the proposed method in the cross-validation experiments as well as training data in the comprehensive prediction.
3 METHODS
Suppose that we are given drug candidate compounds and we want to predict unknown interactions between the compounds and target proteins on a genome-wide scale. The proposed method consists of two steps: (i) prediction of potential pharmacological effects from chemical structures of given compounds and (ii) inference of unknown drug–target interactions based on the pharmacological effect similarity in the framework of supervised bipartite graph inference. The details of each step of the proposed method are described below.
3.1 Prediction of pharmacological effects from compound chemical structures
If pharmacological information is available for given compounds, this process can be skipped. In this subsection, we assume that given compounds do not have any pharmacological information.
3.1.1 Formulation of the problem
Let us now consider the situation where chemical structure data is available for all the N compounds {xi}i=1N, while the pharmacological data is available for the first n compounds {yi}i=1n and unavailable for the remaining (N − n) compounds {yi}i=n+1N. We refer to the first n compounds as training set, and we refer to the remaining N − n compounds as prediction set below.
For the prediction set, we want to predict a pharmacological profile y (K-dimensional binary vector) from a chemical structure x (chemical graph). A straightforward approach would be to apply a binary classification method such as SVM in order to individually predict whether each element yk in y is 1 or 0. However, this strategy needs to construct K individual classifiers for K pharmacological keywords, which will require prohibitive computational burden, because K is quite huge in practical applications (K is 17 109 in this study).
Note that the inputs of the supervised bipartite graph inference method in the next step are similarity scores for compounds and proteins. Therefore, we propose to consider predicting the pharmacological similarity scores involving compounds rather than predicting the pharmacological profile itself directly. The key idea here is to reformulate the problem of predicting unknown high-dimensional binary vectors for the prediction set by the problem of predicting unknown similarity scores sphar(yi, yj) involving the prediction set.
Let schem(·, ·) and sphar(·, ·) be chemical structure and pharmacological effect similarity functions, respectively. When we compute the chemical structure similarity scores for {xi}i=1N, we obtain an N × N similarity matrix C, where (C)ij=schem(xi, xj) (1≤i, j≤N). On the other hand, when we compute the pharmacological similarity scores for {yi}i=1n, we obtain an N × N similarity matrix P, where (P)ij=sphar(yi, yj) (1≤i, j≤n) (n<N). Note that P contains in fact missing values for all entries (P)ij with max(i, j)>n. We want to estimate the missing part of P using full similarity matrix C, taking into account a form of correlation between the two similarity functions.
In this study, we express each similarity matrix by splitting the matrix into four parts. We denote by Ctt (resp. Ptt) the n × n similarity matrix for the training set versus itself, Cpt (resp. Ppt) the (N − n) × n similarity matrix for the prediction set versus the training set and Cpp (resp. Ppp) the (N − n)×(N − n) similarity matrix for the prediction set versus itself:
| (2) |
Note that Cpt and Cpp are known, while Ppt and Ppp are unknown. The goal is to predict Ppt and Ppp from C and Ptt.
3.1.2 Algorithm
The ordinary regression model between an explanatory variable x and a response variable y can be formulated as y=f(x)+ϵ, where f is a regression function and ϵ is a noise term. By analogy we propose to regard (x, x′) as an explanatory variable and sphar(y, y′) as a response variable in our context.
Assuming the underlying feature space in which each x can be represented by m features u(1)(x), u(2)(x),…, u(m)(x), we formulate a variant of the regression model as follows:
| (3) |
where u(x)=(u(1)(x), u(2)(x),…, u(m)(x))⊤. We refer to this model as similarity matrix regression model.
We consider features that possess an expansion of the form
| (4) |
where β=(β1, β2,…, βn)⊤ is a weight vector and n is the number of compounds in the training set.
In order to represent the set of features for all the compounds, we define feature score matrices Ut(x)=[u(x1), u(x2),…, u(xn)]⊤ for the training set and Up(x)=[u(xn+1), u(xn+2),…, u(xN)]⊤ for the prediction set. In the matrix form, we can actually compute the feature score matrices as Ut=CttB for the training set and Up=CptB for the prediction set, where B=[β(1), β(2),…, β(m)].
We consider predicting sphar(y, y′) by the inner products of the feature vectors of x and x′ based on the regression model (3). Since all the compound–compound similarities in the feature space can be represented as ŝphar(yi, yj)=u(xi)⊤ u(xj) for 1≤i,j≤N, the missing entries in P are to be estimated as
Here, we want to find the n × m weight matrix B such that 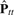 fits Ptt as much as possible. If we set A = BB⊤, this problem can be replaced by finding A which minimizes the difference between Ptt and
fits Ptt as much as possible. If we set A = BB⊤, this problem can be replaced by finding A which minimizes the difference between Ptt and 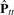 . It means that, this enables us to avoid considerable computational burden for computing B itself, even if m is infinite. Therefore, we attempt to find A (=BB⊤) which minimizes
. It means that, this enables us to avoid considerable computational burden for computing B itself, even if m is infinite. Therefore, we attempt to find A (=BB⊤) which minimizes
| (5) |
where ∥ · ∥F indicates the Frobenius norm. We can rewrite the above equation in the trace form as
| (6) |
From setting 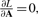 , the solution is analytically obtained by
, the solution is analytically obtained by
| (7) |
Therefore, we can compute the feature-based similarity matrix 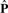 involving the prediction set as follows:
involving the prediction set as follows:
| (8) |
3.2 Inference of drug–target interactions
We perform the inference of potential drug–target interactions based on pharmacological information about compounds and genomic information about proteins in the framework of supervised bipartite graph inference. Among several algorithms for the supervised bipartite graph inference mentioned in the introduction section, we use an algorithm based on distance learning (Yamanishi et al., 2008), because this method is known to work the best in terms of prediction accuracy and computational efficiency (Lodhi and Yamanishi, 2010).
The procedure of the method for drug–target interaction prediction in this context is briefly explained as follows:
Embed compounds and proteins on the known interaction network into a unified feature space, where interacting compounds and proteins are close to each other.
Learn a correlation model between the pharmacological space and the unified feature space with respect to compounds, and learn a correlation model between the genomic space and the unified feature space with respect to proteins.
Map any compounds onto the unified feature space based on the pharmacological similarities, and map any proteins onto the unified feature space based on the genomic sequence similarities.
Predict potential compound–protein interactions by connecting compounds and proteins which are closer than a threshold in the unified feature space, following the spirit of the nearest neighbor.
The details of each step can be found in the original article.
The resulting prediction score for any new compound y and protein z in the fourth process is formulated as
| (9) |
where ny (resp. nz) is the number of compounds (resp. proteins) in the training set, sphar(·, ·) is pharmacological similarity function for compounds, sgeno(·, ·) is genomic sequence similarity function for proteins and αij(i=1,…, ny, j = 1,…, nz) are the parameters learned. If g(y, z) is higher than a threshold, compound y and protein z are predicted to interact to each other.
The pharmacological similarity for compounds and the genomic sequence similarity for proteins are used as inputs of the bipartite graph inference method. The use of sphar(·, ·) is a unique feature of this study, while the use of schem(·, ·) corresponds to the previous study (Yamanishi et al., 2008). Note that the method is also able to infer potential interactions involving new target candidate proteins as well as new drug candidate compounds, but we focus on predicting potential interactions involving new drug candidate compounds, because the objective of this article is to investigate the effect of introducing pharmacological information about new drug candidate compounds.
4 RESULTS
4.1 Relationship between chemical and pharmacological spaces with respect to drug targets
We investigated the relationship between the chemical space and the pharmacological space about the same drugs. Each panel in Figure 1 shows the scatter-plot of pharmacological effect similarity scores against chemical structure similarity scores for drugs targeting enzymes, ion channels, GPCRs and nuclear receptors, respectively. The Pearson's correlation coefficients are 0. 321, 0.420, 0.344 and 0.391, respectively (the corresponding P-value is almost zero in each case).
Fig. 1.

Scatter-plots of pharmacological effect similarity scores and chemical structure similarity scores for drugs targeting enzyme, ion channel, GPCR and nuclear receptor, respectively.
It seems that chemical structure similarities are correlated with pharmacological effect similarities to some extent. However, there are many exceptions. For example, there exist many drug pairs which share high structure similarity but do not have similar pharmacological effects. These results suggest that chemical structures similarity does not always correspond to pharmacological effect similarity.
We investigated the relationship between the chemical space, the pharmacological space and the topology of drug–target interactions networks. We constructed the drug–target interaction network for each protein class using a bipartite graph representation (Yildirim et al., 2007). In the bipartite graph, the heterogeneous nodes correspond to either drugs or target proteins, and edges correspond to interactions between them. The edge is placed between a drug node and a target node if the protein is a known target of the drug.
Figure 2 shows the distributions of chemical structure similarity scores and pharmacological effect similarity scores against the network distance for drugs targeting enzymes, ion channels, GPCRs and nuclear receptors. The top four panels in Figure 2 show the box-plots of drug–drug chemical structure similarities, and the bottom four panels in Figure 2 show the box-plots of drug–drug pharmacological similarities. The network distance means the shortest path between drugs on the bipartite graph representation of each drug–target interaction network. From the figure, we observe several tendencies.
Fig. 2.

Distributions of chemical structure similarity scores (top four panels) and pharmacological effect similarity scores (bottom four panels) against the network distance of drugs targeting enzymes, ion channels, GPCRs and nuclear receptors.
Firstly, the larger the network distance between drugs, the smaller the variability of chemical structure similarities and pharmacological similarities, respectively. Also, the larger the network distance between drugs, the lower the scores of the chemical structure similarities and the drug pharmacological similarities, respectively. These observations suggest that two drugs sharing high chemical structure similarity or high pharmacological similarity tend to interact with similar target proteins.
Secondly, the above tendency is much clearer in the pharmacological similarity than in the chemical structural similarity. It seems that most pharmacological similarity scores are almost zero at larger distances, while many chemical similarity scores are relatively high even at larger distances. The difference of the distributions between ‘distance 2’ and ‘distance 4’ is important, because ‘distance 2’ corresponds to drug-drug pairs which share the same target proteins, while ‘distance 4’ corresponds to drug–drug pairs which do not share the same target proteins. These observations suggest that pharmacological similarity is more correlated with drug targets than with chemical structure similarity, and the pharmacological similarity information is a more useful source for drug–target identification.
4.2 Performance evaluation of the proposed method
We tested the three different inputs: (i) chemical structure similarity, (ii) true pharmacological similarity, and (iii) predicted pharmacological similarity on their abilities to reconstruct four classes of drug–target interactions involving enzymes, ion channels, GPCRs and nuclear receptors. Note that input (i) corresponds to the previous method (Yamanishi et al., 2008), and input (ii) and input (iii) correspond to the proposed method in this study. Input (ii) reflects the situation where all compounds in the prediction set have pharmacological information, so we can skip the process of pharmacological effect prediction. Input (iii) reflects the situation where all compounds in the prediction set do not have any pharmacological information.
We performed the following 5-fold cross-validation procedure: drugs in the gold standard set were split into five subsets of roughly equal size, each subset was then taken in turn as a test set, and we performed the training on the remaining four sets. To obtain robust results and accurate comparison, we kept the same experimental conditions, where the same training drugs and test drugs are used across the three different inputs in each cross-validation. We repeated the above cross-validation experiment five times.
Table 1 shows the averages of the AUC [area under the receiver operating curve (ROC)], sensitivity, specificity and PPV (positive predictive value). The ROC (Gribskov and Robinson, 1996) is the plot of true positives as a function of false positives based on various thresholds, where true positives are correctly predicted interactions and false positives are predicted interactions that are not present in the gold standard interactions. The upper one percentile in the prediction score is chosen as a threshold for computing sensitivity, specificity and PPV, because high-confidence prediction results are interesting in practical applications.
Table 1.
Statistics of the prediction performance
| Class | Statistics |
Input |
||
|---|---|---|---|---|
| Chemical | True | Predicted | ||
| structure | pharmacological | pharmacological | ||
| similarity | similarity | similarity | ||
| Enzyme | AUC | 0.821 | 0.892 | 0.845 |
| Sensitivity | 0.239 | 0.356 | 0.245 | |
| Specificity | 0.993 | 0.995 | 0.993 | |
| PPV | 0.358 | 0.527 | 0.369 | |
| Ion | AUC | 0.692 | 0.812 | 0.731 |
| channel | Sensitivity | 0.134 | 0.137 | 0.142 |
| Specificity | 0.996 | 0.996 | 0.997 | |
| PPV | 0.704 | 0.714 | 0.742 | |
| GPCR | AUC | 0.811 | 0.827 | 0.812 |
| Sensitivity | 0.147 | 0.172 | 0.164 | |
| Specificity | 0.994 | 0.996 | 0.995 | |
| PPV | 0.519 | 0.614 | 0.581 | |
| Nuclear | AUC | 0.814 | 0.835 | 0.830 |
| receptor | Sensitivity | 0.067 | 0.057 | 0.077 |
| Specificity | 0.995 | 0.994 | 0.996 | |
| PPV | 0.560 | 0.480 | 0.640 | |
The AUC (ROC score) is the area under the ROC, normalized to 1 for a perfect inference and 0.5 for a random inference. The sensitivity is defined as TP/(TP+FN), the specificity is defined as TN/(TN+FP) and the PPV (positive predictive value) is defined as TP/(TP+FP), where TP, FP, TN, FN are the number of true positives, false positives, true negatives and false negatives, respectively.
It seems that the true pharmacological similarity-based method outperforms the chemical structure similarity-based method in all the four protein classes. Especially, the use of pharmacological information is effective in the case of enzyme and ion channel data. It seems that the predicted pharmacological similarity-based method also outperforms the chemical similarity-based method, but the performance is a little worse than that of the true pharmacological similarity-based method. In practical applications, it is rare to obtain the detailed pharmacological information about all compounds to be tested, so the result suggests that the predicted pharmacological information is useful for identification of unknown drug–target interactions even when pharmacological information is not available for compounds of interest. These results serve to highlight the significant performance of the proposed method.
We also made a simple check of the effectiveness of grouping the keywords into the five tag groups. Figure 3 shows the AUC scores of the predicted pharmacological similarity-based method for the five tag groups (caution, interaction, patient, pharmaceutical effect and property) and the combination of the five groups, respectively, where ‘caut’, ‘inte’, ‘pati’, ‘phar’, ‘prop’ and ‘comb’ indicate the five tag groups and the combination, respectively. The low predictive performances of the inte profile is that the number of drugs having the inte keywords is much fewer than those of other types of keywords. It is notable that the remaining four types of keywords (caut, pati, phar and prop) outperformed the comb profiles, indicating the usefulness of discriminating the context of the keywords. It is natural, for example, that the drugs for high blood pressure and the drugs that cause high blood pressure have to be distinguished. These results suggest that appropriate selection of informative keywords and discriminating context will improve the predictive performance.
Fig. 3.

Barplot of AUC score for the five tag groups (caution, interaction, patient, pharmaceutical effect and property) and their combination.
4.3 Comprehensive prediction for unknown drug–target interactions
After confirming the usefulness of our method, we conducted a comprehensive prediction of interactions between all possible compounds and proteins for the four classes of target proteins studied: enzymes, ion channels, GPCRs and nuclear receptors. In the inference process for these predictions, we used all the known drugs and target proteins in the gold standard data as training data, and predicted potential interactions for all compounds in KEGG LIGAND and all the other drugs in KEGG DRUG (the drugs are absent from the gold standard data). Note that there remain many marketed drugs whose target proteins have not been identified yet. The total number of compounds including drugs in the prediction set is 15 383 in each case. Note that most of the compounds and drugs in the prediction set are not assigned any pharmacological information, so the pharmacological effect prediction is required. All the prediction results for each target protein class can be obtained from the web-supplement. Because of space limitations, we focused on the results for enzyme data below.
We focused on the top 1000 scoring predictions for the enzyme data. We investigated the validity of the predicted pairs based on the databases (e.g. KEGG BRITE, SuperTarget, DrugBank), because they contain information about interactions involving compounds which do not have any pharmacological information. Recall that in the Section 2 we constructed the gold standard set for drug–target interactions involving drugs for which the pharmacological information (by JAPIC package inserts) is available. As a result, we confirmed that 223 out of the top 1000 predictions are now annotated in at least one database. On the other hand, in the case of comprehensive prediction based on chemical structure information only, we confirmed that 140 out of the top 1000 predictions are now annotated in at least one database. We take this result as strong evidence supporting the practical relevance of our approach. Table 2 shows 10 examples of high scoring compound–protein pairs which were not predicted by chemical structure similarity but predicted by pharmacological similarity.
Table 2.
Examples of compound–protein pairs predicted by the proposed method for enzyme data
| Pair | Annotation | |
|---|---|---|
| 1 | C04000 | Benzyl 2-methyl-3-oxobutanoate |
| 5743 | prostaglandin-endoperoxide synthase 2 | |
| 2 | C04000 | Benzyl 2-methyl-3-oxobutanoate |
| 5742 | prostaglandin-endoperoxide synthase 1 | |
| 3 | D05868 | Sodium phenylbutyrate (USAN) |
| 5742 | prostaglandin-endoperoxide synthase 1 | |
| 4 | C07773 | Ambenonium |
| 43 | acetylcholinesterase (Yt blood group) | |
| 5 | D05619 | Prodolic acid (USAN) |
| 5742 | prostaglandin-endoperoxide synthase 1 | |
| 6 | D05868 | Sodium phenylbutyrate (USAN) |
| 5743 | prostaglandin-endoperoxide synthase 2 | |
| 7 | D02587 | Metildigoxin (JP15) |
| 476 | ATPase, Na+/K+ transporting, alpha 1 polypeptide | |
| 8 | C02505 | 2-Phenylacetamide |
| 5743 | prostaglandin-endoperoxide synthase 2 | |
| 9 | C15513 | Benzyl acetate |
| 5743 | prostaglandin-endoperoxide synthase 2 | |
| 10 | C02505 | 2-Phenylacetamide |
| 5742 | prostaglandin-endoperoxide synthase 1 | |
Because of space limitation, all the prediction pairs are put on the supplemental website.
Next, we manually investigated the validity of the predicted pairs which were not confirmed in the databases, based on the literatures. We take some analgesic and antipyretic agents as examples, as shown in Figure 4. Salicylamide (D01811) and acetaminophen (D00217) are both known to act on prostaglandin-endoperoxide synthase 1/2 (PTGS1/2) (Aronoff et al., 2003). Based on these known interactions, some compounds are suggested to interact with PTGS1/2: ethenzamide (D01466), actarit (D01395), N-acetylphenylethylamine (C06746) and N-ethylphenylacetamide (C11487). Among these, D01466 is also an analgesic and antipyretic agent (Darias et al., 2006), although we could not find the target in the databases we used. On the other hand, D01395 is an anti-rheumatic agent (Ye et al., 2008). The JAPIC entry including D01811 describes that this drug also has an effect on rheumatism (Frankl , 1953). We could not find any information about the pharmaceutical effects for other two compounds (C06746 and C11487), but they are structurally similar with the other drugs (D01811, D00217, D01466 and D01395). Therefore, it seems possible that these compounds act on PTGS1/2.
Fig. 4.

Examples of the proposed drug–target interactions. Four boxes in the center of the figure are the target proteins, and bold lines indicate the known drug–target interactions. Solid lines represent the proposed interactions based on the resemblance to the known interacting drugs indicated by the dashed lines. Black stars indicate the interactions predicted by the previous method. White stars indicate the interactions additionally predicted by the proposed method.
On the other hand, PTGS1 has some other interacting analgesic and antipyretic drugs, such as mofezolac (D01718) (Goto et al., 1998), from which tangeretin (C10190) (Hirano et al., 1995) is suggested as another potential drug. The structural commonality between these two compounds seems only that they both contain some O-methyl groups on aromatic rings, therefore this result might not be convincing. As the other questionable example, sodium lactate (D02183) is suggested to act on PTGS2 based on the known interacting drug sodium salicylate (D00566), an analgesic agent (Preston et al., 1989). However, this result seems not convincing at all, because their common substructures are only sodium ion and carboxylate group, and D02183 is an electrolyte replenisher.
The other group of analgesic and antipyretic drugs may possibly share a different target protein. Fluocinolone acetonide (D01825) (Emerit et al., 1983) and fluocinonide (D00325) (Schlessinger et al., 2006) are known to act on human cytosolic calcium-dependent phospholipase A2 (PLA2G4A), which is involved in lipid metabolism and related to various signal transductions (Balsinde et al., 1999). From resemblance to these two drugs, triamcinolone acetonide (D00983) (Keele, 1969) and diflorasone diacetate (D01327) (Bluefarb et al., 1976) are suggested to act on PLA2G4A. These four drugs are all corticosteroids, and are all known to act as analgesic and antipyretic drugs. Therefore we assume these results are convincing.
There are other possible drug–target interactions that belong to different therapeutic categories. For example, Metildigoxin (D02587) is predicted to have an interaction with a human Na+/K+ transporting ATPase (ATP1A1), based on the reported interaction of digoxin (D00298). D00298 is a digitalis-like cardiotonic substance that acts directly on heart muscle (Cumberbatch et al., 1981). D02587 is the methylated derivative of D00298, and many reports suggest that D02587 has no significant difference from D00298 in terms of their effects on heart functions (Kaufmann et al., 1981). Therefore, there is no wonder the two compounds share the same target protein.
5 DISCUSSION AND CONCLUSION
In this article, we investigated the relationship between the chemical space, the pharmacological space and the topology of drug–target interaction networks, and showed that drug–target interactions are more correlated with pharmacological effect similarity than with chemical structure similarity. We then developed a new statistical method to predict unknown drug–target interactions from chemical structure information, genomic sequence information and pharmacological effect information simultaneously on a large scale. The originality of the proposed method lies in prediction of pharmacological effects from chemical structures of given compounds, and its use for identification of unknown drug–target interactions in the framework of supervised bipartite graph inference. To our knowledge, this is the first report to predict drug–target interactions from the integration of chemical, genomic and pharmacological spaces in a unified framework.
One previous research related with this study is the use of side-effect similarity for drug–target identification (Campillos et al., 2008). However, the method is applicable only to marketed drugs for which detailed side-effect information is available. Therefore, newly detectable interactions were limited to the linkage between known marketed drugs assigned with side-effect information and known target proteins. To overcome these problems, we established a procedure to obtain pharmacological information about not only marketed drugs but also any drugs or any drug candidate compounds based on their chemical structures. The proposed procedure makes it possible to perform screening of any chemical compounds against many target candidate proteins.
In practice, there are four possible classes for predictable compound–protein pairs: (i) new drug candidate compounds versus known target proteins, (ii) known drugs versus new target candidate proteins, (iii) new drug candidate compounds versus new target candidate proteins, and (iv) known drugs versus known target proteins, where compounds and proteins with interaction partner information are called ‘known’, otherwise called ‘new’. Note that in this study we focus on class (i), because the objective of this article is to investigate the effect of introducing pharmacological information about new drug candidate compounds. Recently, the bipartite local model approach has been proposed to detect missing interactions between known drugs and known target proteins based on chemical and genomic data (Bleakley and Yamanishi, 2009). The approach works similarly with other bipartite graph inference methods for classes (i) and (ii) in terms of accuracy, but the approach with an aggregation scheme is quite powerful for class (iv) (Bleakley and Yamanishi, 2009), so the approach with pharmacological information could detect missing interactions in class (iv) with high accuracy.
From a technical viewpoint, the performance of our method could be improved by using more sophisticated similarity functions for compounds and proteins, such as kernel functions designed for genomic sequences and chemical structures (Schölkopf et al., 2004). In this study, we evaluated the drug pharmacological similarity based on all available pharmacological keywords categorized into each tag in the package insert of each drug. There remain many unimportant keywords to be filtered and there might exist some correlation between related keywords or hierarchy among medical vocabulary. To deal with these problems, the use of sophisticated text mining approaches is an important research direction.
The proposed method is expected to be useful for virtual screening of chemical libraries. To detect new biological findings and find potentially useful drug leads, we are currently working with collaborators on biological assays. We believe that our method is able to increase research productivity toward genomic drug discovery.
ACKNOWLEDGEMENTS
Computational resources were provided by the Bioinformatics Center, Institute for Chemical Research and the Super Computer Laboratory, Kyoto University.
Funding: Ministry of Education, Culture, Sports, Science and Technology of Japan, the Japan Science and Technology Agency; Japan Society for the Promotion of Science; the bi-national JSPS/INSERM grant Japan-France Research Cooperative Program.
Conflict of Interest: none declared.
REFERENCES
- Aronoff DM, et al. Inhibition of prostaglandin h2 synthases by salicylate is dependent on the oxidative state of the enzymes. J. Pharamacol. Exp. Thera. 2003;304:589–595. doi: 10.1124/jpet.102.042853. [DOI] [PubMed] [Google Scholar]
- Balsinde J, et al. Regulation and inhibition of phospholipase. Annu. Rev. Pharmacol. Toxicol. 1999;39:175–189. doi: 10.1146/annurev.pharmtox.39.1.175. [DOI] [PubMed] [Google Scholar]
- Bleakley K, Yamanishi Y. Supervised prediction of drug-target interactions using bipartite local models. Bioinformatics. 2009;25:2397–2403. doi: 10.1093/bioinformatics/btp433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluefarb SM, et al. Diflorasone diacetate: vasoconstrictor activity and clinical efficacy of a new topical corticosteroid. J. Int. Med. Res. 1976;4:454–461. doi: 10.1177/030006057600400613. [DOI] [PubMed] [Google Scholar]
- Bock JR, Gough DA. Virtual screen for ligands of orphan g protein-coupled receptors. J. Chem. Inf. Model. 2005;45:1402–1414. doi: 10.1021/ci050006d. [DOI] [PubMed] [Google Scholar]
- Butina D, et al. Predicting ADME properties in silico: methods and models. Drug Discov. Today. 2002;7:S83–S88. doi: 10.1016/s1359-6446(02)02288-2. [DOI] [PubMed] [Google Scholar]
- Byvatov E, et al. Comparison of support vector machine and articial neural network systems for drug/nondrug classification. J. Chem. Inf. Comput. Sci. 2003;43:1882–1889. doi: 10.1021/ci0341161. [DOI] [PubMed] [Google Scholar]
- Campillos M, et al. Drug target identification using side-effect similarity. Science. 2008;321:263–266. doi: 10.1126/science.1158140. [DOI] [PubMed] [Google Scholar]
- Cumberbatch M, et al. The early and late effects of digoxin treatment on the sodium transport, sodium content and Na+K+-ATPase or erythrocytes. Br. J. Clin. Pharmacol. 1981;11:565–570. doi: 10.1111/j.1365-2125.1981.tb01172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darias V, et al. Synthesis and preliminary pharmacological study of thiophene analogues of the antipyretic and analgesic agent ethenzamide. Arch. Pharm. 2006;325:83–87. doi: 10.1002/ardp.19923250206. [DOI] [PubMed] [Google Scholar]
- Dobson CM. Chemical space and biology. Nature. 2004;432:824–828. doi: 10.1038/nature03192. [DOI] [PubMed] [Google Scholar]
- Emerit I, et al. Suppression of tumor promoter phorbolmyristate acetate-induced chromosome breakage by antioxidants and inhibitors of arachidonic acid metabolism. Mutat. Res. 1983;110:327–335. doi: 10.1016/0027-5107(83)90149-5. [DOI] [PubMed] [Google Scholar]
- Erhan D, et al. Collaborative ltering on a family of biological targets. J. Chem. Inf. Model. 2006;46:626–635. doi: 10.1021/ci050367t. [DOI] [PubMed] [Google Scholar]
- Faulon JL, et al. Genome scale enzyme-metabolite and drug-target interaction predictions using the signature molecular descriptor. Bioinformatics. 2008;24:225–233. doi: 10.1093/bioinformatics/btm580. [DOI] [PubMed] [Google Scholar]
- Frankl R. Intravenous salicylamide therapy in rheumatic diseases. Munch. Med. Wochensch. 1953;95:512–513. [PubMed] [Google Scholar]
- Goto K, et al. Analgesic effect of mofezolac, a non-steroidal anti-inflammatory drug, against phenylquinone-induced acute pain in mice. Prostaglandins Other Lipid Mediat. 1998;56:245–254. doi: 10.1016/s0090-6980(98)00054-9. [DOI] [PubMed] [Google Scholar]
- Gribskov M, Robinson NL. Use of receiver operating characteristic (ROC) analysis to evaluate sequence matching. Comput. Chem. 1996;20:25–33. doi: 10.1016/s0097-8485(96)80004-0. [DOI] [PubMed] [Google Scholar]
- Gunther S, et al. SuperTarget and Matador: resources for exploring drug-target relationships. Nucleic Acids Res. 2008;36:D919–D922. doi: 10.1093/nar/gkm862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori M, et al. Development of a chemical structure comparison method for integrated analysis of chemical and genomic information in the metabolic pathways. J. Am. Chem. Soc. 2003;125:11853–11865. doi: 10.1021/ja036030u. [DOI] [PubMed] [Google Scholar]
- Hirano T, et al. Citrus flavone tangeretin inhibits leukaemic hl-60 cell growth partially through induction of apoptosis with less cytotoxicity on normal lymphocytes. Br. J. Cancer. 1995;72:1380–1388. doi: 10.1038/bjc.1995.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob L, Vert J.-P. Protein-ligand interaction prediction: an improved chemogenomics approach. Bioinformatics. 2008;24:2149–2156. doi: 10.1093/bioinformatics/btn409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–D485. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann B, et al. Pharmacokinetics of metildigoxin and digoxin in geriatric patients with normal and elevated serum creatinine levels. Clin. Pharmacokinet. 1981;6:463–468. doi: 10.2165/00003088-198106060-00004. [DOI] [PubMed] [Google Scholar]
- Keele C.A. Sites and modes of action of antipyretic-analgesic drug. Proc. R. Soc. Med. 1969;62:535–539. [PMC free article] [PubMed] [Google Scholar]
- Lodhi H, Yamanishi Y. Chemoinformatics and Advanced Machine Learning Perspectives: Complex Computational Methods and Collaborative Techniques. Hershey, PA: IGI Global; 2010. [Google Scholar]
- Nagamine N, Sakakibara Y. Statistical prediction of protein-chemical interactions based on chemical structure and mass spectrometry data. Bioinformatics. 2007;23:2004–2012. doi: 10.1093/bioinformatics/btm266. [DOI] [PubMed] [Google Scholar]
- Preston SJ, et al. Comparative analgesic and anti-inflammatory properties of sodium salicylate and acetylsalicylic acid (aspirin) in rheumatoid arthritis. Br. J. Clin. Pharmacol. 1989;27:607–611. doi: 10.1111/j.1365-2125.1989.tb03423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rarey M, et al. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- Scheiber J, et al. Mapping adverse drug reactions in chemical space. J. Med. Chem. 2009;52:3103–3107. doi: 10.1021/jm801546k. [DOI] [PubMed] [Google Scholar]
- Schlessinger J, et al. An open-label adrenal suppression study of 0.1% fluocinonide cream in pediatric patients with atopic dermatitis. Arch. Dermatol. 2006;142:1568–1572. doi: 10.1001/archderm.142.12.1568. [DOI] [PubMed] [Google Scholar]
- Schölkopf B, et al. Kernel Methods in Computational Biology. Cambridge, MA: MIT Press; 2004. [Google Scholar]
- Schomburg I, et al. BRENDA, the enzyme database: updates and major new developments. Nucleic Acids Res. 2004;32:D431–433. doi: 10.1093/nar/gkh081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TF, Waterman MS. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Stockwell BR. Chemical genetics: ligand-based discovery of gene function. Nat. Rev. Genet. 2000;1:116–125. doi: 10.1038/35038557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, et al. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36:D901–D906. doi: 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanishi Y. Supervised bipartite graph inference. In: Koller D, et al., editors. Advances in Neural Information Processing Systems 21. Cambridge, MA: MIT Press; 2009. pp. 1841–1848. [Google Scholar]
- Yamanishi Y, et al. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics. 2008;24:i232–i240. doi: 10.1093/bioinformatics/btn162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, et al. Injectable actarit-loaded solid lipid nanoparticles as passive targeting therapeutic agents for rheumatoid arthritis. Int. J. Pharmaceutics. 2008;352:273–179. doi: 10.1016/j.ijpharm.2007.10.014. [DOI] [PubMed] [Google Scholar]
- Yildirim MA, et al. Drug-target network. Nat. Biotechnol. 2007;25:1119–1126. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]