

Abstract
Motivation: There has recently been a notable shift in biomedical information extraction (IE) from relation models toward the more expressive event model, facilitated by the maturation of basic tools for biomedical text analysis and the availability of manually annotated resources. The event model allows detailed representation of complex natural language statements and can support a number of advanced text mining applications ranging from semantic search to pathway extraction. A recent collaborative evaluation demonstrated the potential of event extraction systems, yet there have so far been no studies of the generalization ability of the systems nor the feasibility of large-scale extraction.
Results: This study considers event-based IE at PubMed scale. We introduce a system combining publicly available, state-of-the-art methods for domain parsing, named entity recognition and event extraction, and test the system on a representative 1% sample of all PubMed citations. We present the first evaluation of the generalization performance of event extraction systems to this scale and show that despite its computational complexity, event extraction from the entire PubMed is feasible. We further illustrate the value of the extraction approach through a number of analyses of the extracted information.
Availability: The event detection system and extracted data are open source licensed and available at http://bionlp.utu.fi/.
Contact: jari.bjorne@utu.fi
1 INTRODUCTION
In response to the explosive growth of biomedical scientific literature, there has recently been significant interest in the development of automatic methods for analyzing domain texts (Chapman and Cohen, 2009). In the previous decade of work on automatic information extraction (IE) from biomedical publications, efforts have focused in particular on the basic task of recognizing entity mentions in text, such as gene, protein or disease names (Kim et al., 2004; Smith et al., 2008; Yeh et al., 2005) and on the extraction of simple relations of these entities, such as statements of protein–protein interactions (PPI; Krallinger et al., 2008; Nédellec, 2005). State-of-the-art IE methods frequently rely on a detailed analysis of sentence structure (parsing) (Airola et al., 2008; Miwa et al., 2009), and several studies have addressed the development and adaptation of parsing methods to biomedical domain texts (Hara et al., 2007; Lease and Charniak, 2005; McClosky, 2009; Rimell and Clark, 2009).
The research focus on biomedical text analysis has brought forth notable advances in many areas. Automatic protein and gene Named Entity Recognition (NER) with performance exceeding 90% F-score1 was demonstrated to be feasible in the recent BioCreative community evaluation (Smith et al., 2008). Similarly, significant improvement has been made in PPI extraction (Airola et al., 2008; Chowdhary et al., 2009; Miwa et al., 2009) and there is an active collaboration between database curators and method developers to integrate PPI methods into curation pipelines (Chatr-aryamontri et al., 2008). Finally, methods for a variety of biomedical text processing tasks ranging from sentence splitting (Tomanek et al., 2007) to full parsing (McClosky, 2009) have been introduced with performance approaching or matching the performance of similar methods on general English texts.
Building on such text analysis methods, several IE systems and services have been created for retrieving interaction information from PubMed (http://www.pubmed.com). Varying levels of parsing and other NLP methods have been used to detect biological entities of interest and their relationships. Most previous efforts have focused on pairwise PPI, with extracted pairs often represented as merged interaction networks. The MEDIE and InfoPubmed systems (Ohta et al., 2006) offer access to deep syntactic analysis and entity relation extraction results from the entire PubMed through subject-verb-object search patterns. The Chilibot system looks for pairwise relationships based on co-occurrence and uses the presence of interaction keywords to type them (Chen and Sharp, 2004). The TextMed system, based on the LYDIA project (Lloyd et al., 2005), uses shallow parsing and co-occurrence information to generate pairwise entity relationship networks from PubMed citations. The Ali Baba system likewise visualizes relationships from PubMed abstracts as graphs (Palaga et al., 2009). IHOP hyperlinks PubMed abstracts together through shared protein and gene mentions (Hoffmann and Valencia, 2004). Finally, GoPubMed uses Gene Ontology (http://www.geneontology.org/) and medical subject headings (MeSH) (http://www.nlm.nih.gov/mesh/) to provide a knowledge-based search for relevant citations (Doms and Schroeder, 2005).
Supported in part by the maturation of basic technologies for biomedical text analysis and the availability of richly annotated text corpora (Kim et al., 2008; Pyysalo et al., 2007), there has recently been notable movement in the biomedical IE community toward more detailed and expressive representations of extracted information. In particular, event representations that can capture different types of associations of arbitrary numbers of entities and events in varying roles have been applied as an alternative to the simple relation representation. While the applicability of the results produced by IE methods employing relation representations is closely tied to the specific relation type targeted (i.e. PPI or gene–disease), event representations have wider potential in supporting applications ranging from PPI to semantic search and pathway extraction (Kim et al., 2009). Showing wide interest in the approach, 24 teams participated in the first community-wide competitive evaluation of event extraction methods, the BioNLP'09 Shared Task on Event Extraction (http://www-tsujii.is.s.u-tokyo.ac.jp/GENIA/SharedTask/; Kim et al., 2009). The number is comparable to the 26 teams participating in the PPI task of the established BioCreative II community evaluation (Krallinger et al., 2008).
In this study, building on the best performing system in the BioNLP'09 shared task (Björne et al., 2009) and J.Björne et al. (submitted for publication) which remains competitive with even the most recent advances (Miwa et al., 2010), we join together state-of-the-art methods for biomedical text parsing, protein/gene name recognition and IE into a unified system capable of event extraction from unannotated text. We apply this system to a random 1% sample of citations from the PubMed literature database, providing the first estimate of the results of event extraction from the entire PubMed data. We further analyze the performance of the key components of the system, thus providing the first evaluation of the ability of state-of-the-art event extraction systems to generalize to PubMed scale.
2 SYSTEM AND METHODS
The event extraction system presented in this article follows the model of the BioNLP'09 Shared Task on event extraction in its representation of extracted information. The BioNLP'09 Shared Task was the first large-scale evaluation of biomedical event detection systems (Kim et al., 2009). The task introduced an event representation and extraction task based on the GENIA event corpus annotation (Kim et al., 2008). The primary extraction targets in the defined task are nine fundamental biomolecular event types (Table 1) and the participants in these events. In this article, the term event refers to events as defined by the Shared Task annotation scheme.
Table 1.
Targeted event types with brief example statements expressing an event of each type
| Event type | Example |
|---|---|
| Gene expression | 5-LOX is expressed in leukocytes |
| Transcription | promoter associated with IL-4 genetranscription |
| Localization | phosphorylation and nuclear translocation of STAT6 |
| Protein catabolism | I kappa B-alphaproteolysis by phosphorylation. |
| Phosphorylation | BCL-2 was phosphorylated at the G(2)/M phase |
| Binding | Bcl-wforms complexes with Bax and Bak |
| Regulation | c-Met expression is regulated by Mitf |
| Positive regulation | IL-12inducedSTAT4 binding |
| Negative regulation | DN-RacsuppressedNFAT activation |
In the examples, the word or words marked as triggering the presence of the event are shown in italics and event participants underlined. The event types are grouped by event participants, with the first five types taking one theme, binding events multiple themes and the regulation types theme and cause participants.
Several aspects of the event representation differentiate the event extraction task from the body of domain IE studies targeting, e.g. PPI and gene–disease relations, including previous domain shared tasks (Krallinger et al., 2008; Nédellec, 2005). While domain IE has largely focused on a relation model representing extracted information as entity pairs, the event model allows for a more expressive way of capturing statement semantics. Events can have an arbitrary number of participants whose roles in the event (e.g. theme or cause) are specified, making it possible to capture n-ary associations and statements where some participants occur in varying roles or are only occasionally mentioned. Further, events are modeled as primary objects of annotation and bound to specific statements in text (triggers), allowing events to participate in other events and facilitating further analysis such as the identification of events stated in a negated or speculative context. Finally, events following the Shared Task model are given GENIA Event ontology types drawn from the community-standard Gene Ontology, giving each event well-defined semantics. Using events to represent information contained in natural language sentences, for the first time it is now possible to accurately describe in a formal fashion the multitude of different biological phenomena depicted in research articles.
In the rest of this section, we describe the steps of the event extraction pipeline (Fig. 1).
Fig. 1.

Event extraction. A multiphased system is used to generate an event graph, a formal representation for the semantic content of the sentence. Before event detection, sentences are parsed (A) to generate a suitable syntactic graph to be used in detecting semantic relationships. Event detection starts with identification of named entities (B) with BANNER (parses are not used at this step). Once named entities have been identified, the trigger detector (C) uses them and the parse for predicting triggers, words which define potential events. The edge detector (D) predicts relationship edges (event arguments) between triggers and named entities. Finally, the resulting semantic graph is divided into individual events by (E) duplicating trigger nodes and regrouping argument edges.
2.1 Named entity recognition
NER is a fundamental requirement for IE: the analysis of IE systems normally takes the form of associations between references to entities, and most applications require the references to be sufficiently specific to identify particular real-world entities, i.e. entity names. Consequently, NER is a well-studied subtask in IE. Also in the biomedical domain NER has been considered in a wealth of studies, including several shared tasks which have demonstrated significant recent advances in NER (Table 2).
Table 2.
NER system performance
| System | Corpus | F-score | References |
|---|---|---|---|
| JNLPBA (best) | GENIA term | 72.6% | Kim et al. (2004) |
| BioCreative I (best) | GENETAG | 83.2% | Yeh et al. (2005) |
| BioCreative II (best) | GENETAG | 87.2% | Smith et al. (2008) |
| BANNER | GENETAG | 86.4% | Leaman and Gonzalez (2008) |
Performance shown for the best performing systems at various shared tasks and the BANNER system used in this work.
Note that while the GENIA term corpus used in the JNLPBA task requires differentiation between, e.g. protein and gene entities, GENETAG only marks a single gene/RNA/protein type, contributing to the measured differences.
While overall only a fraction of the systems participating in shared tasks are publicly available, some systems competitive with the state-of-the-art have been made available. In this study we apply the BANNER NER system of Leaman and Gonzalez (2008), which in its current release achieves results close to the best published on the standard GENETAG dataset (Table 2) and was reported to have the best performance in a recent study comparing publicly available taggers (Kabiljo et al., 2009).
BANNER follows the major trends in recent domain NER in being based on a model automatically learned from annotated training data, specifically using the conditional random field (CRF) model. BANNER applies a rich set of features found beneficial in recent domain studies to represent its input, including the part-of-speech tags and base forms of the input words, word prefix and suffix features, and basic word form features. We use a recent release of the BANNER system that further includes features derived from lookup from a broad-coverage dictionary of gene and protein names as well as a number of post-processing modules performing, for example, local abbreviation detection to improve performance.
As a machine learning system, BANNER could be trained on a variety of different corpus resources tagged for genes and proteins. As there are a number of difficult issues in the joint use of annotated corpora relating to differences in coverage and tagging criteria (Wang et al., 2009), we chose to train the system on the GENETAG corpus (Tanabe et al., 2005). In addition to being one of the largest manually annotated resources for gene and protein NER and the reference standard used for the BioCreative evaluations, GENETAG has been specifically constructed to include a heterogeneous set of sentences from PubMed, a property expected to provide good generalization performance to large-scale tagging of documents from various subdomains.
2.2 Parsing
Our event detection system relies on the availability of full dependency parses in the Stanford dependency (SD) scheme (de Marneffe and Manning, 2008a; Fig. 1A). We use the Charniak–Johnson (Charniak and Johnson, 2005) parser with the improved biomedical parsing model of McClosky (2009) (http://bllip.cs.brown.edu/download/bioparsingmodel-rel1.tar.gz). This parser achieved the highest published parsing accuracy on the GENIA Treebank (Tateisi et al., 2005) and is thus arguably the best parser available for text in PubMed abstracts (McClosky, 2009). The Penn Treebank scheme analyses given by the Charniak–Johnson parser are subsequently processed using the Stanford conversion tool (http://nlp.stanford.edu/software/stanford-parser-2008-10-26.tgz; de Marneffe et al., 2006), resulting in the final analyses in the collapsed dependencies with propagation of conjunct dependencies version of the SD scheme, as defined by de Marneffe and Manning (2008a).
2.3 Event detection
At the core of our approach to event extraction are graph representations of sentence syntax and semantics. The syntactic graph corresponds to a dependency analysis of sentence structure, and the semantic graph represents the event structure, with nodes representing named entities and events, and edges corresponding to event arguments (Fig. 1). The syntactic graph is generated from the text by the parser and the protein/gene entities of the semantic graph are detected by the NER system. The goal of the core event extraction system, described in detail in Björne et al. (2009) and J.Björne et al. (submitted for publication) is then to generate the semantic graph given a sentence with marked named entities and syntactic analysis. The system considers the sentences independently since, based on an analysis of the BioNLP'09 Shared Task dataset, only 4.8% of events cross sentence boundaries.
The event extraction system is based on supervised machine learning, i.e. it performs predictions for unknown cases based on a model automatically learned from manually annotated training data, here derived from that provided in the BioNLP'09 Shared Task. For the machine learning phases of the system (trigger and event detection), we use the standard approach of dividing the problem into individual examples (for each potential trigger or event argument), which are then classified into a number of classes. For each example, information about the sentence is converted into a number of individual features, each describing a particular aspect of the text. The classifier, in our case a support vector machine (SVM), then uses correlations between all of these features to give a classification for each example.
As can be seen in Figure 1, the dependency parse is very close to the semantic graph that we aim to extract, and consequently it is the most important source of features. Notable is the use of very large numbers of unique features, enabled by modern large-margin classification methods, such as the SVMs used. For example, the training data for the edge detector consists of 31 792 examples with 295 034 unique features.
Our system has two key machine learning-based classification steps, trigger detection and edge detection, followed by a rule-based event construction step.
2.3.1 Trigger detection
Triggers are the words in the sentence that state the events between the named entities, and trigger detection is thus the first step in event detection. As determined from the BioNLP'09 Shared Task dataset, 92% of all triggers consist of a single word. Therefore, we represent all triggers with their head word, the semantically most relevant word within a particular trigger, typically its syntactic head. Trigger detection is thus the task of identifying those individual words in the sentence that act as a trigger head word. In contrast with NER, where sequential models are typically applied (Smith et al., 2008), the system classifies each word in isolation as one of the nine event types, or a negative, i.e. not corresponding to an event trigger.
Triggers cannot be identified based only on the words themselves, as most potential trigger words do not uniquely correspond to a specific trigger class. For example, only 28% of the instances of the word activates are triggers for positive regulation events in the BioNLP'09 Shared Task dataset and the commonly used word overexpression is equally distributed between gene expression events, positive regulation events and the negative class. For this reason, a large number of features that aim to capture the full semantic context of a potential trigger word are used in the classification.
Features for the trigger detection are based on both the linear order of words and the dependency parse. The largest number of features comes from the dependency parse, from which undirected chains of up to three dependencies are built, starting from the potential trigger word. The words themselves are also sliced into two or three letter N-grams that aim to capture similarities between closely related words or inflected forms.
2.3.2 Edge detection
Each edge in the semantic graph corresponds to an event argument and edge detection thus follows trigger detection. Edge detection is cast as a multiclass classification task, since event arguments may have multiple types (cause, theme). One example is defined for each possible directed pairwise combination of trigger and entity nodes, and this example will then be classified as belonging to one of the argument type classes or as negative. Each edge example is classified independently, even though the BioNLP'09 Shared Task event scheme imposes conditions on valid combinations of argument types for each type of event. These conditions are enforced in the event construction phase.
In contrast with trigger detection, the linear order of words is not considered and the features are entirely based on the dependency parse, more specifically on the shortest path of dependencies that link together the two trigger or entity nodes under consideration. To describe the path as features, it is divided into multiple dependency N-grams, each consisting of 2–4 consecutive dependencies between sentence words. Features built from previous classification steps, i.e. ones based on the named entities and triggers, are also an important part of the edge detection feature set.
2.3.3 Event construction
The semantic graph created in the trigger and edge detection steps (Fig. 1D) contains strictly one event node for each trigger word. The final events are created by duplicating these nodes when necessary and separating their edges into valid combinations based on the syntax of the sentences and the conditions on argument type combinations stated in the BioNLP'09 Shared Task event scheme. This step is rule based and as such does not require training data.
2.3.4 Machine learning method
As the machine learning method applied in all stages of the core event extraction system, we use the SVMmulticlass (Tsochantaridis et al., 2005) implementation (http://svmlight.joachims.org/svm_multiclass.html) of an SVM. SVMs perform competitively at a number of tasks in natural language processing and are suited for tasks involving multidimensional, partially redundant feature sets such as that applied in the event extraction system.
3 RESULTS
We apply the event extraction system to a 1% sample of the 2009 distribution of the PubMed literature database. The full PubMed dataset contains 17.8 million citations, which we downsampled at random to create a dataset of 177 648 citations. To assure that results are representative of the full PubMed database, we performed no document selection or filtering. Consequently, 81 516 of the citations in the sample (46%) contain only a title but no abstract, and the earliest citation in the sample is for an article published in 1867 (PMID 17230723). While many of the citations in the sample are thus likely to have limited utility for biomolecular event extraction, their inclusion assures unbiased results and a fair test of the true generalization ability of the applied methods.
In total, the system extracted 168 949 events from 29 781 citations in the PubMed sample. The number of extracted events for the nine event types is presented in Table 3. The BANNER NER system marked 365 204 gene/protein entities in 54 051 citations in the sample, averaging two mentions per citation overall and almost seven per citation for those containing at least one tagged entity. By this estimate less than a third of PubMed citations contain gene/protein mentions. When the extracted events are broken up by publication year, a strong trend for increasing number of citations containing gene/protein mentions and events is visible, with 25% of citations from the last 10 years containing at least one event (Fig. 2). Since the extracted events are of the protein/gene-specific BioNLP'09 Shared Task types, this trend can be seen to reflect the growing prominence of molecular biology. Based on the predicted named entities and events, the present release of PubMed can be estimated to contain more than 5 million citations with gene/protein mentions, totaling over 35 million mentions overall and nearly 3 million citations containing events of the Shared Task types, totaling over 16 million such events overall. The number of citations with events has increased consistently with the growth of PubMed (Fig. 2) until the year 2000. After this the total amount of citations grows more rapidly, perhaps reflecting PubMed's expanded coverage of life science topics since then (Benton, 1999).
Table 3.
Frequency of the nine event types in the output of the system on the PubMed sample
| Event Type | Count (%) |
|---|---|
| Gene expression | 48 144 (28.5) |
| Positive regulation | 43 155 (25.5) |
| Binding | 24 159 (14.3) |
| Negative regulation | 21 833 (12.9) |
| Regulation | 13 330 (7.9) |
| Localization | 10 766 (6.4) |
| Phosphorylation | 3 852 (2.3) |
| Transcription | 2 492 (1.5) |
| Protein catabolism | 1 218 (0.7) |
| TOTAL | 168 949 (100.0) |
Fig. 2.

Total number of citations and citations with tagged gene/protein mentions and events in the sample by year.
While not the primary result of this study, the extraction output can be used to support analysis of some large-scale trends in PubMed. As an example, Figure 3 shows the number of citations per year with mentions of insulin, immunoglobulin G (IgG) and tumor necrosis factor alpha (TNF-α), the three most common named entities identified, and their associated events. We note that citations for insulin show a long-term growing trend, perhaps reflecting the considerable resources directed toward diabetes research. The decreasing number of article abstracts mentioning IgG, despite its centrality in many experimental applications, might be seen to indicate its waning as a primary subject of research, considering average gene quotation frequencies over time (Hoffmann and Valencia, 2003). The number of citations mentioning tumor necrosis factor alpha has grown explosively since it was first cloned and named in 1984, showing continued and growing interest in this apoptosis-related cytokine centrally associated with multiple pathways and implicated in cancer.
Fig. 3.

Number of citations with tagged mentions of insulin, IgG and TNF-alpha (normalized for capitalization and hyphenization), as well as extracted events of these proteins. The counts are cumulative for every five years to smooth the curves.
3.1 Evaluation
The event extraction system (without the NER component) achieves an F-score of 52.86% (precision 58.13% and recall 48.46%) on the BioNLP'09 Shared Task test set. The Shared Task data, based on the GENIA corpus, is composed of PubMed citations relevant to biological reactions concerning transcription factors in human blood cells (Kim et al., 2008). The GENIA corpus is focused on a particular subdomain and thus not a representative sample of the entire PubMed, the focus of this study. Additional analysis is, therefore, necessary to evaluate the extraction result. A particular point of interest is the ability of the system to perform on input data that, compared with the GENIA corpus, has far fewer events per sentence and thus deviates from the distribution on which the system was originally trained.
Evaluating the recall of the system would require fully annotating a large enough fraction of the PubMed sample for all named entities and events. Annotating sentences for positive and negative events is, however, a time and labor-intensive process. For example, annotating the GENIA event corpus consisting of 9 372 sentences required 1.5 years with five part-time annotators and two coordinators (Kim et al., 2008). Such an annotation is thus not practical for this study, particularly since relevant events are very rare in a random sample of PubMed not focused on any particular subdomain. In contrast, manually inspecting the system output for errors is a comparatively easy task and allows us to determine the precision of the system output. We examine a random sample of 100 predicted named entities and 100 predicted events and determine their correctness.
In the BioNLP'09 Shared Task data events are annotated only between genes and proteins, leaving out e.g. the multitude of signaling interactions between proteins and small inorganic molecules such as Ca2+. Our aim in this work is to recover as many of the biomolecular interactions stated in the texts as possible, so we extend the criteria for what is considered a named entity and an event. For named entities, we consider as positives cells, cellular components and molecules that take part in biochemical interactions. For events, we consider the event to be correct if all its named entity arguments are correct and the trigger word in the text is correctly detected.
In the manual evaluation of the 100 predicted named entities, we estimate the precision of the named entity detection step (the BANNER system) to be 87%. This compares well with BANNER performance on the GENETAG corpus with precision of 89% (for an f-score of 86%). The precision of the 100 predicted events was 64%, a figure close to the 58% (for f-score of 53%) established on the BioNLP'09 Shared Task data. While recall cannot be directly measured, as discussed above, considering that the event extraction system was trained on example-rich data that favors making positive predictions, it can be expected not to decrease substantially from results established on subdomain corpora.
The results of this manual analysis indicate that the performance of the named entity and event detection components does generalize from the subdomain corpus data to a representative unbiased sample of the entire PubMed. It should, however, be noted that evaluating automatically generated predictions after the fact is more prone to a positive bias than annotation of plain text with no predictions.
3.2 Event network
One of the most promising applications for large-scale event mining is the generation of interaction networks. Unlike networks constructed from binary interactions, an event network defines the types and directions of the relationships, the polarity (positive or negative) of regulatory relationships and the mechanisms involved (e.g. phosphorylation). Sufficiently accurate event graphs can be used for inferring complex regulatory relationship networks and other biologically relevant tasks.
Figure 4 illustrates a sample network constructed from events extracted by our system around interleukin-4. To build the network, we merge the individual predicted events into a single graph, loosely following the approach employed by Saeys et al. (2009). We determine two protein mentions to be the same if their names match after lowercasing and removal of whitespace and hyphens. All mentions of the same protein are represented in the graph by a single node. Event argument edges connect the proteins through event trigger nodes.
Fig. 4.

Extracted event network around interleukin-4. This graph shows a subset of the predicted event network, including only named entities with at least 50 extracted instances. The round event nodes are (P)ositive regulation, (N)egative regulation, (R)egulation, gene (E)xpression, (B)inding, p(H)osphorylation and (L)ocalization. For clarity, single-argument events (E, B, H and L) are displayed only when they also act as arguments of regulation events.
The entire graph extracted from the PubMed sample has one major connected component comprising 88 477 (38%) of the total of 232 760 nodes. The remaining nodes form a large number of considerably smaller connected components, the largest of which contains a mere 95 nodes.
3.3 Topic analysis
As a final analysis of the extraction results, we studied the topics of event-containing citations. Over 90% of records in PubMed are manually indexed with a number of descriptors chosen from Medical Subject Headings (MeSH), a hierarchical thesaurus in which the descriptors are arranged primarily in general—specific hierarchy. The descriptors assigned to a PubMed citation record express the main topics discussed in the respective article and allow queries within specific subtopics, reducing the variance and sparsity of simple keyword search. In the following, we investigate the connection between MeSH descriptors and event types, establishing the topical areas in PubMed likely to contain citations relevant to event extraction.
We measured the degree of dependence between a MeSH descriptor d and an event type e using pointwise mutual information
where P(d, e) is defined as the fraction of citations indexed by the descriptor d and containing at least one event of type e, out of all citations that contain at least one event. Similarly, P(d) (respectively P(e)) is the fraction of citations containing the descriptor d (respectively event of type e) out of all citations that contain at least one event. The measure calculates the ratio of joint probability of d and e to the probability of their co-occurrence by chance. To deal with sparsity problems, we first expanded, for each citation, the set of its original MeSH descriptors indexed in PubMed with all descriptors that are more general in the MeSH hierarchy. This allows us to find more general descriptors, rather than the specific ones indexed in PubMed.
For each of the nine event types, we built a list of five most related descriptors, that is, descriptors with the highest pointwise MI. To avoid unnecessarily specific descriptors, we only considered those descriptors that were present in at least 10% of citations with an event of the given type, and discarded descriptors that were hyponyms (more specific) to another descriptor already in the list. The resulting lists are given in Table 4. These illustrate that the descriptors obtained are indeed relevant to the respective event types, except for the two obviously too general descriptors Technology, Industry, and Agriculture and Information Services, which we discarded in all subsequent analyses. Apart from validating the IE system, although very indirectly, these MeSH descriptor lists can be used to focus PubMed searches to citations likely containing the relevant event types.
Table 4.
Top related MeSH descriptors for the nine event types
| Event type | Five most related MeSH descriptors |
|---|---|
| Gene expression | Gene expression regulation; RNA; gene expression; |
| cytokines; immunohistochemistry | |
| Positive regulation | Intracellular signaling peptides and proteins; |
| phosphotransferases; transcription factors; | |
| cytokines; gene expression regulation | |
| Negative regulation | Molecular mechanisms of pharmacological action; |
| intracellular signaling peptides and proteins; | |
| therapeutic uses; phosphotransferases; | |
| tumor cells, cultured | |
| Binding | Protein binding; information services; |
| physicochemical phenomena; chemistry techniques; | |
| analytical receptors; cell surface | |
| Regulation | Gene expression regulation; RNA, messenger; |
| transcription factors; protein kinases; peptide | |
| hormones | |
| Localization | Endocrine system, protein precursors; nerve tissue |
| proteins; hormones, hormone substitutes and | |
| hormone antagonists; organelles | |
| Transcription | RNA; gene components; gene expression; base |
| sequence; transcription factors | |
| Phosphorylation | Organic chemistry phenomena; tyrosine |
| adaptor proteins, signal transducing; | |
| phosphotransferases (alcohol group acceptor) | |
| phosphoproteins | |
| Protein catabolism | Hydrolases; macromolecular substances; |
| technology, industry and agriculture; | |
| physicochemical processes; metabolism |
Of the 177 648 citations in the PubMed sample, 66 227 (37.3%) are indexed by at least one of the descriptors in Table 4, or its hyponym (we will refer to these citations as MeSH-relevant). In contrast, only 12 405 (7.3%) of the 168 949 events identified by the system were extracted from the 62.7% MeSH-irrelevant citations, demonstrating that the MeSH terms in Table 4 are indeed strong predictors of citations containing relevant events.
Intuitively, it can be expected that MeSH-irrelevant citations are also likely to contain a higher proportion of false positive named entities and events. To verify this hypothesis, we measure the proportions of true positive (TP) named entities and events among MeSH-relevant citations and contrast them to the proportions in MeSH-irrelevant citations. The results of this analysis, performed on the same set of 100 random events and 100 random entities introduced in Section 3.1, are presented in Table 5. For both named entities and events, the proportion of TPs (precision) is notably lower in MeSH-irrelevant citations. In case of named entities, the difference is 20.6 percentage points (significant with P = 0.009, two-tailed Fisher's test) and in case of events, the difference is full 35.9 percentage points (significant with P = 0.028, two-tailed Fisher's test). These results suggest that MeSH descriptors may provide features for the event extraction system with a high predictive power and could be, for instance, used to generate likely negative examples for further retraining of the extraction system. Thus, the broad manual annotation of the MeSH descriptors can enhance detailed automated event extraction.
Table 5.
Comparison of named entity and event detection precision between MeSH-relevant and MeSH-irrelevant citations
| Citation | TP | FP | Precision (%) | |
|---|---|---|---|---|
| Named entities | MeSH-relevant | 66 | 5 | 93.0 |
| MeSH-irrelevant | 21 | 8 | 72.4 | |
| Events | MeSH-relevant | 58 | 29 | 66.7 |
| MeSH-irrelevant | 4 | 9 | 30.8 | |
3.4 Computational requirements
The computational requirements of the system components, in terms of time and space, are detailed in Table 6.
Table 6.
Processing requirements for different components as measured for the sample and estimated for the whole PubMed
| Sample |
PubMed |
|||
|---|---|---|---|---|
| Component | Time (h) | Space (MB) | Time (day) | Space (GB) |
| NER (BANNER) | 18 | 272 | 75 | 27 |
| Parsing | 53 | 830 | 222 | 81 |
| Event extraction | 27 | 276 | 114 | 27 |
| TOTAL | 98 | 1378 | 411 | 135 |
Space requirements are stated for uncompressed XML files.
NER using BANNER is a comparatively light-weight processing step in the pipeline. Processing the entire dataset consumed <18 h on a desktop-level computer, averaging more than three citations per second. NER tagging could thus be run for the entire PubMed database in ∼75 days on a single machine, or a matter of days on a modest cluster.
Full dependency parsing is the most resource-intensive step in the pipeline. The parsing time of the Charniak–Johnson parser for one sentence is in our case 0.81 s, with an additional 0.15 s taken by the SD scheme conversion. Parsing all 935 186 sentences in the PubMed sample would thus take 249 processor hours (2.84 processor years projected for the entire PubMed). However, only sentences with at least one recognized named entity, and thus a potential target for the IE system, need to be parsed, considerably decreasing the number of sentences that must be parsed to 199 941 and the parsing time to 53 h (222 days projected for the entire PubMed).
We note that the parsing process is not a straightforward technical undertaking. The Charniak–Johnson parser takes about 10 s to load the parsing model files and, in order for this fixed time penalty not to accumulate, it is necessary to divide the parsing task to large batches rather than parsing a single abstract, or even single sentence at a time. On the other hand, there were 26 sentences in the PubMed sample that caused the parser to process interminably without producing an analysis. It was necessary to detect these cases, terminate the parser and restart the process. Of the 199 915 sentences parsed by the Charniak–Johnson parser, further 37 sentences were not successfully processed by the SD conversion tools. The final number of successfully parsed sentences was thus 199 878. It must be stressed that this number represents a highly respectable 99.97% of sentences successfully parsed, demonstrating the high reliability achieved by the current state-of-the-art in syntactic parsing.
Finally, the event extraction step took 27 processor hours (114 processor days projected for the entire PubMed), thus averaging roughly one citation per 2 s for the 54 051 citations with at least one detected named entity. The total processing time of the pipeline was 98 processor hours (411 processor days projected for the entire PubMed), or, one PubMed citation per 2 s.
4 DISCUSSION AND CONCLUSIONS
We have presented to the best of our knowledge the first application of event-style biomedical interaction extraction to a large-scale real-world dataset, 1% of the PubMed citation database. We combined the event detection system of J.Björne et al. (submitted for publication), the winning system of the BioNLP'09 Shared Task, with the efficient Charniak–Johnson parser (Charniak and Johnson, 2005) equipped with the biomedical domain model of McClosky (2009) and the BANNER named entity detector (Leaman and Gonzalez, 2008), creating a system capable of extracting events from unannotated biomedical text. Successful processing of 1% of PubMed in 98 processor hours demonstrates that the computational requirements, although considerable, are well within reach of generally available computational resources.
Analysis of the 1% PubMed sample dataset produced over 160 000 biomedical events. Based on a manual analysis of a subset of these predicted events, the precision of the system was 64%, indicating that the event detection system performance generalized well to real world conditions represented by a random, unbiased sample of PubMed. Analysis of the resulting event graph showed that the generated events form a highly connected interaction network. It is especially in this regard that extraction of events, rather than n-ary relations, holds a great promise: the complexity of interaction networks can only be utilized effectively when detailed information about the type, direction and polarity of the various interactions is available.
Even though a mere 1% of PubMed was analyzed in this study, the result is a large dataset of over 160 000 events extracted by a state-of-the-art event extraction system as well as 177 000 PubMed citations processed with the essential NLP tools. In most respects, this dataset is representative of the entire PubMed and allows a number of subsequent analyses to be performed even before the entire PubMed is processed by the system. As a practical contribution to the emerging field of biomedical event detection, we publish the dataset in a flexible XML format as well as the widely adopted BioNLP'09 Shared Task format. The event detection system used in this study and described in more detail in Björne et al. (2009) and J.Björne et al. (submitted for publication) is published under an open source license. Both are available at http://bionlp.utu.fi/.
Having shown its feasibility, the future work will focus on processing the entire PubMed and, as with this study, making the results available for analysis to the community.
ACKNOWLEDGEMENTS
We would like to thank Robert Leaman for providing us with advance access and assistance in using the most recent release of BANNER. The computational resources for this study were provided by CSC - IT Center for Science, Ltd, a joint computing center for Finnish academia and industry.
Funding: Funding was provided by University of Turku, Academy of Finland, Turku Centre for Computer Science and Grant-in-Aid for Specially Promoted Research (Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan).
Conflict of Interest: none declared.
Footnotes
1F-score is the harmonic mean of (p)recision and (r)ecall, i.e. 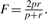 .
.
REFERENCES
- Airola A, et al. All-paths graph kernel for protein-protein interaction extraction with evaluation of cross-corpus learning. BMC Bioinformatics. 2008;9(Suppl. 11):S2. doi: 10.1186/1471-2105-9-S11-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton N. Scope expands for PubMed® and MEDLINE®. NLM Technical Bulletin. 1999;311 [Google Scholar]
- Björne J, et al. Proceedings of the BioNLP 2009 Workshop Companion Volume for Shared Task. New York, NY, USA: Association for Computational Linguistics; 2009. Extracting complex biological events with rich graph-based feature sets; pp. 10–18. [Google Scholar]
- Chapman WW, Cohen KB. Current issues in biomedical text mining and natural language processing. J. Biomed. Inform. 2009;42:757–759. doi: 10.1016/j.jbi.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Charniak E, Johnson M. Proceedings of the 43rd Annual Meeting of the Association for Computational Linguistics (ACL'05). New York, NY, USA: Association for Computational Linguistics; 2005. Coarse-to-fine n-best parsing and maxent discriminative reranking; pp. 173–180. [Google Scholar]
- Chatr-aryamontri A, et al. MINT and IntAct contribute to the second BioCreative challenge: serving the text-mining community with high quality molecular interaction data. Genome Biol. 2008;9(Suppl. 2):S5. doi: 10.1186/gb-2008-9-s2-s5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Sharp BM. Content-rich biological network constructed by mining PubMed abstracts. BMC Bioinformatics. 2004;5:147. doi: 10.1186/1471-2105-5-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhary R, et al. Bayesian inference of protein-protein interactions from biological literature. Bioinformatics. 2009;25:1536–1542. doi: 10.1093/bioinformatics/btp245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Marneffe M.-C, et al. Generating typed dependency parses from phrase structure parses. Proceedings of LREC-06. 2006:449–454. [Google Scholar]
- de Marneffe M.-C, Manning C. The Stanford typed dependencies representation. COLING Workshop on Cross-framework and Cross-domain Parser Evaluation. 2008a [Google Scholar]
- de Marneffe M.-C, Manning C. Technical report. Stanford University; 2008b. Stanford typed dependencies manual. [Google Scholar]
- Doms A, Schroeder M. GoPubMed: exploring PubMed with the Gene Ontology. Nucleic Acids Res. 2005;33:W783–W786. doi: 10.1093/nar/gki470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, et al. IWPT '07: Proceedings of the 10th International Conference on Parsing Technologies. Morristown, NJ: Association for Computational Linguistics; 2007. Evaluating impact of re-training a lexical disambiguation model on domain adaptation of an HPSG parser; pp. 11–22. [Google Scholar]
- Hoffmann R, Valencia A. Life cycles of successful genes. Trends Genet. 2003;19:79–81. doi: 10.1016/s0168-9525(02)00014-8. [DOI] [PubMed] [Google Scholar]
- Hoffmann R, Valencia A. A gene network for navigating the literature. Nat. Genet. 2004;36:664. doi: 10.1038/ng0704-664. [DOI] [PubMed] [Google Scholar]
- Kabiljo R, et al. A realistic assessment of methods for extracting gene/protein interactions from free text. BMC Bioinformatics. 2009;10:233. doi: 10.1186/1471-2105-10-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.-D, et al. Introduction to the bio-entity recognition task at JNLPBA. In: Collier N, et al., editors. Proceedings of the International Joint Workshop on Natural Language Processing in Biomedicine and its Applications (JNLPBA) Geneva, Switzerland: 2004. pp. 70–75. [Google Scholar]
- Kim J.-D, et al. Corpus annotation for mining biomedical events from literature. BMC Bioinformatics. 2008;9:10. doi: 10.1186/1471-2105-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.-D, et al. Proceedings of the BioNLP 2009 Workshop Companion Volume for Shared Task. Boulder, Colorado: ACL; 2009. Overview of BioNLP'09 shared task on event extraction; pp. 1–9. [Google Scholar]
- Krallinger M, et al. Overview of the protein-protein interaction annotation extraction task of BioCreative II. Genome Biol. 2008;9(Suppl. 2):S4. doi: 10.1186/gb-2008-9-s2-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaman R, Gonzalez G. BANNER: an executable survey of advances in biomedical named entity recognition. Pac. Symp. Biocomput. 2008;13:652–663. [PubMed] [Google Scholar]
- Lease M, Charniak E. Parsing biomedical literature. In: Dale R, et al., editors. Proceedings of the Second International Joint Conference on Natural Langage Processing (IJCNLP'05) Vol. 3651. Springer; 2005. pp. 58–69. Lecture Notes in Computer Science. [Google Scholar]
- Lloyd L, et al. Lydia: a system for large-scale news analysis. 12th Symposium of String Processing and Information Retrieval (SPIRE '05) 2005;3772:161–166. of Lecture Notes in Computer Science. [Google Scholar]
- McClosky D. PhD Thesis. Department of Computer Science, Brown University; 2009. Any Domain Parsing: Automatic Domain Adaptation for Natural Language Parsing/ [Google Scholar]
- Miwa M, et al. Protein-protein interaction extraction by leveraging multiple kernels and parsers. Int. J. Med. Inform. 2009;78:e39–e46. doi: 10.1016/j.ijmedinf.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Miwa M, et al. Event extraction with complex event classification using rich features. J. Bioinform. Comput. Biol. 2010;8:131–146. doi: 10.1142/s0219720010004586. [DOI] [PubMed] [Google Scholar]
- Nédellec C. Learning language in logic - genic interaction extraction challenge. In: Cussens J, Nédellec C, editors. Proceedings of the 4th Learning Language in Logic Workshop (LLL05) 2005. pp. 31–37. [Google Scholar]
- Ohta T, et al. Proceedings of the COLING/ACL 2006 Interactive Presentation Sessions. Sydney, Australia: Association for Computational Linguistics; 2006. An intelligent search engine and GUI-based efficient MEDLINE search tool based on deep syntactic parsing; pp. 17–20. [Google Scholar]
- Palaga P, et al. EDBT. Vol. 360. New york, NY, USA: Association of Computational Linguistics; 2009. High-performance information extraction with AliBaba; pp. 1140–1143. of ACM International Conference Proceeding Series. [Google Scholar]
- Pyysalo S, et al. BioInfer: a corpus for information extraction in the biomedical domain. BMC Bioinformatics. 2007;8 doi: 10.1186/1471-2105-8-50. Available at: http://www.biomedcentral.com/1471-2105/8/50/citation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimell L, Clark S. Porting a lexicalized-grammar parser to the biomedical domain. J. Biomed. Inform. 2009;42:852–865. doi: 10.1016/j.jbi.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Saeys Y, et al. Proceedings of the 3rd Machine Learning in Systems Biology workshop (MLSB) 2009. Integrated network construction using event based text mining; pp. 105–14. Available at: http://jmlr.csail.mit.edu/proceedings/ [Google Scholar]
- Smith L, et al. Overview of BioCreative II gene mention recognition. Genome Biol. 2008;9(Suppl. 2):S2. doi: 10.1186/gb-2008-9-s2-s2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe L, et al. GENETAG: a tagged corpus for gene/protein named entity recognition. BMC Bioinformatics. 2005;6(Suppl. 1):S3. doi: 10.1186/1471-2105-6-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateisi Y, et al. Proceedings of the IJCNLP 2005, Companion volume. Korea: 2005. Syntax annotation for the GENIA corpus; pp. 222–227. [Google Scholar]
- Tomanek K, et al. Proceedings of the 10th Conference of the Pacific Association for Computational Linguistics. Melbourne, Australia: 2007. Sentence and token splitting based on conditional random fields; pp. 49–57. [Google Scholar]
- Tsochantaridis I, et al. Large margin methods for structured and interdependent output variables. J. Mach. Learn. Res. 2005;6:1453–1484. [Google Scholar]
- Wang Y, et al. Investigating heterogeneous protein annotations toward cross-corpora utilization. BMC Bioinformatics. 2009;10:403. doi: 10.1186/1471-2105-10-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh A, et al. BioCreAtIvE task 1A: gene mention finding evaluation. BMC Bioinformatics. 2005;6(Suppl. 1):S2. doi: 10.1186/1471-2105-6-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]