

Abstract
Motivation: Haplotype inference is an important step for many types of analyses of genetic variation in the human genome. Traditional approaches for obtaining haplotypes involve collecting genotype information from a population of individuals and then applying a haplotype inference algorithm. The development of high-throughput sequencing technologies allows for an alternative strategy to obtain haplotypes by combining sequence fragments. The problem of ‘haplotype assembly’ is the problem of assembling the two haplotypes for a chromosome given the collection of such fragments, or reads, and their locations in the haplotypes, which are pre-determined by mapping the reads to a reference genome. Errors in reads significantly increase the difficulty of the problem and it has been shown that the problem is NP-hard even for reads of length 2. Existing greedy and stochastic algorithms are not guaranteed to find the optimal solutions for the haplotype assembly problem.
Results: In this article, we proposed a dynamic programming algorithm that is able to assemble the haplotypes optimally with time complexity O(m × 2k × n), where m is the number of reads, k is the length of the longest read and n is the total number of SNPs in the haplotypes. We also reduce the haplotype assembly problem into the maximum satisfiability problem that can often be solved optimally even when k is large. Taking advantage of the efficiency of our algorithm, we perform simulation experiments demonstrating that the assembly of haplotypes using reads of length typical of the current sequencing technologies is not practical. However, we demonstrate that the combination of this approach and the traditional haplotype phasing approaches allow us to practically construct haplotypes containing both common and rare variants.
Contact: danhe@cs.ucla.edu
1 INTRODUCTION
Obtaining haplotypes, or the sequence of alleles on each chromosome, is an important step for many types of analyses of genetic variation in the human genomes. In particular, haplotype inference is required for the application of many imputation algorithms (Marchini et al., 2007), which are now widely applied in the analysis of genome-wide association studies.
The standard approach for obtaining haplotype information involves collecting genotype data from a population of individuals. Genotype data contains information on the set of alleles at each locus, but lacks information on which chromosome a particular allele occurs on. Computational methods are then applied to these genotype data to infer the haplotypes (Browning and Browning, 2008; Halperin and Eskin, 2004; Stephens et al., 2001). These methods take advantage of the fact that alleles at neighboring loci in the genomes are correlated or are ‘in linkage disequilibrium’ (LD), as well as the fact that in any given region only a few common haplotypes account for the majority of the genetic variations in the population. Due to their reliance on LD, these methods have difficulty inferring haplotypes with rare variants and have no ability to infer haplotypes for alleles that are unique to an individual.
Recently, the development of high-throughput sequencing (HTS) technology has enabled an alternative strategy to obtain haplotypes. Since each sequence read is from a single chromosome, if a read covers two variant sites, all of the alleles present in the read must be from the same haplotype. Using this insight, it is possible to assemble the two haplotypes for a chromosome from the collection of such reads by joining reads that share alleles at common variants. The problem is referred to as ‘haplotype assembly’ (Lancia et al., 2001), which is challenging in the following two aspects:
Reads are sampled from either of the two haplotypes and no information is given about which one they come from. The reads need to be separated for the two haplotypes in the assembly process.
Errors in reads significantly increase the difficulty of the problem and it has been shown that the problem is NP-hard even for reads of length 2 (Cilibrasi et al., 2005; Lancia et al., 2001).
A simple greedy heuristic method (Levy et al., 2007) (which we call the Greedy algorithm), concatenates the reads with minimum conflicts and is fast but not very accurate when reads contain errors. Other stochastic algorithms, such as HASH (Bansal et al., 2008), which is a Markov chain Monte Carlo (MCMC) algorithm, and HapCut (Bansal and Bafna, 2008), which is a combinatorial approach, have been shown to be much more accurate than the Greedy algorithm on the HuRef diploid genome sequence (Levy et al., 2007).
However, both HASH and HapCut algorithms use stochastic strategies and therefore are not guaranteed to find optimal solutions for the haplotype assembly problem. In this article, we propose a dynamic programming algorithm that is able to assemble the haplotypes optimally with time complexity O(m × 2k × n), where m is the number of reads, k is the length of the longest read and n is the total number of heterozygous sites in the haplotypes. Since this time complexity is exponential in k, we reduce the problem to the maximum satisfiability (MaxSAT) problem for cases where k is large. MaxSAT conversion is a well-known strategy for many computational biology problem such as SNP Tagging (Choi et al., 2008). The converted MaxSAT problem can often be solved optimally in a reasonable amount of time with an MaxSAT solver. Our experiments show that the MaxSAT approach can solve 99.98% instances of the converted haplotype assembly problem optimally. We also show for the first time that the current best-known solution is only 1.1% from the optimal solution and our solution is the best result that has yet been achieved.
Taking advantage of the efficiency and optimality of our method, we are able to perform simulation experiments to evalute the feasibility of assembling haplotypes using sequence reads with the length typical of the current high-throughput technologies. The current sequencing technologies are able to collect paired-end reads, where sequences of two segments are obtained separated by an approximate distance (insert length). Our experiments show that the insert length and in particular the variability in the insert length play a crucial role in our ability to assemble haplotypes. Using data from HapMap (International HapMap Consortium, 2007) we demonstrate that using current HTS technologies, the assembly of reads into haplotypes is impractical. However, we show that combining haplotype assembly from sequencing with traditional approaches for inferring haplotypes using genotypes can effectively recover haplotypes for both common and rare alleles.
2 RELATED WORK
The haplotype assembly problem was first introduced by Lancia et al. (2001). They show that the problem is computationally challenging when reads contain errors, since the reads can not be partitioned perfectly into two disjoint sets. Therefore, various combinatorial objective functions have been proposed (Lancia et al., 2001; Lippert et al., 2002) to define the best reconstruction of haplotypes such as minimum fragment removal (MFR), minimum error correction (MEC), minimum SNP removal (MSR), minimum implicit SNP removal (MISR), minimum implicit fragment removal (MIFR). Out of these objective functions, MEC, which is the number of conflicts between the sequence reads and the constructed haplotypes, is the most difficult one to optimize. The haplotype assembly problem with MEC as the object function is NP-hard even for gapless reads of length 2, while polynomial algorithms exist for solving the problem with MFR and MSR as the objective function (Cilibrasi et al., 2005; Lippert et al., 2002). Several heuristic and stochastic methods (Bansal and Bafna, 2008; Bansal et al., 2008; Levy et al., 2007; Panconesi and Sozio, 2004; Wang et al., 2005) have been proposed to optimize MEC for gapped reads. In this article we also focus on minimizing MEC. Therefore, the ‘haplotype assembly’ problem can be defined as following: given a set of reads that may contain errors, reconstruct the pair of haplotypes by partitioning the reads to either haplotype such that the MEC is minimized.
The Greedy heuristic algorithm (Levy et al., 2007), which concatenates the reads with minimum conflicts, is able to construct optimal haplotypes very quickly if the reads are error-free. When there are errors in the reads, the Greedy algorithm usually outputs much worse results than the optimal solution. HASH (Bansal et al., 2008) and HapCut (Bansal and Bafna, 2008) algorithms are both based on the idea of building a graph from the read matrix, where each row corresponds to a read and each column corresponds to a position of the haplotype. In the graph, each column is a node and an edge between two nodes is created if there is a read spanning the corresponding two columns. The weights of the edges are determined by the number of reads that are consistent with the haplotypes minus the number of reads that are in conflict with the haplotypes in the two columns. The HASH algorithm uses graph cut computations to construct the Markov chain used for sampling the haplotype space. HapCut uses Max-Cut computations in an associated graph to greedily move towards the optimal MEC solution. Both HASH and HapCut algorithms obtain much more accurate haplotypes than the Greedy algorithm. Since convergence of Markov chain is slow, HapCut is much faster than HASH with almost the same accuracy.
Although HASH and HapCut achieve reasonably good results, they are stochastic and therefore can not guarantee optimal solutions. In this article we propose a dynamic programming algorithm that is able to find the optimal solution with time complexity O(m × 2k × n) for gapless reads, where m is the number of reads, k is the length of the longest read and n is the total number of SNPs in the haplotypes. The time complexity implies that this algorithm is most effective when k is relatively small. Since only heterozygous sites in the reads are considered for assembly, and the number of heterozygous sites, or variants, is small, and these sites are often far from one another, most of the reads only cover a small number of heterozygous sites. The dynamic programming algorithm is computationally practical for up to reads of length 15, where length is defined as the number of heterozygous sites covered by the read. We later show that in the HuRef data, >90% of the reads are of length less than 15, which indicates the dynamic programming algorithm is practical for the majority of the data. In our experiments, the run time of the dynamic programming algorithm we propose here is comparable to that of the HapCut algorithm. Furthermore, to handle reads of length greater than 15, we propose to take advantage of recent advances in the field of logical reasoning, by modeling the haplotype problem as an MaxSAT problem. We show that modern MaxSAT solvers are powerful enough to solve such haplotype problems optimally in practice.
3 METHODS
3.1 The Haplotype assembly problem
We will follow the notation by Bansal and Bafna (2008) for the haplotype assembly problem. Given a reference genome sequence and the set of reads containing sequence from both chromosome, after aligning all the reads to the reference genome (Li et al., 2008), the homozygous sites (columns in the alignment with identical values) are discarded since they are not informative. The heterozygous sites (columns in the alignment with different values) correspond to alleles that differ between chromosomes and they are labeled as 0 or 1 arbitrarily. A matrix X of size m × n can be built from the alignment, where m is the number of reads and n is the number of heterozygous sites. The i-th read is described as a ternary string Xi∈{0, 1,−}n, where ‘−’ indicates a gap, namely that the allele is not covered by the fragment [again following the notation of Bansal and Bafna (2008) for clarity]. The start position and end position of a read are the first and last positions in the corresponding row that are not ‘−’, respectively. Therefore, the ‘−’s in the head and tail of each row will not be considered as part of the corresponding read. However, there can be ‘−’s inside each read, which correspond to either missing data for single reads or gaps connecting a pair of single reads (called paired-end reads). Reads without ‘−’ are called gapless reads; otherwise they are called gapped reads. Assuming a read's end position is j, start position is i, the length of the read is defined as j−i+1. We also assume all the reads have already been correctly aligned to the reference genome by some mapper, which may not be true since the mapper may introduce mapping errors and the reads may come from repeat-rich regions. However, the mapping process is out of the scope of this article and we thus do not evaluate the effects of mapping errors on the quality of our haplotype assembly solution.
The haplotypes can be represented as an unordered pair of binary strings H=(h1, h2), each of length n. Since all the sites are heterozygous, h2 is the bit-wise complement of h1. An example of the read matrix is shown in Table 1. As we can see in this example, each read corresponds to one row where ‘−’ indicates missing information. Reads often contain errors. For example, if we only consider reads 1,2,3, we can partition them perfectly into two sets ({read1, read3}, {read2}) and reconstruct the haplotypes as H=(h1={0000}, h2={1111}) by assigning reads 1 and 3 to h1 and assigning read2 to h2. However, read4 is in conflict with this partition and there is no perfect partition for reads 1,2,3 and 4.
Table 1.
An example of read matrix that consists of 10 reads spanning 13 positions
| Reads | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Read1 | 0 | 0 | - | - | - | - | - | - | - | - | - | - | - |
| Read2 | - | - | 1 | 1 | - | - | - | - | - | - | - | - | - |
| Read3 | 0 | 0 | 0 | 0 | - | - | - | - | - | - | - | - | - |
| Read4 | - | - | 1 | 0 | 1 | - | - | - | - | - | - | - | - |
| Read5 | - | - | 0 | - | - | 0 | - | - | - | - | - | - | - |
| Read6 | - | - | - | 0 | - | - | - | - | - | - | 1 | 1 | - |
| Read7 | - | - | - | - | 0 | 0 | 0 | - | - | - | - | - | - |
| Read8 | - | - | - | - | 0 | 1 | 1 | 0 | - | - | - | - | - |
| Read9 | - | - | - | - | - | - | - | - | 1 | 1 | - | - | - |
| Read10 | - | - | - | - | - | - | - | 1 | 1 | 0 | - | - | 0 |
Therefore, in the presence of errors, we need to reconstruct the haplotypes such that some objective function is minimized. The objective function we use is MEC, which is the minimum number of changes, or corrections, that need to be made in the read matrix such that the resulting matrix admits a perfect bi-partition, where each corrected read maps to either haplotype perfectly. Alternatively speaking, for any pair of complementary haplotypes, the set of reads can be partitioned into two subsets that satisfy the following property: if both subsets of the reads are mapped to the two haplotypes at their corresponding intervals indicated by their starting positions and lengths, where one subset is mapped to only one of the haplotypes, the number of errors or mismatches for the mapping is minimized. This minimum number of errors is the MEC score of the reads for the pair of haplotypes. In our example, if we only consider reads 1,2,3 and 4, we can change read4 from (101) to (001) such that now we can obtain a perfect bi-partition ({read1, read3, read4}, {read2}) with reconstructed haplotypes H′=(h1={00001}, h2={11110}). The number of changes we made is obviously 1. Therefore the ‘haplotype assembly’ problem is identical to finding a pair of haplotypes H such that the MEC score of the reads in the read matrix is minimized. For example, the MEC score for reads 1,2,3 and 4 is 1 and the corresponding optimal pair of haplotypes is H′. This example is very simple, however, in reality, the number of reads can be very large and it has been shown that the ‘haplotype assembly’ problem is NP-hard even for gapless reads of length 2.
Notice that the optimal haplotypes that minimize the MEC score may not be exactly the same as the real haplotypes. However, our objective function for the haplotype assembly problem makes the maximal parsimony assumption common in many computational biology problems (Fitch, 1977; Gusfield, 2003). Therefore, the optimal solution is the most biologically meaningful solution for our problem. Another factor that may affect the quality of the reconstructed haplotypes is sequencing error. If the errors are consistent across reads, the reconstructed haplotypes may maintain these errors and may be incorrect. However, the MEC criteria attempts to discover haplotypes that minimizes the number of errors in the reconstruction. This is because if the errors are contained in only the minority of the reads, by minimizing the possible errors, the reconstructed haplotypes can still capture the reads that were sequenced correctly and thus avoid the sequencing errors.
3.2 Dynamic programming algorithm
To obtain the optimal solution for the haplotype assembly problem, a naive approach is to enumerate all binary strings, each of which represents a possible haplotype, and then assign the reads to each pair of possible haplotypes to minimize the conflicts with the reads. Given the length of haplotypes as n, the number of reads as m, this naive approach requires O(m × 2n) complexity and is therefore infeasible for large n. However, the problem can be solved optimally using a dynamic programming algorithm as we show here. The basic idea of the dynamic programming algorithm is to store the optimal MEC for partial haplotypes (the prefixes for full-length haplotypes) ending with every possible length k binary strings. Then the algorithm extends the partial haplotypes by one bit repeatedly until full-length haplotypes are obtained.
We define r as a length, k binary string and s(i, r) as the MEC score for partial haplotypes starting at position 0 and ending at position i+k−1 with suffix r, where the partial haplotypes are the prefixes of full-length haplotypes. s(i, r) is obtained by considering only the reads whose starting positions are no greater than i and it solves a subproblem of the full-length haplotype assembly where all reads are considered. We build a dynamic programming matrix and at each position i we store s(i, r) for all r. The best MEC is the minimum s(n−k, r) over all r, where n is the full length of the haplotypes. Given the definition of MEC (minimum number of changeseq flips needed), we can initialize s(0, r) by considering the reads that start at position 0, and for each read compute the number of mismatches between the read and r and the read and the complement of r. The partial haplotypes at position i can be obtained by extending the partial haplotypes at position i−1 with either a 0 or 1. s(i, r1) can be computed from s(i−1, r2) and the newly introduced errors between r1 and all reads starting at position i, where the length k−1 suffix of r2 is the same as the length k−1 prefix of r1. The recursion is illustrated in the following formula:
| (1) |
where b is a binary bit of either 0 or 1, r[0, k−2] is the length k−1 prefix of r, (b, r[0, k−2]) is a length-k binary string generated by concatenating b with r[0, k−2], h(i, r) is the minimum of the total number of disagreements between r or the complement of r and all reads starting at position i. Notice that, for h(i, r), we consider both r and the complement of r because each read can be assigned to either the current haplotype or its complement (depending on which assignment produces smaller disagreements). The assignments producing the minimum disagreements are then selected. Each ‘−’ matches both 0 and 1, so a mismatch only happens between non- ‘−’ symbols.
Starting from the solution that leads to a minimal value of s(n−k, r) over all r, we can trace back the dynamic programming matrix to reconstruct the haplotypes that minimize the MEC score. The time complexity of the dynamic programming algorithm is O(m × 2k × n), where m is the number of reads, k is the length of the longest read and n is the total number of SNPs in the haplotypes. Here, for illustrative purpose, we assume all reads are of length k. In reality, the reads are of different length and the time complexity of the dynamic programming algorithm becomes O(m × 2kmax × n), where kmax is the maximal number of alleles contained among all reads.
We also observe that we can split up the reads into sets where there is no read that spans any two sets. We call such a set a block. The set of reads can thus be partitioned into many blocks. Since no read spans any two blocks, which means those blocks are independent, we can reconstruct haplotypes for each block in parallel using the dynamic programming algorithm developed above and then concatenate the solutions for each block to construct the complete haplotypes.
Next, we show a simple example for the dynamic programming algorithm. We take read 1–read 4 from Figure 1 as an example. We need to order them according to their start positions.
- At position i=0: we have read 1 and read 3, k=length(read 3)=4. Therefore, we compute s(i, r) for all length k=4 binary strings r, using only read 1 and read 3:
- s(0, 0000)=h(0, 0000)=0,
- s(0, 0001)=h(0, 0001)=1,
- s(0, 0010)=h(0, 0010)=1,
- …,
- s(0, 1111)=h(0, 1111)=0.
At position i=1: there is no read starting at position 1.
- At position i=2: we have read 2 and read 4 and
- k=length (read 4)=3. Again, we compute s(i, r) for all length k=3 binary strings r, using only read 2 and read 4. Since we do not have read starting at position 1, to simplify the computation, in Formula 1, s(i−1, (b, r[0, k−2])) becomes s(i−2, (b1, b2, r[0, k−2]) where b1∈{0, 1} and b2∈ {0, 1} are single binary bits. Therefore, we have:
- s(2, 000)=min(s(0, 0000)+h(2,000), s(0, 0100)+h(2,000), s(0, 1100)+h(2,000), s(0, 1000)+h(2,000))=1,
- s(2, 001)=min(s(0, 0000)+h(2,001), s(0, 0100)+h(2,001), s(0, 1100)+h(2,001), s(0, 1000)+h(2,001))=1,
- …,
- s(2, 111)=min(s(0, 0011)+h(2,111), s(0, 0111)+h(2,111), s(0, 1111)+h(2,111), s(0, 1011)+h(2,111))=1.
Therefore, the optimal MEC is min(s(2, r)) for all length k=3 binary strings r and the optimal MEC is 1.
Fig. 1.

(a) The number of short reads, all reads and (b) the length of haplotypes for each chromosome. The threshold for short reads is 15. The length of haplotypes is the number of heterozygous sites in each chromosome.
Notice that when we trace back to obtain the optimal haplotypes, it is not necessarily the case that there is only one pair of optimal haplotypes. In the above example, s(2, 000)=1 and it is from s(0, 0000). By tracing back from s(2, 000), we get the optimal haplotype pair ({00000}, {11111}). We also have s(2, 001)=1 and it is from s(0, 0000). By tracking back from s(2, 001) we get the optimal haplotype pair ({00001, 11110}). Both the pairs have optimal MEC of 1. When there are multiple optimal solutions, data on multiple individuals may be used to infer the most likely haplotypes for each ambiguous individual.
3.3 MaxSAT conversion for haplotype assembly
So far, we only discussed a dynamic programming algorithm for single reads. Consider reads 5, 6 and 10 in Figure 1. In each of these reads, there are two continuous strings connected by ‘−’, which indicates missing information. These reads are called paired-end reads and are generated by modern sequencing technologies. The problem becomes much more complicated when paired end reads are considered since paired-end reads usually span a long fragment, which can be as long as a few hundred positions. Although paired-end read can be considered as a special case of a single read, the dynamic programming algorithm introduced above becomes impractical, since we need to enumerate all positions the paired-end read covers. As concluded above, the time complexity of the dynamic programming algorithm is O(m × 2kmax × n), where kmax is the maximum number of alleles contained among all reads. When paired-end reads are considered, kmax could be as large as a few hundred, making the dynamic programming approach impractical. Even single reads can be too long to enumerate some of the positions. We set a threshold for kmax such that the enumeration of all length kmax binary strings is computationally feasible. We call the single reads and the paired-end reads of length greater than the threshold as long reads and the other reads as short reads.
We solve the haplotype assembly problem when long reads are also considered by conversion to MaxSAT. The MaxSAT problem is an optimization version of the well-known Boolean satisfiability problem (SAT) (Biere et al., 2009). Given a set of clauses (a clause is a disjunction of Boolean literals), the MaxSAT problem asks for a complete assignment of all variables that maximizes the number of clauses the assignment satisfies. For example, consider the following set of four clauses:
The assignment x1=false, x2=false, x3=false satisfies three clauses and is optimal. In this work, we consider a variant of MaxSAT known as partial MaxSAT. Partial MaxSAT allows some clauses to be labeled as hard—i.e. their satisfiability is mandatory in any solution. The objective of the problem is to find an assignment that satisfies all hard clauses and satisfies the most number of non-hard (i.e. soft) clauses. For more discussion on MaxSAT and its variations, please see (Li and Manyá, 2009).
In our conversion of the haplotype assembly problem to partial MaxSAT, we define the following boolean variables:
hi, 0≤i<n, represents the binary symbol at position i in the haplotype to be constructed.
rj, 0≤j<m, represents the assignment of read j to a haplotype. The value rj=0 indicates that read j is assigned to the considered haplotype, while the value rj=1 indicates that the read is assigned the complement haplotype.
eij, 0≤i<n, 0≤j<m, represents whether a correction is needed for position i of read j with respect to the considered haplotype. The value eij=1 indicates that a correction is needed, while the value eij=0 indicates that no correction is needed at that position.
Given these variables, we can define the set of clauses that describes the relationship between hi, rj and eij. These clauses essentially specify that there is an error whenever the value at position i of read j does not match with the value at position i of the haplotype that the read is assigned to. Let read[i][j] represent the value at position i of read j (i.e. the value of cell[i][j] of the read matrix). We can formally define a set of clauses for each non-‘−’ entry in the read matrix as follows:
Note that ⇔ is the logical equivalence operator and that (x ⇔ y ⇔ z) is a shorthand notation for the clauses (x ν y ν z), (¬ x ν ¬ y ν z), (¬ x ν y ν ¬ z), (x ν ¬ y ν ¬ z). The above clause definition can be understood as follows. If read[i][j] is 0, then the error eij is defined to be hi ⇔ ¬ rj. That is, there should be an error if (and only if) (i) hi is 1 and the read is assigned to the considered haplotype or (ii) hi is 0 and the read is assigned to the complement haplotype. The case when read[i][j] is 1 can be understood in a similar way. As these clauses describe how the errors are calculated, they should be hard clauses in our MaxSAT problem (they should not be violated by any solution).
Since we would like to find an assignment that minimizes error, we add to our MaxSAT problem the unit clause (¬ eij) for each non-‘−’ position in the read matrix. Every unit clause that an assignment falsifies (i.e. every error introduced) will incur a penalty of 1 to that assignment. These unit clauses are soft clauses that might be falsified by the optimal solution. The optimal solution to this MaxSAT problem is simply any assignment that respects the error calculation rules and introduces the least amount of error.
This concludes our conversion of the haplotype assembly problem to partial MaxSAT problem. We may use any partial MaxSAT solver to solve the resulting problem. In this work, we used the solvers called Clone (Pipatsrisawat et al., 2008) and WBO (Manquinho et al., 2009) to solve the resulting MaxSAT problems. We will report the experimental results in the next section.
4 RESULTS
4.1 HuRef experiments
We first examine the performance of the dynamic programming algorithm on the filtered HuRef data from (Levy et al., 2007) overall 22 chromosomes and directly compare our method to previous approaches (Bansal and Bafna, 2008). The data consists of 32 million DNA fragments generated by Sanger sequencing and contains a total of 1.85 million heterozygous variants for the 22 chromosomes. We show the number of short reads, all reads and number of heterozygous sites for each chromosome, where the threshold for short reads is 15 as shown in Figure 1. As we can see, the number of reads for each chromosome is very large. More than 90% of the reads are short reads. Haplotypes for each chromosome are also very long, making the haplotype assembly problem computationally intensive. The average number of reads that span each heterozygous site is between six and seven.
The whole-genome sequence data consists of many disconnected blocks where no read spans the boundary of two blocks. Therefore, we can split the sequence data independently into many blocks and then solve the haplotype assembly problem for each block. The global MEC score is the sum of the scores from each block and the optimal haplotypes are the concatenation of the haplotypes from each block. We show the read matrix for the first block of chromosome 22 in Figure 2 as an example, where we have around 2300 reads spanning a block of length around 400.
Fig. 2.

Graphical representation of the read matrix for the first block of Chromosome 22, where the reads are sorted by their starting positions. The rows are the reads and the columns are the haplotype positions. The black dots are the non-‘−’ cells for the short reads and the red dots are the non-‘−’ cells for the long reads. The red lines are the gap cells of the paired-end reads.
4.1.1 HuRef experiments on only short reads
We first compare the results of the dynamic programming algorithm with the results of Greedy and HapCut, on short reads, namely single reads and the paired-end reads of length less than 15 only. As we showed in Figure 1, most of the reads are very short. However, there are still tens of thousands of reads of length more than 15. For example, in the block shown in Figure 2, there are around 400 long reads and the maximal length of the reads is around 200. We run all three algorithms on short reads only and the results are shown in Table 2. As we can see, on average, HapCut improves the MEC score of Greedy by 30%, while our dynamic programming algorithm shows for the first time that the solution from HapCut is only 1.1% from the optimal solution. The run time of the dynamic programming algorithm is reasonably fast and comparable to the HapCut algorithm. For example, for the first block of Chromosome 22 in Figure 2, HapCut runs for 20 s while the dynamic programming algorithm runs for 24 s. The run times are even closer for small size blocks and ∼90% blocks are such small size blocks. Both algorithms run for more than 10 h and finish in roughly the same time on a computational cluster for all the 22 chromosomes.
Table 2.
The MEC scores computed by Greedy, HapCut and dynamic programming (DP), MaxSAT conversion, on short reads only and on all reads, respectively, for each chromosome
| On short reads |
On all reads |
|||||
|---|---|---|---|---|---|---|
| Greedy | HapCut | DP | Greedy | HapCut | MaxSAT | |
| Chromosome 1 | 21 355 | 15 312 | 15 292 | 29 518 | 19 687 | 19 584 |
| Chromosome 2 | 16 067 | 11 251 | 11 107 | 22 706 | 14 615 | 14576 |
| Chromosome 3 | 11 909 | 8223 | 8181 | 16 696 | 10 702 | 10 647 |
| Chromosome 4 | 12 518 | 8820 | 8775 | 17 509 | 11 525 | 11 304 |
| Chromosome 5 | 11 621 | 8017 | 7944 | 16 432 | 10 536 | 10 528 |
| Chromosome 6 | 10 624 | 7487 | 7369 | 15 295 | 9842 | 9826 |
| Chromosome 7 | 11 668 | 8531 | 8423 | 17 188 | 11244 | 11 187 |
| Chromosome 8 | 10 501 | 7343 | 7311 | 14 535 | 9741 | 9025 |
| Chromosome9 | 10 199 | 7350 | 7312 | 13 512 | 9222 | 9201 |
| Chromosome 10 | 10 263 | 7313 | 7236 | 15 076 | 9846 | 9778 |
| Chromosome 11 | 8825 | 6224 | 6196 | 12 667 | 8200 | 8183 |
| Chromosome 12 | 8641 | 6337 | 6155 | 12 453 | 8218 | 8176 |
| Chromosome 13 | 6412 | 4396 | 4341 | 8848 | 5822 | 5761 |
| Chromosome 14 | 6634 | 4567 | 4532 | 9070 | 5879 | 5845 |
| Chromosome 15 | 9289 | 6653 | 6623 | 13291 | 9311 | 9285 |
| Chromosome 16 | 8574 | 6160 | 6093 | 12365 | 8259 | 8207 |
| Chromosome 17 | 7088 | 5034 | 4955 | 10195 | 6525 | 6459 |
| Chromosome 18 | 4973 | 3526 | 3398 | 8324 | 4991 | 4943 |
| Chromosome 19 | 5549 | 3996 | 3907 | 7939 | 5319 | 5288 |
| Chromosome 20 | 4136 | 2909 | 2891 | 5563 | 3739 | 3723 |
| Chromosome 21 | 3877 | 2903 | 2796 | 5607 | 3888 | 3881 |
| Chromosome 22 | 4424 | 3267 | 3250 | 6685 | 4495 | 4479 |
| Sum | 205 147 | 145 619 | 144 087 | 291 474 | 191 606 | 189 886 |
4.1.2 HuRef experiments on all reads
As mentioned in the previous section, in order to solve the haplotype assembly problem containing reads of all lengths, we convert the problem into a partial MaxSAT problem and use partial MaxSAT solvers to solve it. In this work, we consider two MaxSAT solvers: Clone (Pipatsrisawat et al., 2008) and WBO (Manquinho et al., 2009).
Out of 47 758 blocks, Clone was able to solve all but eight blocks optimally. WBO, on the other hand, solved all but 34 blocks optimally. These two solvers report the same optimal solutions for the blocks they both solved. Interestingly, even for the eight blocks that Clone could not solve optimally, it still reported solutions with lower MEC scores than those obtained from the HapCut algorithm.1 We compared our results with the results of Greedy and HapCut. The results are shown in Table 2. As we can see, on average, HapCut improves the MEC score of Greedy by 34%, while our MaxSAT conversion method again shows that the solution of HapCut is very close to the optimal solution. Although there are eight blocks that we could not solve optimally, the remaining 99.98% of the blocks were optimally solved. Therefore, it is reasonable to believe that the overall solution we obtained is very close to optimal. The run time of the MaxSAT solver on our cluster machine is ∼15 h, which is comparable to that of HapCut.
4.2 Designing haplotype assembly protocols
While previously developed haplotype assembly approaches have been successfully applied to the HuRef data, it is not clear how applicable these approaches would be using current high-throughput genotyping technologies that have much shorter read lengths, yet higher coverage than the HuRef data. We take advantage of the efficiency of our algorithm to perform simulations in order to design sequencing protocols for current high-throughput technology in order to effectively obtain haplotypes. Unlike the reads for HuRef data, which are sequenced with the Sanger-based whole-genome shotgun sequencing and therefore are very long (each segment is thousands of base pairs long including both homozygous and heterozygous sites), here we consider the reads generated by the HTS technology (Wheeler et al., 2008). The reads generated by HTS are usually very short (each segment is around 30–100 bp including both homozygous and heterozygous sites).
The basic parameters of sequencing technology that we explore are the sequence coverage ratio (the number of times that each base pair in the sequence is covered), the insert length of the paired-end reads (the distance between the two segments of the paired-end reads), the variance of this insert length and the read length. We explore how these parameters affect the haplotype assembly. We perform our experiments over individual genotype data from HapMap (International HapMap Consortium, 2007). For a single individual, we concatenate the heterozygous SNPs to construct a true haplotype. For the individual we downloaded, there are 505 065 heterozygous SNPs. We then mimic the sequencing process by randomly generating paired-end reads with varying parameters including coverage ratio, insert length of the reads and standard deviation of the insert length. The insert length follows a Gaussian distribution with a mean of 1000. Assume the genome length is n, the sequence read length is l, the coverage ratio is c, the number of reads to be generated then is 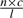 . The starting positions of the reads are randomly selected within the range of the whole genome, therefore they may cover both heterozygous and homozygous SNPs. The segment length of the sequence paired-end read is 36 (including both heterozygous and homozygous sites), which is a reasonable value given current technology. To evaluate the effects of these parameters on the assembly process, we first divide the haplotypes into blocks with distances >1 SD above the sum of the mean of the insert length (we use 1000). The reads are then very unlikely to span two blocks due to the distance between them. The length of a read in the read matrix is the number of heterozygous SNPs the read covers. Since we generate only paired-end reads in our simulation, which consist of two segments, if only one segment of a read covers heterozygous SNPs, the resulting read in the read matrix will be considered as a single read, otherwise it is considered as a paired-end read. The insertion of a paired-end read corresponds to the gap of the read in the read matrix. Although the mean of the insert length is 1000, the corresponding gap length in the read matrix is very small because only heterozygous SNPs are considered for assembly. Therefore, almost all the reads are very short. To illustrate this, we vary the coverage ratio as 10, 20, 30, 40 times, the standard deviation of insert length as 5, 50, 500. For all combinations of parameter settings, the ratios of short reads, namely reads of length less than 15, out of all reads, are all greater than 99.99%, indicating that our dynamic programming algorithm is indeed very practical and can be considered as optimal.
. The starting positions of the reads are randomly selected within the range of the whole genome, therefore they may cover both heterozygous and homozygous SNPs. The segment length of the sequence paired-end read is 36 (including both heterozygous and homozygous sites), which is a reasonable value given current technology. To evaluate the effects of these parameters on the assembly process, we first divide the haplotypes into blocks with distances >1 SD above the sum of the mean of the insert length (we use 1000). The reads are then very unlikely to span two blocks due to the distance between them. The length of a read in the read matrix is the number of heterozygous SNPs the read covers. Since we generate only paired-end reads in our simulation, which consist of two segments, if only one segment of a read covers heterozygous SNPs, the resulting read in the read matrix will be considered as a single read, otherwise it is considered as a paired-end read. The insertion of a paired-end read corresponds to the gap of the read in the read matrix. Although the mean of the insert length is 1000, the corresponding gap length in the read matrix is very small because only heterozygous SNPs are considered for assembly. Therefore, almost all the reads are very short. To illustrate this, we vary the coverage ratio as 10, 20, 30, 40 times, the standard deviation of insert length as 5, 50, 500. For all combinations of parameter settings, the ratios of short reads, namely reads of length less than 15, out of all reads, are all greater than 99.99%, indicating that our dynamic programming algorithm is indeed very practical and can be considered as optimal.
To evaluate how well the haplotype assembly can be done w.r.t. the sequencing protocols, we next construct a graph from the read matrix. Each heterozygous SNP is a vertex in the graph and we draw an edge between two vertices if their corresponding SNPs are covered by the same read. We construct such a graph using all the generated reads and consider the connected components in this graph since we have no information on how to phase heterozygous sites in different connected components relative to each other. The number of optimal solutions will be exponential in the number of connected components. Therefore, the smaller the number of connected components is, the better we can assemble the haplotypes. We count the average number of connected components each block contains. We also compute the average size of the connected components. We show the experimental results in Table 3. As we can see, the number of connected components in each block decreases as coverage ratio increases and as SD increases. Meanwhile, the average size of connected components also increases. However, to reduce the number of connected components each block contains to one such that we can fully reconstruct each block, we need to use a very high-coverage ratio such as 100. Thus for any reasonable coverage ratio that would be collected in a sequencing study, haplotype assembly will not be able to assemble haplotypes because there will not be enough reads to connect all of the variants into complete haplotypes.
Table 3.
Average number of connected components contained in each block and average size of the connected components whose size is greater than 1 for different (coverage ratio, SD) settings
| (10, 5) | (10, 50) | (10, 500) | (20, 5) | (20, 50) | (20, 500) | (30, 5) | (30, 50) | (30, 500) | (40, 5) | (40, 50) | (40, 500) | (100, 500) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Num | 8 | 7 | 10 | 8 | 6 | 6 | 8 | 6 | 4 | 8 | 5 | 3 | 1 |
| Size | 2 | 2 | 4 | 2 | 3 | 8 | 2 | 3 | 13 | 2 | 4 | 17 | 31 |
However, the strategy of haplotype assembly can be combined with traditional haplotype inference techniques (Halperin and Eskin, 2004; Stephens et al., 2001) to infer haplotypes. The basic idea is that genotypes are obtained from the sequence data by performing SNP calling (Li et al., 2008) in the sequence reads. The majority of the common variants will be present in the reference datasets such as the HapMap (International HapMap Consortium, 2007) or the 1000 Genomes Project (1000 Genomes Project, 2010) and for these variants, haplotypes can be inferred using traditional techniques by leveraging the haplotypes from the reference dataset. We note that by using a reference dataset, we can predict haplotypes (or phase) at the common sites even for a single individual given the genotypes at the common sites for the individual.
Then the remaining variants (mostly rare variants) can be attached to the haplotypes by considering reads that span both the rare variant and a common allele for which the haplotypes have been inferred. The inference of the haplotypes can be performed using a modified dynamic programming algorithm that forces the haplotypes at the common variants to match the haplotype inferred from the genotypes.
We take the phased haplotype and treat them as a pair of very long reads with a gap at each site, which is not present in the reference sample. We have two ways to place these ‘reads’, namely assign one of the phased haplotype to one of the final haplotypes, say, h0, the other phased haplotype to h1, or the other way around. For each placement, we then apply the dynamic programming algorithm on the set of paired-end reads to infer the rare variants missed by the phased haplotypes. We need to use a modified dynamic programming algorithm where the phased haplotypes also need to be taken into consideration for the MEC since we initially have placed them. The placement with the minimum MEC will be the optimal placement and the corresponding reconstructed haplotypes are the optimal haplotypes. Weights can also be applied to the dynamic programming algorithm when the MEC is computed. The weights can be determined according to our belief of the relative accuracies of the traditional techniques and the HTS technology respectively. Then when the MEC is summed over the paired-end reads and the phased haplotypes, different weights are assigned to the number of errors from the paired-end reads and the phased haplotypes accordingly.
We can estimate how effective this approach would be by considering how often any given variant is covered by a read, which also covers an additional variant. We show such probabilities in Table 4 as well as the probabilities that the variants are covered by more than one read. As we can see, the probability increases as the coverage ratio or the standard deviation increases. With 40 times coverage and 500 bp SD, the probability of an SNP being attached to other SNPs at least once is as high as 92%, and at least twice is also high as 83%. Therefore, with even a moderate amount of coverage, most variants are covered by at least one read to another variant when the SD of the insert length is big enough. Thus, the combined strategy of using a traditional approach to infer haplotypes using the genotypes at the common variants combined with assembly of the rare variants using the sequence reads is a practical approach for inferring haplotypes.
Table 4.
The probability (%) of an SNP attached to other SNPs more than once, twice, three times, four times, for different (coverage ratio, SD) settings
| (10, 5) | (10, 50) | (10, 500) | (20, 5) | (20, 50) | (20, 500) | (30, 5) | (30, 50) | (30, 500) | (40, 5) | (40, 50) | (40, 500) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ≥1 | 41 | 56 | 65 | 45 | 66 | 82 | 46 | 70 | 89 | 47 | 73 | 92 |
| ≥2 | 35 | 38 | 39 | 41 | 54 | 62 | 43 | 62 | 75 | 44 | 66 | 83 |
| ≥3 | 29 | 26 | 23 | 38 | 44 | 44 | 41 | 54 | 61 | 43 | 60 | 71 |
| ≥4 | 23 | 19 | 16 | 35 | 35 | 32 | 39 | 46 | 48 | 41 | 54 | 60 |
5 DISCUSSION
In this article, we proposed a dynamic programming algorithm for the ‘haplotype assembly’ problem, which is able to assemble the haplotypes optimally with time complexity O(m × 2k × n), where m is the number of reads, k is the length of the longest read and n is the total number of SNPs in the haplotypes. Our experiments show for the first time that the current best-known solutions are very close to the optimal solution.
The most difficult part of the haplotype assembly problem is to handle the long reads. Long reads can span up to a few hundred positions. To handle these cases, we convert the problem to an MaxSAT problem, which can be solved optimally by an MaxSAT solver. We show that our MaxSAT solver is able to solve 99.98% of the problem instances optimally. For the remaining 0.02%, the MaxSAT solver also reports better results than HapCut does. Therefore, the overall solution we obtained is very close to the optimal.
Although the empirical results of our methods did not show a major advance over existing methods, we believe it is technically important and also interesting to have optimal algorithms for the haplotype assembly problem.
Our analysis on individual genotype data from HapMap shows that it is impractical to fully assemble the haplotypes as the coverage ratio needed is too high. However, combined with a traditional haplotype inference approach, our algorithm is able to infer haplotypes containing both rare and common SNPs, including SNPs that are unique to individuals.
Funding: D.H. and E.E. are supported by National Science Foundation (grants 0513612, 0731455, 0729049 and 0916676); NIH (grants K25-HL080079 and U01-DA024417). University of California, Los Angeles subcontract of contract N01-ES-45530 from the National Toxicology Program and National Institute of Environmental Health Sciences to Perlegen Sciences (in part).
Footnotes
1Clone is an any time algorithm that reports the current best solution as soon as it is discovered.
REFERENCES
- 1000 Genomes Project. A deep catalog of human genetic variation. 2010 Available at http://www.1000genomes.org/ (last accessed date April 23, 2010) [Google Scholar]
- Bansal V, Bafna V. HapCUT: an efficient and accurate algorithm for the haplotype assembly problem. Bioinformatics. 2008;24:i153. doi: 10.1093/bioinformatics/btn298. [DOI] [PubMed] [Google Scholar]
- Bansal V, et al. An MCMC algorithm for haplotype assembly from whole-genome sequence data. Genome Res. 2008;18:1336. doi: 10.1101/gr.077065.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biere A, et al. Frontiers in Artificial Intelligence and Applications. Vol. 185. Nieume Hemweg, Amsterdam: IOS Press; 2009. Handbook of Satisfiability. [Google Scholar]
- Browning B, Browning S. Haplotypic analysis of Wellcome Trust Case Control Consortium data. Hum. genet. 2008;123:273–280. doi: 10.1007/s00439-008-0472-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi A, et al. Proceedings of the 8th International Workshop on Algorithms in Bioinformatics. Vol. 5251. Berlin, Heidelberg: Springer; 2008. Efficient genome wide tagging by reduction to SAT; pp. 135–147. of Lecture Notes in Computer Science. [Google Scholar]
- Cilibrasi R, et al. Proceedings of the 5th International Workshop on Algorithms in Bioinformatics. Vol. 3692. Berlin, Heidelberg: Springer; 2005. On the complexity of several haplotyping problems; pp. 128–139. Lecture Notes in Computer Science. [Google Scholar]
- Fitch W. On the problem of discovering the most parsimonious tree. Am. Nat. 1977;111:223–257. [Google Scholar]
- Gusfield D. Proceedings of the Combinatorial Pattern Matching Conference. Vol. 2161. Berlin, Heidelberg: Springer; 2003. Haplotype inference by pure parsimony; pp. 144–155. of Lecture Notes in Computer Science. [Google Scholar]
- Halperin E, Eskin E. Haplotype reconstruction from genotype data using imperfect phylogeny. Bioinformatics. 2004;20:1842–1849. doi: 10.1093/bioinformatics/bth149. [DOI] [PubMed] [Google Scholar]
- International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPS. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancia G, et al. Proceedings of the 9th Annual European Symposium on Algorithms. Lecture Notes in Computer Science. Berlin, Heidelberg: Springer; 2001. SNPs problems, complexity, and algorithms; pp. 182–193. [Google Scholar]
- Levy S, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Manyá F. MaxSAT, hard and soft constraints. Handbook of Satisfiability. 2009;185:613–631. [Google Scholar]
- Li H, et al. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippert R, et al. Algorithmic strategies for the single nucleotide polymorphism haplotype assembly problem. Brief. Bioinform. 2002;3:23. doi: 10.1093/bib/3.1.23. [DOI] [PubMed] [Google Scholar]
- Manquinho V, et al. Proceedings of the 12th International Conference on Theory and Applications of Satisfiability Testing. Berlin, Heidelberg: Springer; 2009. Algorithms for weighted Boolean optimization; pp. 495–508. Lecture Notes in Computer Science. [Google Scholar]
- Marchini J, et al. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007;39:906–13. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- Panconesi A, Sozio M. Lecture Notes in Computer Science. Vol. 3240. Berlin, Heidelberg: Springer; 2004. Fast hare: a fast heuristic for single individual SNP haplotype reconstruction; pp. 266–277. [Google Scholar]
- Pipatsrisawat K, et al. Solving weighted Max-SAT problems in a reduced search space: a performance analysis. Journal on Satisfiability, Boolean Modeling and Computation. 2008;4:191–217. [Google Scholar]
- Stephens M, et al. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Gene. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, et al. Haplotype reconstruction from SNP fragments by minimum error correction. Bioinformatics. 2005;21:2456. doi: 10.1093/bioinformatics/bti352. [DOI] [PubMed] [Google Scholar]
- Wheeler D, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]