

Abstract
Motivation: Tumorigenesis is an evolutionary process by which tumor cells acquire sequences of mutations leading to increased growth, invasiveness and eventually metastasis. It is hoped that by identifying the common patterns of mutations underlying major cancer sub-types, we can better understand the molecular basis of tumor development and identify new diagnostics and therapeutic targets. This goal has motivated several attempts to apply evolutionary tree reconstruction methods to assays of tumor state. Inference of tumor evolution is in principle aided by the fact that tumors are heterogeneous, retaining remnant populations of different stages along their development along with contaminating healthy cell populations. In practice, though, this heterogeneity complicates interpretation of tumor data because distinct cell types are conflated by common methods for assaying the tumor state. We previously proposed a method to computationally infer cell populations from measures of tumor-wide gene expression through a geometric interpretation of mixture type separation, but this approach deals poorly with noisy and outlier data.
Results: In the present work, we propose a new method to perform tumor mixture separation efficiently and robustly to an experimental error. The method builds on the prior geometric approach but uses a novel objective function allowing for robust fits that greatly reduces the sensitivity to noise and outliers. We further develop an efficient gradient optimization method to optimize this ‘soft geometric unmixing’ objective for measurements of tumor DNA copy numbers assessed by array comparative genomic hybridization (aCGH) data. We show, on a combination of semi-synthetic and real data, that the method yields fast and accurate separation of tumor states.
Conclusions: We have shown a novel objective function and optimization method for the robust separation of tumor sub-types from aCGH data and have shown that the method provides fast, accurate reconstruction of tumor states from mixed samples. Better solutions to this problem can be expected to improve our ability to accurately identify genetic abnormalities in primary tumor samples and to infer patterns of tumor evolution.
Contact: tolliver@cs.cmu.edu
Supplementary information:Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Genomic studies have dramatically improved our understanding of the biology of tumor formation and treatment. In part, this has been accomplished by harnessing tools that profile the genes and proteins in tumor cells, revealing previously indistinguishable tumor sub-types that are likely to exhibit distinct sensitivities to treatment methods (Golub et al., 1999; Perou et al., 2000; Sorlie et al., 2001, 2003). As these tumor sub-types are uncovered, it becomes possible to develop novel therapeutics more specifically targeted to the particular genetic defects that cause each cancer (Atkins and Gershell, 2002; Bild et al., 2006; Pegram et al., 2000). While recent advances have had a profound impact on our understanding of the tumor biology, the limits of our understanding of the molecular nature of cancer obstruct the burgeoning efforts in ‘targeted therapeutics’ development. These limitations are apparent in the high failure rate of the discovery pipeline for novel cancer therapeutics (Kamb et al., 2007) as well as in the continuing difficulty of predicting which patients will respond to a given therapeutic. A striking example is the fact that traztuzumab, the targeted therapeutic developed to treat HER2-amplified breast cancers, is ineffective in many patients who have HER2-overexpressing tumors and yet effective in some who do not (Paik et al., 2008). Furthermore, sub-types typically remain poorly defined—e.g. the ‘basal-like’ breast cancer sub-type, for which different studies have inferred very distinct genetic signatures (Perou et al., 2000; Sorlie et al., 2001; Sotiriou et al., 2003)—and yet many patients do not fall into any known sub-type. Our belief, then, is that clinical treatment of cancer will reap considerable benefit from the identification of new cancer sub-types and genetic signatures.
One promising approach for better elucidating the common mutational patterns by which tumors develop is to recognize that tumor development is an evolutionary process and apply phylogenetic methods to tumor data to reveal these evolutionary relationships. Much of the work on tumor evolution models flows from the seminal efforts of Desper et al. (1999) on inferring oncogenetic trees from array comparative genomic hybridization (aCGH) profiles of tumor cells. The strength of this model stems from the extraction of ancestral structure from many probe sites per tumor, potentially utilizing measurements of the expression or copy number changes across the entire genome. However, this comes at the cost of overlooking the diversity of cell populations within tumors, which can provide important clues to tumor progression but are conflated with one another in tissue-wide assays such as aCGH.
The cell-by-cell approaches, such as Pennington et al. (2007); Shackney et al. (2004), use this heterogeneity information but at the cost of allowing only a small number of probes per cell. In recent work, Schwartz and Shackney (2010) proposed bridging the gap between these two methodologies by computationally inferring cell populations from tissue-wide gene expression samples. This inference was accomplished through ‘geometric unmixing,’ a mathematical formalism of the problem of separating components of mixed samples in which each observation is presumed to be an unknown convex combination1 of several hidden fundamental components. Other approaches to inferring common pathways include mixture models of oncogenetic trees (Beerenwinkel et al., 2005), principle component analysis (PCA)-based methods (Hglund et al., 2001), conjunctive Bayesian networks (Gerstung et al., 2009) and clustering (Liu et al., 2006).
Unmixing falls into the class of methods that seek to recover a set of pure sources from a set of mixed observations. Analogous problems have been coined ‘the cocktail problem,’ ‘blind source separation’ and ‘component analysis’ and various communities have formalized a menagerie of models with distinct statistical assumptions. In a broad sense, the classical approach of PCA (Pearson, 1901) seeks to factor the data under the constraint that, collectively, the fundamental components form an orthonormal system. Independent component analysis (ICA; Comon, 1994) seeks a set of statistically independent fundamental components. These methods, and their ilk, have been extended to represent non-linear data distributions through the use of kernel methods (see Schölkopf and Smola, 2002; Schölkopf et al., 1998, for details), which often confound modeling with black-box data transformations. Both PCA and ICA break down as pure source separators when the sources exhibit a modest degree of correlation. Collectively, these methods place strong independence constraints on the fundamental components that are unlikely to hold for tumor samples, where we expect components to correspond to closely related cell states.
The structure of our present inference problem, that of extracting multiple correlated fundamental components, has motivated the development of new methods for unmixing genetic data. Similar unmixing methods were first developed for tumor samples by Billheimer and colleagues (Etzioni et al., 2005) to improve the power of statistical tests on tumor samples in the presence of contaminating stromal cells. Similarly, a hidden Markov model approach to unmixing was developed by Lamy et al. (2007) to correct for stromal contamination in DNA copy number data. These recent advances demonstrate the feasibility of unmixing-based approaches for separating cell sub-populations in tumor data. Outside the bioinformatics community, geometric unmixing has been successfully applied in the geo-sciences (Ehrlich and Full, 1987) and in hyper-spectral image analysis (Chan et al., 2009).
The recent work by Schwartz and Shackney (2010) applied the hard geometric unmixing model (see Section 2.1.1) to gene expression data with the goal of recovering expression signatures of tumor cell sub-types, with the specific goal of facilitating phylogenetic analysis of tumors. The results showed promise in identifying meaningful sub-populations and improving phylogenetic inferences. They were, however, hampered by limitations of the hard geometric approach, particularly the sensitivity to experimental error and outlier data points caused by the simplex fitting approach. An example of simplex fitting in the plane is shown in Figure 1, illustrating why the strict containment model used in Chan et al. (2009); Ehrlich and Full (1987); Schwartz and Shackney (2010) is extremely sensitive to the noise in data. In the present work, we introduce a soft geometric unmixing model (see Section 2.1.2) for tumor mixture separation, which relaxes the requirement for strict containment using a fitting criterion that is robust to noisy measurements. We develop a formalization of the problem and derive an efficient gradient-based optimization method. We develop this method specifically for analyzing tissue-wide DNA copy number data as assessed by aCGH data. We demonstrate the value of the soft unmixing model by comparison to a hard unmixing method on synthetic and real aCGH data. We apply our method to an aCGH dataset taken from Navin et al. (2010) and show that the method identifies state sets corresponding to known sub-types consistent with much of the analysis performed by the authors.
Fig. 1.

(A) The minimum area fit of a simplex containing the sample points in the plane (shown in black) using the program in Section 2.1.1. On noiseless data, hard geometric unmixing recovers the locations of the fundamental components at the vertices. (B) However, the containment simplex is highly sensitive to noise and outliers in the data. A single outlier, circled above, radically changes the shape of the containment simplex fit (light gray above). In turn, this changes the estimates of basis distributions used to unmix the data. We mitigate this short coming by developing a soft geometric unmixing model (see Section 2.1.2) that is comparatively robust to noise. The soft fit (shown dark gray) is geometrically very close to the generating sources as seen on the left.
2 APPROACH
The data are assumed to be given as g genes sampled in s tumors or tumor sections. The samples are collected in a matrix, M∈ℜg×s, in which each row corresponds to an estimate of gene copy number across the sample population obtained with aCGH. The data in M are processed as raw or baseline normalized raw input, rather than as log ratios. The ‘unmixing’ model, described below, asserts that each sample mi, a column of M, is well approximated by a convex combination of a fixed set C = [c0|…|ck] of k+1 unobserved basis distributions over the gene measurements. Further, the observed measurements are assumed to be perturbed by additive noise in the log domain, i.e.
where Fi is the vector of coefficients for the convex combination of the (k + 1) basis distributions and η the additive zero mode i.i.d. noise.
2.1 Algorithms and assumptions
Given the data model above, the inference procedure seeks to recover the k + 1 distributions over gene-copy number or expression that ‘unmix’ the data. The procedure contains three primary stages
Compute a reduced representation xi for each sample mi;
Estimate the basis distributions Kmin in the reduced coordinates and the mixture fractions F;
Map the reduced coordinates Kmin back into the ‘gene space,’ recovering C.
The second step in the method is performed by optimizing the objective in Section 2.1.1 or the robust problem formulation in Section 2.1.2.
Obtaining the reduced representation: We begin our calculations by projecting the data into a k-dimension vector space (i.e. the intrinsic dimensionality of a (k + 1)−vertex simplex). We accomplish this using PCA (Pearson, 1901), which decomposes the input matrix M into a set of orthogonal basis vectors of maximum variance and retain only the k components of highest variance. PCA transforms the g × s measurement matrix M into a linear combination as MT = XV + A, where V is a matrix of the principal components of M, X provides a representation of each input sample as a linear combination of the components of V and A is a s × g matrix in which each row contains g copies of the mean value of the corresponding row of MT. Thus, the matrix X provides a reduced-dimension representation of M, and becomes the input to the sample mixture identification method in Stage 2. V and A are retained to allow us to later construct estimated aCGH vectors corresponding to the inferred mixture components in the original dimension g.
Assuming the generative model of the data above, PCA typically recovers a sensible reduced representation, as low magnitude log additive noise induces ‘shot-noise’ behavior in the subspace containing the simplex with small perturbations in the orthogonal complement subspace. An illustration of this stage of our algorithm can be found in Figure 2.
Fig. 2.

An illustration of the reduced coordinates under the unmixing hypothesis: points (show in gray) sampled from the 3—simplex embedded are ℜ3 and then perturbed by log-normal noise, producing points shown in black with sample correspondence given the green arrows. Note that the dominant subspace remains in the planar variation induced by the simplex, and a 2D reduced representation for simplex fitting is thus sufficient.
Sample mixture identification: Stage 2 invokes either a hard geometric unmixing method that seeks the minimum volume simplex enclosing the input point set X (Program 1) or a soft geometric unmixing method that fits a simplex to the points balancing the desire for a compact simplex with that for containment of the input point set (Program 2). For this purpose, we place a prior over simplexes, preferring those with small volume that fit or enclose the point set of X. This prior captures the intuition that the most plausible set of components explaining a given dataset are those that can explain as much as possible of the observed data while leaving in the simplex as little empty volume, corresponding to mixtures that could be but are not observed, as possible.
Upon completion, Stage 2 obtains estimates of the vertex locations Kmin, representing the inferred cell types from the aCGH data in reduced coordinates, and a set of mixture fractions describing the amount of each observed tumor sample attributed to each mixture component. The mixture fractions are encoded in a (k + 1) × s matrix F, in which each column corresponds to the inferred mixture fractions of one observed tumor sample and each row corresponds to the amount of a single component attributed to all tumor samples. We define Fij to be the fraction of component i assigned to tumor sample j and Fj to be vector of all mixture fractions assigned to a given tumor sample j. To ensure that the observations are modeled as convex combinations of the basis vertices, we require that F1 = 1.
Cell type identification: The reduced coordinate components from Stage 2, Kmin, are projected up to a g × (k + 1) matrix C in which each column corresponds to one of the k + 1 inferred components and each row corresponds to the approximate copy number of a single probe in a component. We perform this transformation using the matrices V and A produced by PCA in Stage 1 with the formula C = VT Kmin + A, augmenting the average to k + 1 columns.
Finally, the complete inference procedure is summarized in the following pseudocode:
Given tumor sample matrix M, the desired number of mixture components k, and the strength of the volume prior γ:
Factor the sample matrix M such that MT = XV + A;
Produce the reduced k−dimensional representation by retaining the top k components in X:
Minimize Program 1, obtaining an estimate of the simplex Kmin0;
Minimize Program 1 starting at Kmin0, obtaining Kmin and F;
Obtain the centers C in gene space as C = A + VT Kmin
2.1.1 Hard geometric unmixing
Hard geometric unmixing is equivalent to finding a minimum volume (k + 1)−simplex containing a set of s points {X} in ℜk. A non-linear program for hard geometric unmixing can be written as follows:
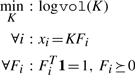 |
(1) |
where logvol measures the volume of simplex defined by the vertices K ≐ [v0|…|vk] and F ⪰ 0 requires that ∀ij. Fij ≥ 0. Collectively, the constraints ensure that each point be expressed exactly as a unique convex combination of the vertices. Exact non-negative matrix factorization (NNMF), see Lee and Seung (1999), can be seen as a relaxation of hard geometric unmixing. Exact NNMF retains the constraint Fi ⪰ 0 while omitting the constraint that the columns sum to unity—thus admitting all positive combinations rather than the restriction to convex combinations as is the case for geometric unmixing.
Approximate and exponential-time exact minimizers are available for Program 1. In our experiments, we use the approach of Chan et al. (2009), which sacrifices some measure of accuracy for efficiency.
2.1.2 Soft geometric unmixing
Estimates of the target distributions, derived from the fundamental components (simplex vertices), produced by hard geometric unmixing are sensitive to the wide-spectrum noise and outliers characteristic of log-additive noise (i.e. multiplicative noise in the linear domain). The robust formulation below tolerates noise in the sample measurements mi and subsequently in the reduced representations xi, improving the stability of these estimates. The sensitivity of hard geometric unmixing is illustrated in Figure 1. The motivation for soft geometric unmixing is to provide some tolerance to experimental error and outliers by relaxing the constraints in Program 1, allowing points to lie outside the boundary of the simplex fit to the data. We extend Program 1 to provide a robust formulation as follows:
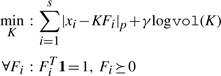 |
(2) |
where the term |xi − KFi|p penalizes the imprecise fit of the simplex to the data and γ establishes the strength of the minimum-volume prior. Optimization of Program 2 is seeded with an estimate produced from Program 1 and refined using the MATLAB's fminsearch with analytical derivatives for the logvol term and an LP-step that determines mixture components Fi and the distance to the boundary for each point outside the simplex.
We observe that when taken as whole, Program 2 can be interpreted as the negative log likelihood of a Bayesian model of signal formation. In the case of array CGH data, we choose p = 1 (i.e. optimizing relative to an ℓ1 norm), as we observe that the errors may be induced by outliers and the ℓ1 norm would provide a relatively modest penalty for a few points far from the simplex. From the Bayesian perspective, this is equivalent to relaxing the noise model to assume i.i.d. heavy-tailed additive noise. To mitigate some of the more pernicious effects of log-normal noise, we also apply a total variation-like smoother to aCGH data in our experiments. Additionally, the method can be readily extended to weighted norms if an explicit outlier model is available.
2.1.3 Analysis and Efficiency
The hard geometric unmixing problem in Section 2.1.1 is a non-convex objective in the present parameterization, and was shown by Packer (2002) to be NP-hard when k + 1 ≥ log(s). For the special case of minimum volume tetrahedra (k = 3), Zhou and Suri (2000) demonstrated an exact algorithm with time complexity Θ(s4) and a (1+ε) approximate method with complexity O(s+1/ε6). Below, we examine the present definition and show that Programs 1 and 2 have structural properties that may be exploited to construct efficient gradient-based methods that seek local minima. Such gradient methods can be applied in lieu of or after heuristic or approximate combinatorial methods for minimizing Program 1, such as Chan et al. (2009) Ehrlich and Full (1987) or the (1 + ε) method of Zhou and Suri (2000) for simplexes in ℜ3.
We begin by studying the volume penalization term as it appears in both procedures. The volume of a convex body is well known (see Boyd and Vandenberghe, 2004) to be a log concave function. In the case of a simplex, analytic partial derivatives with respect to vertex position can be used to speed the estimation of the minimum volume configuration Kmin. The volume of a simplex, represented by the vertex matrix K = [v0|…|vk], can be calculated as
| (2) |
where ck is the volume of the unit simplex defined on k + 1 points and Γ a fixed vertex-edge incidence matrix such that ΓT K = [v1 − v0|…|vk − v0]. The matrix Q is an inner product matrix over the vectors from the special vertex v0 to each of the remaining k vertices. In the case where the simplex K is non-degenerate, these vectors form a linearly independent set and Q is positive definite (PD). While the determinant is log concave over PD matrices, our parameterization is linear over the matrices K, not Q. Thus, it is possible to generate a degenerate simplex when interpolating between the two non-degenerate simplexes K and K′. For example, let K define a triangle with two vertices on the y-axis and produce a new simplex K′ by reflecting the triangle K across the y-axis. The curve K(α)=αK + (1 − α)K′ linearly interpolates between the two. Clearly, when α=1/2, all three vertices of K(α) are co-linear and thus the matrix Q is not full rank and the determinant vanishes. However, in the case of small perturbations, we can expect the simplexes to remain non-degenerate.
To derive the partial derivative, we begin by substituting the determinant formulation into our volume penalization and arrive at the following calculation:
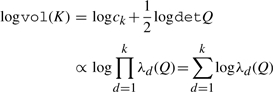 |
therefore, the gradient of logvol(K) is given by
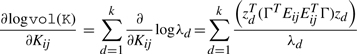 |
where the eigenvector zd satisfies the equality Qzd = λdzd and Eij is the indicator matrix for the entry ij. To minimize the volume, we move the vertices along the paths specified by the negative log gradient of the current simplex volume. The Hessian is derived by an analogous computation, making Newton's method for Program 1, with log barriers over the equality and inequality constraints, a possible optimization strategy.
Soft geometric unmixing (Program 2) trades the equality constraints in Program 1 for a convex, but non-differentiable, term in the objective function ∑i=1s |xi − KFi|p for p|1≤p≤2. Intuitively, points inside the simplex have no impact on the cost of the fit. However, over the course of the optimization, as the shape of the simplex changes points move from the interior to the exterior, at which time they incur a cost. To determine this cost, we solve the non-negative least squares problem for each mixture fraction Fi, minF : (KFi − xi)T (KFi − xi). This step simultaneously solves for the mixture fraction, and for exterior points, the distance to the simplex is determined. The simplex is then shifted under a standard shrinkage method based on these distances.
3 EXPERIMENTAL METHODS
We evaluated our methods using synthetic experiments, allowing us to assess two properties of robust unmixing (1) the fidelity with which endmembers (sub-types) are identified and (2) the relative effect of noise on hard versus robust unmixing. We then evaluate the robust method on a real-world aCGH dataset published by Navin et al. (2010) in which ground truth is not available, but for which we uncover much the structure reported by the authors.
3.1 Methods: synthetic experiments
To test the algorithms given in Section 2, we simulated data using a biologically plausible model of ad-mixtures. Simulated data provides a quantitative means of evaluation as ground truth is available for both the components C and the mixture fractions Fi associated with each measurement in the synthetic design matrix M. The tests evaluate and compare hard geometric unmixing (Section 2.1.1) and soft geometric unmixing (Section 2.1.2) in the presence of varying levels of log-additive Gaussian noise and varying k. By applying additive Gaussian noise in the log domain, we simulate the heteroscedasticity characteristic of CGH measurements (i.e. higher variance with larger magnitude measurements). By varying k, the dimensionality of the simplex used to fit the data, we assess the algorithmic sensitivity to this parameter as well as that to γ governing the strength of the volume prior in Program 2. The sample generation process consists of three major steps: (1) mixture fraction generation (determining the ratio of sub-types present in a sample); (2) end-member (i.e. sub-type) generation; and (3) the sample perturbation by additive noise in the log-ratio domain.
3.1.1 Mixture sampler
Samples over mixture fractions were generated in a manner analogous to the Polya's Urn Process, in which previously sampled simplicial components (e.g. line segments, triangles and tetrahedra) are more likely to be sampled again. This sampling mechanism produces data distributions that are similar to those we see in low-dimensional projections of aCGH data when compared against purely uniform samples over mixtures. An example of a low-dimensional sample set and the simplex that was used to generate the points is shown in Figure 3.
Fig. 3.
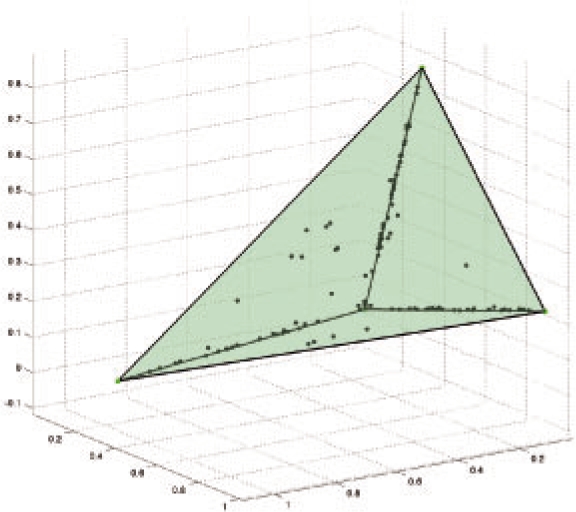
An example sample set generated for Section 3.1.2 shown in the ‘intrinsic dimensions’ of the model. Note that sample points cleave to the lower dimensional substructure (edges) of the simplex.
To generate the mixture fractions Fi for the i-th sample, the individual components in Ctrue are sampled without replacement from a dynamic tree model. Each node in the tree contains a dynamic distribution over the remaining components, each of which is initialized to the uniform distribution. We then sample s mixtures by choosing an initial component according to the root's component distribution and proceed down the tree. As a tree-node is reached, its component distribution is updated to reflect the frequency with which its children are drawn. To generate the i-th sample, the fractional values Fi are initialized to zero. As sample generation proceeds, the currently selected component Cj updates the mixture as Fij ∼ uniform[(1/2)fjp, 1] where fpj is the frequency of j's parent node. For the i-th mixture, this process terminates when the condition 1 ≤ ∑j=1k+1 Fij holds. Therefore, samples generated by long paths in the tree will tend to be homogenous combinations of the components Ctrue, whereas short paths will produce lower dimensional substructures. At the end of the process, the matrix of fractions F is re-normalized so that the mixtures associated with each sample sum to unity. This defines a mixture Fitrue for each sample—i.e. the convex combination over fundamental components generating the sample point.
3.1.2 Geometric sampling of end-members & noise
To determine the locations of the end-members, we specify an extrinsic dimension (number of genes) g, and an intrinsic dimension k (requiring k + 1 components). We then simulate k + 1 components by constructing a g × (k + 1) matrix Ctrue of fundamental components in which each column is an end-member (i.e. sub-type) and each row is the copy number of one hypothetical gene, sampled from the unit Gaussian distribution and rounded to the nearest integer. Samples mi, corresponding to the columns of the data matrix M, are then given by:
| (4) |
where η ∼normal(0, 1) and the mixture fractions Fitrue were obtained as in Section 3.1.1.
3.1.3 Evaluation
We follow Schwartz and Shackney (2010) in assessing the quality of the unmixing methods by independently measuring the accuracy of inferring the components and the mixture fractions. We first match inferred mixture components to true mixture components by performing a maximum weighted bipartite matching of columns between Ctrue and the inferred components Ce, weighted by negative Euclidean distance. We will now assume that the estimates have been permuted according to this matching and continue. We then assess the quality of the mixture component identification by the root mean square distance over all entries of all components between the matched columns of the two C matrices:
| (5) |
where 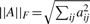 denotes the Frobenius norm of the matrix A.
denotes the Frobenius norm of the matrix A.
We similarly assess the quality of the mixture fractions by the root mean square distance between Ftrue and the inferred fractions Fe over all genes and samples
| (6) |
This process was performed for s = 100 and d = 10000 to approximate a realistic tumor expression dataset and evaluated for k = 3 to k = 7 and for σ = {0, 0.1, 0.2, …, 1.0}, with 10 repetitions per parameter.
4 RESULTS
4.1 Results: synthetic data
The results for the synthetic experiment are summarized in Figure 4. The figure shows the trends in MSE for hard geometric unmixing Section 2.1.1 and soft geometric unmixing Section 2.1.2 on the synthetic data described above. As hard geometric unmixing requires that each sample lie inside the fit simplex, as noise levels increase (larger σ), the fit becomes increasingly inaccurate. Further, the method MVES deteriorates to some degree as order k of the simplex increases. However, soft geometric unmixing degrades more gracefully in the presence of noise if an estimate of the noise level is available with ±0.1 in our current model. The trend of soft unmixing exhibiting lower error and better scaling in k than hard unmixing holds for both components and mixture fractions, although components exhibit a higher average degree of variability due to the scale of the synthetic measurements when compared to the mixture fractions.
Fig. 4.

(A): mean squared error for the component reconstruction comparing hard geometric unmixing (MVES: Chan et al., 2009) and soft geometric unmixing (SGU) introduced in Section 2.1.2 for the experiment described in Section 3.1.2 with variable γ. The plot demonstrates that robust unmixing more accurately reconstructs the ground truth centers relative to hard unmixing in the presence of noise. (B): mean squared error for mixture reconstruction comparing MVES and SGU.
4.2 aCGH data
We further illustrate the performance of our methods on a publicly available primary ductal breast cancer aCGH dataset furnished with Navin et al. (2010). This dataset is of interest in that each tumor sample has been sectored multiple times during biopsy, which is ideal for understanding the substructure of the tumor population. The data consists of 87 aCGH profiles from 14 tumors run on a high-density ROMA platform with 83055 probes. Profiles are derived from 4 to 6 sectors per tumor, with samples for tumors 5–14 sub-partitioned by cell sorting according to the total DNA content, and with healthy control samples for tumors 6, 9, 12 and 13. For full details, the reader is referred to Navin et al. (2010). The processed data consists of log10 ratios, which were exponentiated prior to the PCA step (Stage 1) of the method.
4.2.1 Preprocessing
To mitigate the effects of sensor noise on the geometric inference problem, we apply a total variation (TV) functional to the raw log-domain data. The ℓ1 − ℓ1 −TV minimization is equivalent to a penalized projection onto the over-complete Harr basis preserving a larger degree of the signal variation when compared to discretization methods (e.g. Guha et al., 2006; Olshen et al., 2004) that employ aggressive priors over the data distribution. The procedure seeks a smooth instance x of the observed signal s by optimizing the following functional:
| (7) |
The functional 7 is convex and can be solved readily using the Newton's method with log-barrier functions (Boyd and Vandenberghe, 2004). The solution x can be taken as the maximum likelihood estimate of a Bayesian model of CGH data formation. That is, the above is the negative log-likelihood of a simple Bayesian model of signal formation. The measurements 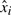 are assumed to be perturbed by the i.i.d. Laplacian noise and the changes along the probe array are assumed to be sparse. Recall that the Laplacian distribution is defined as
are assumed to be perturbed by the i.i.d. Laplacian noise and the changes along the probe array are assumed to be sparse. Recall that the Laplacian distribution is defined as 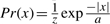 . In all experiments, the strength of the prior λ was set to λ = 10. The data fit this model well, as illustrated in Figure 5. The dimension of the reduced representation k, fixing the number of fundamental components, was determined using the eigengap heuristic during the PCA computation (Stage 1). This rule ceases computing additional principal components when the difference in variances jumps above threshold.
. In all experiments, the strength of the prior λ was set to λ = 10. The data fit this model well, as illustrated in Figure 5. The dimension of the reduced representation k, fixing the number of fundamental components, was determined using the eigengap heuristic during the PCA computation (Stage 1). This rule ceases computing additional principal components when the difference in variances jumps above threshold.
Fig. 5.

Empirical motivation for the ℓ1 − ℓ1−total variation functional for smoothing CGH data. (A) The plot shows the histogram of values found in the CGH data obtained from the Navin et al. (2010) dataset. The distribution is well fit by the high kurtosis Laplacian distribution in lieu of a Gaussian. (B)The plot shows the distribution of differences along the probe array values. As with the values distribution, these frequencies exhibit high kurtosis.
4.2.2 Unmixing analysis and validation
The raw data was preprocessed as described above and a simplex was fit to the reduced coordinate representation using the soft geometric unmixing method (see Section 2.1.2). A 3D visualization of the resulting fit is shown for the Navin et al. (2010) dataset in Figure 6. To assess the performance with increasing dimensionality, we ran experiments for polytope dimensionality k ranging from 3 to 9. Following the eigen-gap heuristic, we chose to analyze the results for k = 6. The γ value was picked according to the estimated noise level in the aCGH dataset and scaled relative to the unit simplex volume (here, γ = 100). The estimated six components/simplex vertices/pure cancer types are labeled C1, C2,…, C6.
Fig. 6.
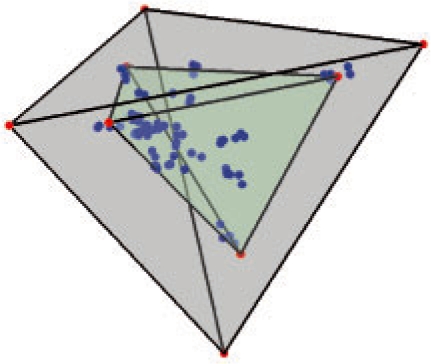
The simplex fit to the CGH data samples from Navin et al. (2010) ductal dataset in ℜ3. The gray tetrahedron was return by the optimization of Program 1 and the green tetrahedron was returned by the robust unmixing routine.
Figure 7 shows mixture fraction assignments for the aCGH data for k = 6. While there is typically a non-zero amount of each component in each sample due to imprecision in assignments, the results nonetheless show distinct subsets of tumors favoring different mixture compositions and with tumor cells clearly differentiated from healthy control samples. The relative consistency within versus between tumors provides a secondary validation that soft unmixing is effective at robustly assigning mixture fractions to tumor samples despite noise inherent to the assay and that produced by sub-sampling cell populations. It is also consistent with observations of Navin et al. (2010)
Fig. 7.

Inferred mixture fractions for six-component soft geometric unmixing applied to breast cancer aCGH data. Data is grouped by tumor, with multiple sectors per tumor placed side-by-side. Columns are annotated below by sector or N for normal control and above by cell sorting fraction (D for diploid, H for hypodiploid, A for aneuploid and A1/A2 for subsets of aneuploid) where cell sorting was used.
It is not possible to know with certainty the true cell components or mixture fractions of the real data, but we can validate the biological plausibility of our results by examining known sites of amplification in the inferred components. We selected 14 benchmark loci frequently amplified in breast cancers through the manual literature search. Table 1 lists the chosen benchmarks and the components exhibiting at least 2-fold amplification of each. Figure 8 visualizes the results, plotting relative amplification of each component as a function of genomic coordinate and highlighting the locations of the benchmark markers. Thirteen of the fourteen benchmark loci exhibit amplification for a subset of the components, although often at minimal levels. The components also show amplification of many other sites not in our benchmark set, but we cannot definitively determine which are true sites of amplification and which are false positives. We further tested for amplification of seven loci reported as amplified by Navin et al. (2010) specifically in the tumors examined here and found that six of the seven are specifically amplified in one of our inferred components: PPP1R12A (C2), KRAS (C2), CDC6 (C2), RARA (C2), EFNA5 (C2), PTPN1 (C3) and LPXN (not detected). Our method did not infer a component corresponding to normal diploid cells as one might expect due to stromal contamination. This failure may reflect a bias introduced by the dataset, in which many samples were cell sorted to specifically select aneuploid cell fractions, or could reflect an inherent bias of the method towards more distinct components, which would tend to favor components with large amplifications.
Table 1.
Benchmark set of breast cancer markers selected for validation of real data, annotated by gene name, genomic locus and the set of components exhibiting amplification at the given marker
| Marker | Locus | Component | Marker | Locus | Component |
|---|---|---|---|---|---|
| MUC1 | 1q21 | C1,C4 | BRCA2 | 13q12.3 | C5 |
| PIK3CA | 3q26.3 | C3,C6 | ESR2 | 14q23 | C1 |
| ESR1 | 6q25.1 | C4 | BRCA1, | 17q21 | C5,C6 |
| EGFR | 7p12 | C5 | ERBB2 | ||
| c-MYC | 8q24 | C1,C3,C5 | STAT5A, | 17q11.2 | C5 |
| PTEN | 10p23 | none | STAT5B | ||
| PGR | 14q23.2 | C6 | GRB7 | 17q12 | C6 |
| CCND1 | 11q13 | C4 | CEA | 19q13.2 | C6 |
Fig. 8.

Copy numbers of inferred components versus genomic position. The average of all input arrays (top) is shown for comparison, with the six components below. Benchmarks loci are indicated by yellow vertical bars.
We repeated these analyses for the hard unmixing with a higher amplification threshold due to the noise levels in the centers. It detected amplification at 11 of the 14 loci, with spurious inferences of deletion at 4 of the 11. For the seven sites reported in Navin et al. (2010), hard unmixing identified five (failing to identify EFNA5 or LPXN) and again made spurious inferences of deletions for three of these sites, an artifact the soft unmixing eliminates. The full results are provided in Supplementary Section S1. The results suggest that hard unmixing produces less precise fits of simplexes to the true data.
We can also provide a secondary analysis based on Navin et al.'s (2010) central result that the tumors can be partitioned into monogenomic (those appearing to show essentially a single genotype) and polygenomic (those that appear to contain multiple tumor sub-populations). We test for monogeniety in mixture fractions by finding the minimum correlation coefficient between mixture fractions of consecutive tumor sectors (ignoring normal controls) maximized over all permutations of the sectors. Those tumors with correlations above the mean over all tumors (0.69) were considered monogenomic and the remainder polygenomic. Navin et al. (2010) assign {1, 2, 6, 7, 9, 11} as monogenomic and {3, 4, 5, 8, 10, 12, 13, 14} as polygenomic. Our tests classify {1, 2, 5, 6, 7, 8, 11} as monogenomic and {3, 4, 10, 12, 13, 14} as polygenomic, disagreeing only in tumors 5 and 8. Our methods are thus effective at identifying true intratumor heterogeneity in almost all cases without introducing spurious heterogeneity. By contrast, hard unmixing identifies only tumors 7 and 8 as polygenomic, generally obscuring true heterogeneity in the tumors (see Supplementary Section S1).
Our long-term goal in this work is not just to identify sub-types, but to describe the evolutionary relationships among them. We have no empirical basis for validating any such predictions at the moment but nonetheless consider the problem informally here for illustrative purposes. To explore the question of possible ancestral relationships among components, we manually examined the most pronounced regions of shared gain across components. Figure 9 shows a condensed view of the six components highlighting several regions of shared amplification between components. The left half of the image shows Components 3, 5 and 1, revealing a region of shared gain across all three components at 9p21 (labeled B). Components 5 and 1 share an additional amplification at 1q21 (labeled A). Components 1 and 5 have distinct but nearby amplifications on chromosome 17, with Component 1 exhibiting amplification at 17q12 (labeled D) and Component 5 at 17q21 (labeled C). We can interpret these images to suggest a possible evolutionary scenario: component 3 initially acquires an amplification at 9p21 (the locus of the gene CDKN2B/p15INK4b), an unobserved descendent of Component 3 acquires secondary amplification at 1q21 (the locus of MUC1), and this descendent then diverges into Components 1 and 5 through acquisition of independent abnormalities at 17q12 (site of PGAP3) or 17q21 (site of HER2). The right side of the figure similarly shows some sharing of sites of amplification between Components 2, 4, and 6, although the amplified regions do not lead to so simple an evolutionary interpretation. The figure is consistent with the notion that Component 2 is ancestral to 4, with Component 2 acquiring a mutation at 5q21 (site of APC/MCC) and Component 4 inheriting that mutation but adding an additional one at 17q21. We would then infer that the amplification at the HER2 locus arose independently in Component 6, as well as in Component 5. The figure thus suggests the possibility that the HER2-amplifying breast cancer sub-type may arise from multiple distinct ancestral backgrounds in different tumors. While we cannot evaluate the accuracy of these evolutionary scenarios, they nonetheless provide an illustration of how the output of this method is intended to be used to make inferences of evolutionary pathways of tumor states.
Fig. 9.

Plot of amplification per probe highlighting regions of shared amplification across components. The lower (blue) dots mark the location of the collected cancer benchmarks set. Bars highlight specific markers of high shared amplification for discussion in the text. Above: A: 1q21 (site of MUC1), B: 9p21 (site of CDKN2B), C: 7q21 (site of HER2), D: 17q12 (site of PGAP3), E: 5q21 (site of APC/MCC).
5 CONCLUSION
We have developed a novel method for unmixing aCGH data to infer copy number profiles of distinct cell states from tumor samples. The method uses ‘soft geometric unmixing’ to provide superior tolerance to experimental noise and outliers compared to the prior work. We have further developed an efficient gradient-based optimization algorithm for this objective function. We have shown through tests on simulated data that the soft unmixing approach dramatically improves accuracy of inference of components and mixture fractions in the presence of high noise or large component numbers relative to a hard unmixing method. We have further verified, with application to a set of real aCGH data from breast cancer patients, that the method is effective at separating components corresponding to distinct subsets of known breast cancer markers. The specific patterns of gain and loss in the components are suggestive of patterns of evolution among the tumor types. Thus, the work demonstrates the potential of tumor sample unmixing applied to aCGH data to infer copy number profiles of cell populations from heterogenous tumor samples. In addition to facilitating studies of tumor evolution, the methods may have value to many other applications of mixture separation from noisy data.
Funding: U.S. National Institutes of Health award # 1R01CA140214.
Conflict of Interest: none declared.
Supplementary Material
Footnotes
1A point p is a convex combination combination of basis points v0,…, vk if and only if the constraints p = ∑i=0k αi vi, ∑i αi = 1 and ∀i : αi ≥ 0 obtain. The fractions αi determine a mixture over the basis points {vi} that produce the location p.
REFERENCES
- Atkins JH, Gershell LJ. From the analyst's couch: selective anticancer drugs. Nat. Rev. Cancer. 2002;2:645–646. doi: 10.1038/nrd842. [DOI] [PubMed] [Google Scholar]
- Beerenwinkel N, et al. Mtreemix: a software package for learning and using mixture models of mutagenetic trees. Bioinformatics. 2005;21:2106–2107. doi: 10.1093/bioinformatics/bti274. [DOI] [PubMed] [Google Scholar]
- Bild AH, et al. Opinion: linking oncogenic pathways with therapeutic opportunities. Nat. Rev. Cancer. 2006;6:735–741. doi: 10.1038/nrc1976. [DOI] [PubMed] [Google Scholar]
- Boyd S, Vandenberghe L. Convex Optimization. New York, NY: Cambridge University Press; 2004. [Google Scholar]
- Chan T, et al. A convex analysis based minimum-volume enclosing simplex algorithm for hyperspectral unmixing. IEEE Trans. Signal Proc. 2009;57:4418–4432. [Google Scholar]
- Comon P. Independent component analysis. Signal Proc. 1994;36:287–314. [Google Scholar]
- Desper R, et al. Inferring tree models for oncogenesis from comparative genome hybridization data. J. Comp. Biol. 1999;6:37–51. doi: 10.1089/cmb.1999.6.37. [DOI] [PubMed] [Google Scholar]
- Ehrlich R, Full W. Use and Abuse of Statistical Methods in the Earth Sciences. Oxford University Press; 1987. Sorting out geology — unmixing mixtures; pp. 33–46. [Google Scholar]
- Etzioni R, et al. Analyzing patterns of staining in immunohistochemical studies: application to a study of prostate cancer recurrence. Cancer Epidemiol Biomarkers Prev. 2005;14:1040–1046. doi: 10.1158/1055-9965.EPI-04-0584. [DOI] [PubMed] [Google Scholar]
- Gerstung M, et al. Quantifying cancer progression with conjunctive Bayesian networks. Bioinformatics. 2009;25:2809–2815. doi: 10.1093/bioinformatics/btp505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub TR, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Guha S, et al. Bayesian hidden Markov modeling of array CGH data. Paper 24. Harvard University; 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hglund M, et al. Multivariate analyses of genomic imbalances in solid tumors reveal distinct and converging pathways of karyotypic evolution. Genes Chromosomes Cancer. 2001 doi: 10.1002/gcc.1129. [DOI] [PubMed] [Google Scholar]
- Kamb A, et al. Why is cancer drug discovery so difficult? Nat. Rev. Drug Discov. 2007;6:115–120. doi: 10.1038/nrd2155. [DOI] [PubMed] [Google Scholar]
- Lamy P, et al. A hidden Markov model to estimate population mixture and allelic copy-numbers in cancers using Affymetrix SNP arrays. BMC Bioinformatics. 2007;8:434. doi: 10.1186/1471-2105-8-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Seung H. Learning the parts of objects by non-negative matrix factorization. Nature. 1999;401:788–791. doi: 10.1038/44565. [DOI] [PubMed] [Google Scholar]
- Liu J, et al. Distance-based clustering of CGH data. Bioinformatics. 2006;22:1971–1978. doi: 10.1093/bioinformatics/btl185. [DOI] [PubMed] [Google Scholar]
- Navin N, et al. Inferring tumor progression from genomic heterogeneity. Genome Res. 2010;20:68–80. doi: 10.1101/gr.099622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshen AB, et al. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- Packer A. NP-hardness of largest contained and smallest containing simplices for v- and h-polytopes. Discrete Comput. Geom. 2002;28:349–377. [Google Scholar]
- Paik S, et al. Her2 status and benefit from adjuvant trastuzumab in breast cancer. N. Engl. J. Med. 2008;358:1409–1411. doi: 10.1056/NEJMc0801440. [DOI] [PubMed] [Google Scholar]
- Pearson K. On lines and planes of closest fit to systems of points in space. Philos. Mag. 1901;2:559–572. [Google Scholar]
- Pegram MD, et al. The molecular and cellular biology of her2/neu gene amplification/overexpression and the clinical development of herceptin (trastuzumab) therapy for breast cancer. Cancer Treat. Res. 2000;103:57–75. doi: 10.1007/978-1-4757-3147-7_4. [DOI] [PubMed] [Google Scholar]
- Pennington G, et al. Reconstructing tumor phylogenies from single-cell data. J. Bioinform. Comput. Biol. 2007;5:407–427. doi: 10.1142/s021972000700259x. [DOI] [PubMed] [Google Scholar]
- Perou CM, et al. Molecular portraits of human breast tumors. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Schölkopf B, Smola AJ. Learning with Kernels: Support Vector Machines, Regularization, Optimization, and Beyond. MIT Press; 2002. [Google Scholar]
- Schölkopf B, et al. Nonlinear component analysis as a kernel eigenvalue problem. Neural Comput. 1998;10:1299–1319. [Google Scholar]
- Schwartz R, Shackney S. Applying unmixing to gene expression data for tumor phylogeny inference. BMC Bioinformatics. 2010;11:42. doi: 10.1186/1471-2105-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackney SE, et al. Intracellular patterns of Her-2/neu, ras, and ploidy abnormalities in primary human breast cancers predict postoperative clinical disease-free survival. Clin. Cancer Res. 2004;10:3042–3052. doi: 10.1158/1078-0432.ccr-0401-3. [DOI] [PubMed] [Google Scholar]
- Sorlie T, et al. Gene expression profiles of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl Acad. Sci. USA. 2001;98:10869–10864. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie T, et al. Repeated observation of breast tumor subtypes in indepednent gene expression data sets. Proc. Natl Acad. Sci. USA. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiriou C, et al. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc. Natl Acad. Sci. USA. 2003;100:10393–10398. doi: 10.1073/pnas.1732912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Suri S. Algorithms for minimum volume enclosing simplex in R3. Proceedings of the Eleventh Annual ACM/SIAM Symposium on Discrete Algorithms. 2000:500–509. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.