

Abstract
Recent years have witnessed an increase in research activity for the detection of structural variants (SVs) and their association to human disease. The advent of next-generation sequencing technologies make it possible to extend the scope of structural variation studies to a point previously unimaginable as exemplified by the 1000 Genomes Project. Although various computational methods have been described for the detection of SVs, no such algorithm is yet fully capable of discovering transposon insertions, a very important class of SVs to the study of human evolution and disease. In this article, we provide a complete and novel formulation to discover both loci and classes of transposons inserted into genomes sequenced with high-throughput sequencing technologies. In addition, we also present ‘conflict resolution’ improvements to our earlier combinatorial SV detection algorithm (VariationHunter) by taking the diploid nature of the human genome into consideration. We test our algorithms with simulated data from the Venter genome (HuRef) and are able to discover >85% of transposon insertion events with precision of >90%. We also demonstrate that our conflict resolution algorithm (denoted as VariationHunter-CR) outperforms current state of the art (such as original VariationHunter, BreakDancer and MoDIL) algorithms when tested on the genome of the Yoruba African individual (NA18507).
Availability: The implementation of algorithm is available at http://compbio.cs.sfu.ca/strvar.htm.
Contact: eee@gs.washington.edu; cenk@cs.sfu.ca
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Human genetic variation can be defined in various size ranges. The smallest type of variation, termed single nucleotide polymorphisms (SNPs), are those at the single base pair level. The International HapMap Project genotyped 270 human individuals for 3.1 million SNPs, and recently the 1000 Genomes Project (http://www.1000genomes.org) was initiated to characterize human genetic variation with lower minor allele frequency by sequencing more than 1000 human genomes. More recently, it was shown that structural variation events significantly contribute to human genome diversity (Feuk et al., 2006; Kidd et al., 2008; Korbel et al., 2007; Tuzun et al., 2005). Many structural variants (SVs) are associated with genetic diseases such as psoriasis (Hollox et al., 2008) and Crohn's disease (McCarroll et al., 2008), prompting an increased interest in structural variation studies (Bashir et al., 2008; Chen et al., 2009; Hormozdiari et al., 2009; Korbel et al., 2007; 2009; Lee et al., 2008; Lee et al., 2009; Sindi et al., 2009; Tuzun et al., 2005). Recently, it was shown that the SV reconstruction is harder than de novo assembly of small genomes and introduced an extensive framework for optimal genome re-sequencing (Du et al., 2009). Recently, we developed a set of combinatorial algorithms to detect structural variation using high-throughput sequencing data with improved sensitivity through interrogating the repeat and duplication rich segments of the human genome. In Hormozdiari et al. (2009), the maximum parsimony structural variation (MPSV) discovery problem was introduced. This MPSV problem asks to compute unique mapping for each discordant paired-end read in the reference genome such that total number of implied (SVs) is minimized. We also previously presented a computational analysis of read depth to characterize segmental duplications and predict absolute copy number and content of duplicated genes (Alkan et al., 2009). See the review from Medvedev et al. (2009) for the details and strengths of different structural variation discovery methods.
One type of structural variation excluded from this analysis is the mobile element transposition. Mobile elements, or transposons, are repetitive elements in the genome that occupy ∼44% of the human genome, including Alu, L1 and SVA elements (Mills et al., 2007). Most of the transposons are fixed in the human lineage; however, around 0.05% of the transposons are still active, and the copy number and loci of these active transposons vary in the genomes of different individuals. Many studies have demonstrated that the mobile elements contribute to genome evolution and human genetic diversity. An interesting case was shown by Bekpen et al. (2009): insertions of an Alu element were posited to cause pseudogenization of the IRGM gene at the split of New World and Old World monkey lineages 35–40 million years ago (mya) by disrupting the open reading frame (ORF). A second transposon integration (ERV9) restored the ORF ∼24 mya in the common ancestor of apes and humans, demonstrating the first report on a ‘resurrected’ gene. The human IRGM gene plays an important role in the immune system (Bekpen et al., 2009) and is associated with Crohn's disease (McCarroll et al., 2008). Mobile element transpositions are associated with the expansion of interspersed segmental duplications (Bailey et al., 2003) and can promote both the creation of segmental duplication in human genomes (Xing et al., 2009) and the alteration of gene transcription by gene trapping and exonization.
Another aspect that most structural variation algorithms ignore is the diploid nature of the human genome [including VariationHunter, BreakDancer (Chen et al., 2009) and GASV (Sindi et al., 2009)]. Each chromosome has exactly two copies; therefore, all loci in the genome (w.l.o.g., except segmental duplications) are represented twice. This means that there cannot be more than two SVs including the ‘normal’ variant signature observed by ‘concordant’ paired-end mapping (Tuzun et al., 2005) at the same locus. However, due to the mapping artifacts in the repetitive segments of the human genome, most algorithms might call multi-allelic SVs within overlapping intervals. In fact, the limitation of two variants per locus information can be used both to reduce false discovery rates and also genotype SVs (homozygous versus hemizygous).
In this article, we present two extensions/improvements to the combinatorial formulation of our original VariationHunter algorithm (Hormozdiari et al., 2009) to detect structural variation using high-throughput sequencing technology. The main contributions of this article are (i) the first mathematically complete formulation and algorithm to identify transposition events (especially mobile element transpositions) and (ii) to remove ambiguity in variation discovery through ‘conflict resolution’ (enforcing at most two variants per locus).
We first show that our method for discovery of mobile element transpositions has high accuracy and precision. In a full-scale simulation, we produce short reads from the genome of J. Craig Venter (very similar to what an Illumina platform would produce). Our method is able to find ∼85% of the known transposon insertions with a precision of >90%. We also demonstrate that our new algorithm (denoted as VariationHunter-CR) has a higher accuracy in comparison with VariationHunter (Hormozdiari et al., 2009), BreakDancer (Chen et al., 2009) and MoDIL (Lee et al., 2009) when tested on a whole-genome shotgun sequence dataset generated from the genome of the Yoruba African individual (NA18507) using the Illumina platform.
2 DEFINITIONS AND THE FORMULATION OF TRANSPOSITION EVENTS
In Tuzun et al. (2005) and Volik et al. (2003), a general framework for detecting structural variation using long paired-end reads was introduced. This framework is based on aligning the paired-end reads to the reference genome and observing the end-reads1 with discordant mapping. The paired-end reads with discordant mapping suggest either deletion insertion or inversion events. For example, an inversion event can be deduced when one of the two end reads of a paired-end read has a different mapping orientation than expected. In the standard library construction of the Illumina platform, in the case of no inversions or duplications, the read that maps to the proximal location is expected to be in the + strand, where its mate should be mapped to a distal location in the − strand. However, if the read pair spans an inversion breakpoint, the mapping orientations of the reads will be observed as either ++ or −− [See the Supplementary Material in Hormozdiari et al. (2009) for a full case study].
As per Hormozdiari et al. (2009), we denote a read pair as pei and the distance between the end coordinate of the proximal read and the start coordinate of the distal read as the GapSize (i.e. the insert-size minus the total length of the reads). An alignment of a read pair to the reference genome is denoted as concordant (Tuzun et al., 2005) if the distance between the aligned end reads is in the range [Δmin, Δmax] and the alignment orientation is +−. The range [Δmin, Δmax] is empirically calculated by analyzing the mapping span size distribution of the read pairs.
The set of discordant paired-end reads is represented as R = {pe1, pe2,…, pen}. Each discordant paired-end read pei may be mapped to multiple locations in the reference genome. The set of all alignments of pei is then defined as Align (pei)={a1pei, a2pei, …,ajpei}.
Note that each alignment of pei to the reference genome (ajpei) is a 5-tuple that represents the map locations of the end reads and their alignment orientation. More formally,
| (1) |
where the pair (Lℓ(pei), Lr(pei)) represents the map location (i.e. both start and end loci) of the left end read of pei, (Rℓ(pei), Rr(pei)) is the mapping location of the right end read of pei, and or(pei) represents the map orientation of both ends. Note that or(pei)∈{+−, ++, –, −+}.
In Hormozdiari et al. (2009), a SV event is defined as SV SV(t, PosL, PosR, Ranmin, Ranmax). SV represents a of SV type t∈{Ins, Del, Inv}2 located between PosL and PosR in the reference genome, and with size estimated between Ranmin and Ranmax.
Another important type of structural variation is the transposition event where a segment of the genome (formally, a transposon) is copied to another location with a small divergence. In the remainder of this article we call such types of SVs as copy events. Examples of common copy events include transpositions of Alu, SVA and L1 elements.
Unfortunately, none of the available methods designed to detect structural variation events (e.g. Bashir et al., 2008; Chen et al., 2009; Hormozdiari et al., 2009; Kidd et al., 2008; Korbel et al., 2007; Lee et al., 2008, 2009; Sindi et al., 2009; Tuzun et al., 2005) considered these copy events, and their focus was mainly on the discovery of deletions, insertions and inversions. A more recent algorithm, HYDRA, includes simple heuristics to detect transposon insertions (Quinlan et al., 2010). Interestingly, even if the goal of a method is to identify only insertions, deletions and inversions in a sequenced genome, the presence of the common copy events will cause many false negative deletion and inversion predictions. Figure 1 clearly demonstrates a common scenario where a copy event is mistakenly identified as a large deletion.
Fig. 1.
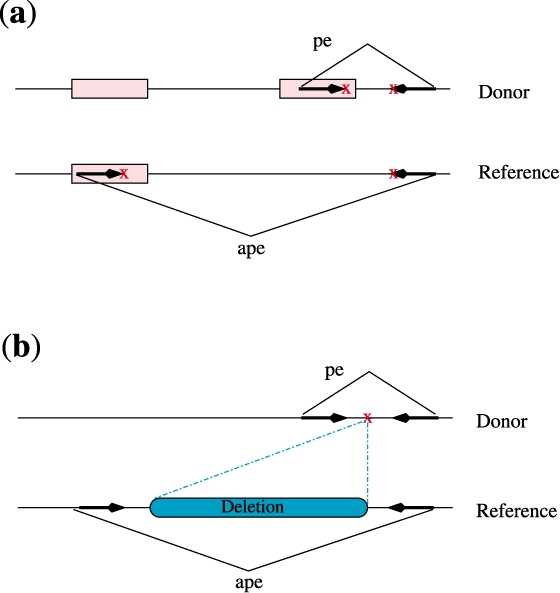
Transposon insertion causing a false negative deletion prediction. A discordant paired-end read alignment due to a copy event shows identical pattern with a discordant paired-end read alignment supporting a deletion event. (a) The presence of a copy event in the donor genome and the supporting discordant paired-end mapping (b) The same discordant mapping as above, however, suggests a large deletion if the annotated repeat element is not considered.
In what follows, we study two classes of copy events and present the set of conditions based on the map locations and orientations of the paired-end alignments that imply a copy event within each of these classes. First, we consider those copy events in which the transposed segment is in direct orientation, and present the set of conditions for all of the four different cases of this class (denoted as Class I). Next, we study the cases for Class II, where the transposon is copied in inverted orientation.
A copy SV of Class I is defined as SVCopy(PosL, PosR, PosBr), where the genomic segment from positions PosL to PosR is copied into location PosBr. Similarly, a copy event 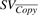 denotes a copy event in inverted orientation.
denotes a copy event in inverted orientation.
One of the following four cases should hold for a paired-end read alignment ape that supports a copy event SVCopy (Class I):
Case 1 (PosBr < PosL and or(ape)=+−) : Δmin < PosBr − Lr(ape) + Rℓ(ape) − PosL < Δmax (Fig. 2a)
Case 2 (PosBr < PosL and or(ape)=−+) : Δmin < Lℓ(ape) − PosBr − Rr(ape) + PosR < Δmax (Fig. 2b)
Case 3 (PosBr > PosR and or(ape)=+−) : Δmin < Rℓ(ape) − PosBr + PosR − Lr(ape) < Δmax (Fig. 2c)
Case 4 (PosBr > PosR and or(ape)=−+) : Δmin < PosBr − Rr(ape)+Lℓ(ape)−PosL < Δmax (Fig. 2d)
Fig. 2.
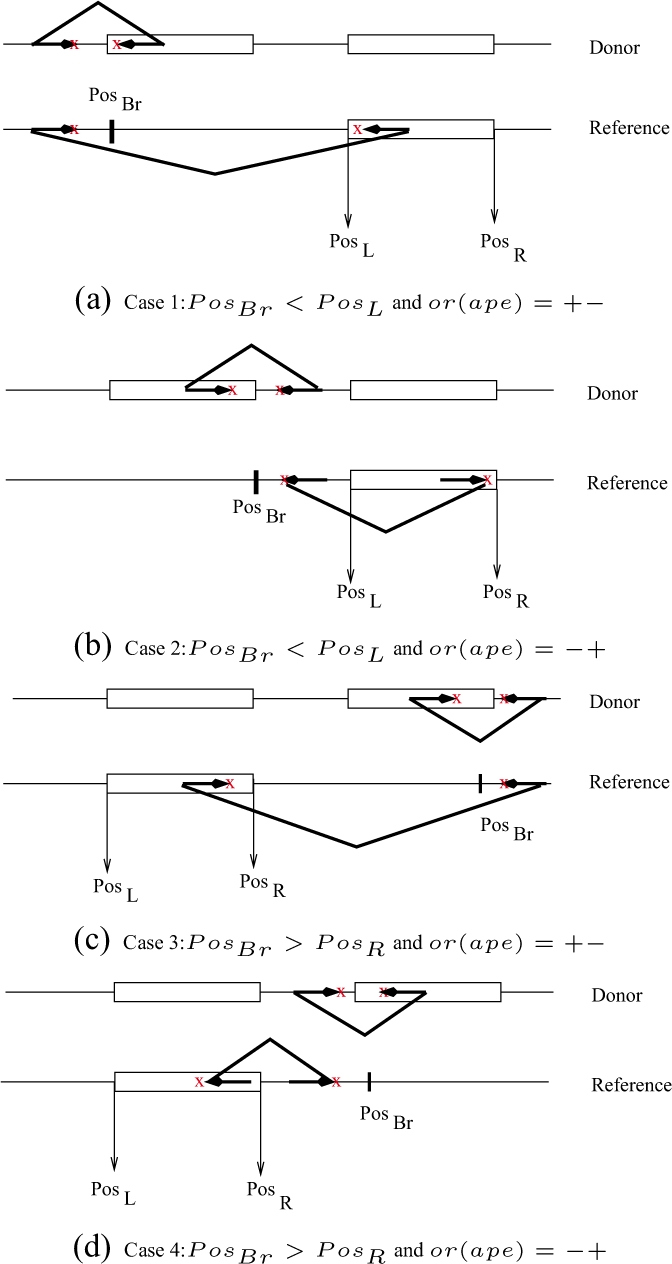
The set of conditions for each case that suggests a copy event in which the transposed segment is copied in direct orientation (Class I). (a) Case 1: PosBr < PosL and or(ape)=+−, (b) Case 2: PosBr < PosL and or(ape)=−+, (c) Case 3: PosBr > PosR and or(ape)=+− and (d) Case 4: PosBr > PosR and or(ape)=−+.
Similarly, one of the following cases should hold for a copy event of Class II:
Case 1 (PosBr < PosR and or(ape)=++) : Δmin < PosBr − Lr(ape)+PosR−Rr(ape) < Δmax (Fig. 3a)
Case 2 (PosBr < PosR and or(ape)=−−) : Δmin < Lℓ(ape) − PosBr+Rℓ(ape) − PosL < Δmax (Fig. 3b)
Case 3 (PosBr > PosR and or(ape)=++) : Δmin < PosBr − Rr(ape) + PosR − Lr(ape) < Δmax (Fig. 3c)
Case 4 (PosBr > PosR and or(ape)=−−) : Δmin < Rℓ(ape) − PosBr+Lℓ(ape)−PosL < Δmax (Fig. 3d)
Fig. 3.
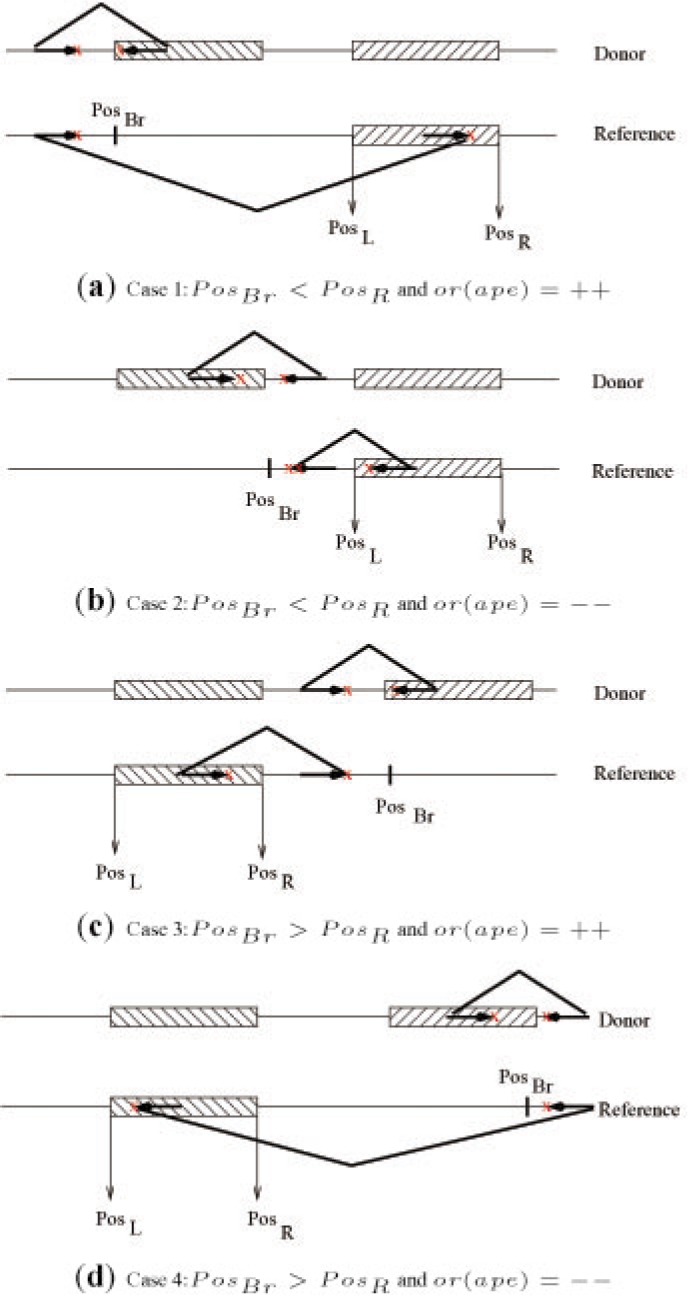
The set of conditions for each case that suggests a copy event in which the transposed segment is copied in inverted orientation (Class II). (a) Case 1: PosBr < PosR and or(ape)=++, (b) Case 2: PosBr < PosR and or(ape)=−−, (c) Case 3: PosBr > PosR and or(ape)=++, (d) Case 4: PosBr > PosR and or(ape)=−−,
3 MAXIMAL VALID CLUSTERS AND COPY EVENT DETECTION
A set of discordant paired-end read alignments that support the same potential SV event is called a ‘valid cluster’ and denoted by VClui = {ai1′pei1, ai2′ pei2,…, aiℓ′peiℓ}.
As per Hormozdiari et al. (2009), a ‘maximal valid cluster’ is defined as a valid cluster where no additional paired-end read alignments can be added such that the cluster remains valid. Note that all paired-end read alignments in maximal valid clusters suggest the same potential structural variation. It was shown that it is sufficient to calculate all maximal valid clusters to solve the MPSV problem. This can be done in polynomial time (with respect to the number of paired-end alignments), where the computation of all valid clusters is unnecessary, and exponential in run time. In Hormozdiari et al. (2009) and Sindi et al. (2009), efficient algorithms to find the maximal valid clusters are presented to predict insertion, deletion and inversion events.
To find all maximal valid clusters for copy events, a naïve method would investigate all O(n3) possibilities of potential copy events, for each of the locations PosBr, PosL and PosR between 1 and n, where n is the genome length. This can be done by first creating a cluster for each possible values of PosBr, PosL and PosR, and then adding those paired-end reads that satisfy the conditions given in Section 2 to the appropriate cluster. Finally, a set of maximal clusters would be selected. The above method guarantees to find all the maximal valid clusters but it would be time consuming in practice. In what follows, we will present a more efficient method to find all the maximal valid clusters provided that the potential positions of copied segments or copied sequences are known.
We define Φ = {(ϕ1ℓ, ϕ1r), (ϕ2ℓ, ϕ2r),…, (ϕtℓ, ϕtr)} as the set of (non-overlapping) segments that can be copied to other locations (Φ can represent the annotated transposons in the reference genome assembly). Note that ∀i≤t, ϕiℓ is the start location of the i-th segment and ϕir is the end location. The coordinates for the intervals are referred to as ‘end-points’ in the rest of the article for simplicity.
For each paired-end read mapping ape with exactly one end-read mapped to a transposon e.g. ϕi = (ϕiℓ, ϕir), there exists a range of locations, or ‘breakpoint intervals’ Briape = [BrL, BrR], where ape supports a copy of subsequence ϕi=(ϕiℓ, ϕir) to any location within Briape in the reference genome. Note that for a given ape and a segment ϕi = (ϕiℓ, ϕir), the breakpoint interval Briape can easily be computed using the set of conditions given in Section 2.
Now we present an efficient algorithm3 to find the maximal valid clusters supporting copy of segment ϕi=(ϕiℓ, ϕir) to any location in genome (the algorithm can trivially be extended for other segments in Φ).
Without loss of generality, we assume that there are total of mi discordant mappings with exactly one end mapped to ϕi (Please note that m=∑i=1t mi, where total number of segments is t). We denote the set of such discordant paired-end read mappings as apei={apei1, apei2,…, apeimi}. In addition the set of breakpoint intervals for the paired-end read alignments in set apei is denoted as Bri={(Bri1L, Bri1R), (Bri2L, Bri2R),…, (BrimiL, BrimiR)}.
It is trivial to see that finding maximal valid clusters for all copies of segment ϕi=(ϕiℓ, ϕir) into any position in the genome is equivalent to finding all maximal intersecting breakpoint intervals for all paired-end read mappings apei. Thus, we are interested in finding all maximal intersections of the breakpoint intervals Bri.
In what follows, we present an O(mi logmi + si) algorithm that outputs all the maximal intersecting intervals (of Bri), where si is the size of the output. This algorithm is the optimal solution to the problem. Please note that an algorithm that finds maximal valid clusters supporting copy of a given segment ϕi with running time of O(mi logmi + si) will yield an O(mlogm+S) algorithm for finding maximal valid clusters for all the segments in Φ when S = ∑i=1t si (since m = ∑i=1t mi).
3.1 Algorithm for finding all maximal intersecting intervals
Given a set of intervals (with cardinality of mi), we want to find all the maximal intersecting intervals. We first sort all end points of mi intervals (2mi coordinates) in ascending order based on their values. We call this sorted list of intervals L where each interval appears twice in the list L. We then scan the sorted list from left to right:
If we observe a point that is at the left end-point of an interval, we insert the interval to a minimum heap data structure, denoted as heap. The priority value of the heap is the right end point of the inserted interval. After each insertion of a new interval to heap, we set a flag newIns = true.
If we observe a point that is at the right end point of an interval, we first check the flag newIns. If it is set to true, we output all the elements in the heap as one maximal cluster and set newIns=false. We then remove the interval from heap since it is guaranteed that the value of the right end point of the interval removed from the heap is the same as the right end point of the interval reached in scanned list L. We continue removing intervals from heap until the priority value of the head element of the heap remains unchanged.
The above algorithm outputs all maximal intersecting intervals that are equivalent to the maximal valid clusters.
3.1.1 Complexity
It can be shown that the running time of the above algorithm is O(milogmi + s), where s is the size of the output. The sorting procedure in the first step of the algorithm takes O(mi log mi). In the worst case, since each removal/insertion operation in the heap takes O(logmi) time, the total run time for the second step is also O(milogmi). It takes O(s) time to write the output. In addition, it was previously proven that finding the maximum clique in an interval graph has a lower bound of Ω(milogmi) (Gupta et al., 1982). Thus, our algorithm gives the optimal solution for finding all maximal clusters.
3.2 Detection of transposon insertion events from maximal valid clusters
Each maximal valid cluster for different SV types (i.e. insertion, inversion, deletion or copy) only suggests a potential structural variation. The fact that each paired-end read can be mapped to multiple locations that are included in multiple maximal valid clusters proves that some of these implied potential variants are incorrect. We previously presented a combinatorial method to select the minimum number of these clusters (equivalent to selecting minimum number of SVs), such that each paired-end read has a map location in at most one selected valid cluster (Hormozdiari et al., 2009). This problem was called MPSV, and an approximation algorithm was given. The same algorithm presented can be used to find transposon insertion events from the maximal valid clusters using the algorithms presented in the previous section.
In the next section, we introduce an extension to MPSV problem and provide an efficient method to solve it. We show that our new method outperforms our previous algorithm in Hormozdiari et al. (2009).
4 MPSV WITH CONFLICT RESOLUTION
As mentioned before, the possibility of multiple map locations for each paired-end read raises the question of resolving which SVs implied by the maximal valid clusters are correct.
The MPSV problem (Hormozdiari et al., 2009) aims to compute a unique mapping for each discordant paired-end read in the reference genome such that the total number of implied SV is minimized (in this section we consider all classes of SV). In Hormozdiari et al. (2009), MPSV was modeled as a combinatorial optimization problem and shown to be NP-complete. An approximation algorithm (denoted as VariationHunter) based on the set-cover problem with O(logn) approximation factor was given. However, the modeling of the MPSV problem imposed no limits on the number of ‘overlapping’ SV predictions. A considerable amount of the predicated calls overlap with each other and a post-processing heuristic to filter some of those overlapping predicted SVs was given [see the Results section of Hormozdiari et al. (2009)]. In this section, we mathematically formulate these ‘conflicts’ and model the structural variation discovery problem as a novel combinatorial optimization problem.
4.1 Conflicting SV clusters in haploid and diploid genome sequences
We motivate the ‘conflict resolution’ for structural variation using a simple example. Given paired-end reads from a haploid genome, a structural variation detection algorithm (such as VariationHunter, MoDIL or BreakDancer) might construct two or more sets of valid clusters that imply multiple conflicting deletion calls (Fig. 4). Assuming the genome is haploid, it is not possible that both valid clusters in Figure 4 can be ‘correct’.
Fig. 4.
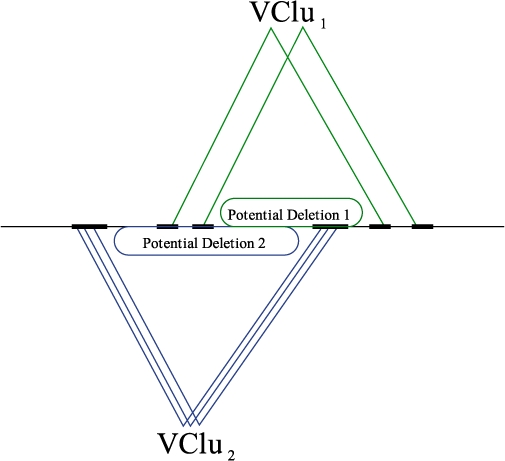
Two valid clusters Vclu1 and Vclu2 are shown to be in conflict in a haploid genome.
We first formalize the MPSV with Conflict Resolution (MPSV-CR) for both haploid and diploid genomes, and then we analyze the complexity of the MPSV-CR problem. Finally, we provide a heuristic solution for MPSV-CR. We call this solution as VariationHunter with Conflict Resolution (VariationHunter-CR).
Assuming a haploid genome, two valid clusters Vclu1 and Vclu2 of paired-end reads are conflicting if and only if there exists a potential scenario of SVs implied by the two valid clusters, such that the existence of one of the events makes the other valid cluster incoherent (Fig. 4). In the Supplementary Material, we present the set of rules to determine whether two valid clusters are in conflict in a haploid genome.
Through the conflict rules we can model the conflict representation of all clusters using a graph denoted as conflict graph. Each cluster is represented a node, and there exists an edge between two nodes if and only if the two corresponding clusters are in conflict with each other. It is not difficult to see that a valid set of SVs is a set of nodes/clusters in which no two nodes within the subset are connected. In another words, the valid solution (without any conflicts) is an independent set of the conflict graph.
One can easily generalize the definition of conflicting clusters to diploid genome sequences. Let Vclu1 and Vclu2 be two conflicting clusters in a haploid genome. However, provided that the genome is diploid, both Vclu1 and Vclu2 might imply a correct SV in different haplotypes. Now consider a third SV cluster, Vclu3 that is in conflict with both Vclu1 and Vclu2. It is trivial that, based on the pigeon hole principle, Vclu1, Vclu2 and Vclu3 cannot simultaneously occur in a diploid genome. In other words, the presence of three different SV clusters that are conflicting pairwise in a haploid genome4 will be a conflict in a diploid genome. The concept of the conflict graphs for haploid genomes can be replaced with conflict hypergraphs for diploid genomes. In the conflict hypergraph each hyperedge connects three nodes, if these three nodes are in conflict with each other.
Now, we define the MPSV-CR problem that does not only aim to minimize the total number of implied SVs, but also guarantees that no pairwise conflicting triplet of the implied SVs exists.
Note that this new version of the MPSV problem does not select any conflicting SVs and thus may not always be able to assign every paired-end read to a particular SV. Thus, the optimization function for MPSV-CR not only tries to minimize the number of SVs predicted, but also maximizes the number of paired-ends that can be assigned to a location in genome.
In what follows, we present the concept of conflicting SV clusters in more detail and give a formal definition of the MPSV-CR problem.
4.2 Formal definition of the MPSV-CR problem
In this section, we formally define the MPSV-CR problem. Let MC = {VClu1, VClu2, …, VClun} be the set of SV clusters and R={pe1, pe2,…, pem} be the collection of discordant paired-end reads. These discordant read pairs can be mapped to multiple locations in the genome, represented by Align(pei)={a1pei, a2pei,…, ajpei}.
In order to formulate the constraints, we define the conflict hypergraph CG as a hypergraph with vertex set V(CG) = MC and a hyperedge set E(CG) as follows. Between every three distinct SV clusters that are pairwise in conflict, there exists a hyperedge in E(CG):
Note that for the case when we only deal with a haploid genome (rather than a diploid genome), the hypergraph is nothing else than a simple graph (denoted by G) where each e ∈ E(G) represents a pair of conflicting SV cluster.
A subset SC ⊂ MC is satisfiable under the constraint hypergraph CG, if ∄e = (VClup, VCluq, VClur) ∈ E(CG): e⊆SC [in the case of a haploid genome a subset SC ⊂ MC satisfiable under the constraint graph G, if ∄e = (VClup, VCluq) ∈ E(G): e⊆SC].
For each satisfiable subset SC and each paired-end read pei, we define the indicator variable δ(SC, pei) as follows:
Intuitively, δ(SC, pei) is the penalty for not assigning the pair-end read pei to a cluster in the satisfiable subset SC. The MPSV-CR problem aims to find the satisfiable set SC′ such that f(SC′)=|SC′|+∑pe∈R δ(SC′, pe) is minimized (i.e. to find a trade-off between the number of SV clusters in a satisfiable set and the number of paired-end reads that are not assigned to any SV clusters.).
4.2.1 Computational complexity of MPSV-CR
Here we prove that MPSV-CR is NP hard, independent from the weights on the cardinality of SC′ and the penalty for unmapped reads [i.e. minimizing the function g(SC′)=k · |SC′| + l·∑pe∈R δ(SC′, pe) for any k > 0 and l > 0 is NP hard]. The NP hardness proof follows a reduction from the minimum set cover problem (See the Supplementary Materials for the detailed proof). In addition, we show an inapproximability result for MPSV-CR problem even when the genome is assumed to be haploid.
Theorem 4.1. —
There is no constant ε > 0 for which MPSV-CR on haploid genome can be approximated within a factor of n1−ε in polynomial time, unless P = NP.
Proof of Theorem 4.1 given in the Supplementary Material.
4.3 An efficient solution to the MPSV-CR problem
In this section, we present a heuristic solution for a special case of the MPSV-CR problem where both k and l (the coefficients in the optimization function g) are set as k = 1, l = 1. This heuristic, named as Max_Assigned_Reads, consists of two phases.
In the first phase, we form a maximal satisfiable set of SV clusters (denoted by MS) in a greedy fashion. Note that a satisfiable set of SV clusters, SC (as noted in the previous section), is called maximal if no other SV cluster VClu can be added to SC such that VClu together with two existing SV clusters in SC form a hyperedge in the conflict hypergraph CG. We start with MS = ∅ and then iteratively add the SV cluster that covers the most number of paired-end reads5 to MS until MS becomes a maximal satisfiable set. Note that we add an SV cluster SVk to MS in an iteration even if SVk does not cover any new discordant paired-end read (provided that MS remains satisfiable). We denote MR as the set of all paired-end reads covered by the SV clusters in MS.
In the second phase of Max_Assigned_Reads, the aim is to select the minimum number of SV clusters from MS that cover all paired-end reads in MR. For this phase, we use a set cover approach similar to Hormozdiari et al. (2009).
In what follows, we give a lower bound on the cardinality of MR.
The analysis of the second phase of Max_Assigned_Reads is similar to Hormozdiari et al. (2009). Let m be the total number of discordant paired-end reads, and let neighbors(VClu)={VClu′|∃e∈E(CG), VClu, VClu′ ∈ e}, deg(VClu)=|neighbors(VClu)| and Δ = max{deg(VClu)|VClu ∈ MC}, i.e. the maximum degree of a vertex in the conflict graph CG.
Theorem 4.2. —
|MR|≥m/(Δ + 1).
Proof. —
Let k be number of iterations of Max_Assigned_Reads. For each i (1 ≤ i ≤ k), we denote VClui as the cluster that is selected at the i-th iteration. We also denote MRi as the set of paired-end reads that are covered by VClui for the first time. Furthermore, we define URi as the set of paired-end reads that are not covered by any of the SV clusters VClu1 through VClui and also is not able to be covered later (as the result of selecting VClui) in the remaining k − i iterations.
At the i-th iteration, the maximum number of reads that could be covered for all neighbors of VClui is at most Δ|MRi|, thus |URi| ≤ Δ|MRi|. Moreover, we have ∑i=1k(URi + MRi) = m and |MR| = ∑i=1k MRi. Hence m/(Δ + 1) ≤ |MR|.
5 EXPERIMENTAL RESULTS
5.1 Implementation
It is established that there are a large number of mobile elements in the human genome (Mills et al., 2007). For example, the reference human genome assembly annotation includes 1 million copies of the Alu element. Considering all known mobile elements (segments in genome) as potential transposon sequences for our algorithm would be very time consuming, and in fact, unnecessary. The consensus sequences for all mobile elements are well studied, and available at the RepBase database (Jurka et al., 2005). We used the consensus sequences of these mobile element families as representative sequences to facilitate faster experimentation. To this end, we create a new sequence, denoted as chrN: we first append a poly-N sequence to each consensus sequence, and then concatenated them, generating chrN. In this article, we considered only Alu and SVA elements in our experiments.
For read mapping, we use mrsFAST (F.Hach et al., unpublished data), a cache-oblivious short read mapper recently developed as an extension of mrFAST (Alkan et al., 2009). mrsFAST maps the paired-end reads to all locations with Hamming distance less than a user-defined threshold ω. In this experiment we set ω = 2.
Given paired-end whole-genome shotgun sequence library R = {pe1, pe2, …pen}, we follow the following steps to obtain the reads (and mapping information) for transposon discovery:
We first map all paired-end reads to chrN using mrsFAST and discard such paired-end reads that cannot be mapped to chrN. We keep the read pairs where one end read is mapped to chrN.
Next, we map the reads we determine in the previous step to the reference genome, and discard all paired end reads with at least one concordant mapping.
Finally we remap the reads from Step 2 to both chrN and the reference genome. As a post-processing step, we select the paired-end alignments where one end is mapped to chrN and the other end is mapped to the reference genome.
5.2 Mobile element insertion discovery in the Venter genome
A list of mobile element insertions in the Venter genome assembly (HuRef) (Levy et al., 2007) in comparison to reference human genome assembly (NCBI build 36) genome was recently published (Xing et al., 2009). We used the available HuRef genome to produce short paired-end reads, similar to reads generated by the Illumina technology (simulating an Illumina sequencing of the Venter genome) to benchmark the sensitivity and specificity of our algorithms. To our knowledge, this is the only dataset for mobile element insertion annotations from the genome of a single individual. We created paired-end reads from the HuRef genome with a read length of 36 bp, and obtained 10-fold sequence coverage. The fragment insert sizes for paired-end reads were chosen randomly that follows a normal distribution very similar to the fragment size distribution in the NA18507 shotgun sequence library generated using the Illumina platform (Bentley et al., 2008).
We used our mobile element insertion discovery algorithm to discover transposons in the autosomes of HuRef (from chr1 to chr22). In our experiments, we focused on Alu, NCAI (Non-classical Alu Insertion)6 and SVA insertions. The results of our experiment are summarized in Table 1. The validated set of mobile element insertions in the HuRef assembly is a union of the published transposon insertions in Xing et al. (2009) and our new transposon predictions not listed in Xing et al. (2009) but are included in HuRef.
Table 1.
Summary of mobile element insertion prediction results in the Venter genome
| Chromosome | Validated | Predicted | Found | Recall (%) | Precision (%) |
|---|---|---|---|---|---|
| Chromosome 1 | 41 Alu | 42 Alu | 40 | 88 | 95 |
| 4 NCAI | |||||
| Chromosome 2 | 59 Alu | 57 Alu | 56 | 91 | 98 |
| 2 NCAI | |||||
| Chromosome 3 | 42 Alu | 40 Alu | 40 | 90 | 100 |
| 1 NCAI | |||||
| 1 SVA | |||||
| Chromosome 4 | 43 Alu | 41 Alu | 40 | 87 | 97 |
| 4 NCAI | 1 NCAI | 1 | |||
| Chromosome 5 | 39 Alu | 44 Alu | 35 | 90 | 80 |
| 1 SVA | 1 SVA | 1 | |||
| Chromosome 6 | 59 Alu | 55 Alu | 53 | 88 | 96 |
| 1 NCAI | |||||
| 1 SVA | 1 SVA | 1 | |||
| Chromosome 7 | 24 Alu | 22 Alu | 20 | 83 | 91 |
| Chromosome 8 | 34 Alu | 33 Alu | 33 | 92 | 100 |
| 2 NCAI | |||||
| Chromosome 9 | 23 Alu | 23 Alu | 21 | 88 | 92 |
| 1 NCAI | 1 NCAI | 1 | |||
| 1 SVA | |||||
| Chromosome 10 | 33 Alu | 32 Alu | 32 | 94 | 100 |
| 2 NCAI | 1 NCAI | 1 | |||
| Chromosome 11 | 35 Alu | 32 Alu | 32 | 85 | 100 |
| 3 NCAI | 1 NCAI | 1 | |||
| 2 SVA | 1 SVA | 1 | |||
| Chromosome 12 | 33 Alu | 34 Alu | 31 | 84 | 91 |
| 4 NCAI | |||||
| Chromosome 13 | 34 Alu | 34 Alu | 34 | 90 | 100 |
| 3 NCAI | |||||
| 2 SVA | 1 SVA | 1 | |||
| Chromosome 14 | 19 Alu | 20 Alu | 19 | 95 | 95 |
| 1 NCAI | |||||
| 2 SVA | 2 SVA | 2 | |||
| Chromosome 15 | 24 Alu | 21 Alu | 20 | 83 | 95 |
| Chromosome 16 | 12 Alu | 12 Alu | 12 | 80 | 100 |
| 3 NCAI | 1 NCAI | 1 | |||
| Chromosome 17 | 9 Alu | 9 Alu | 9 | 77 | 100 |
| 2 NCAI | |||||
| 2 SVA | 1 SVA | 1 | |||
| Chromosome 18 | 22 Alu | 21 Alu | 20 | 91 | 95 |
| Chromosome 19 | 11 Alu | 11 Alu | 11 | 80 | 100 |
| 3 NCAI | 1 NCAI | 1 | |||
| 1 SVA | |||||
| Chromosome 20 | 11 Alu | 13 Alu | 9 | 77 | 71 |
| 2 NCAI | 1 NCAI | 1 | |||
| Chromosome 21 | 7 Alu | 7 Alu | 7 | 100 | 100 |
| Chromosome 22 | 7 Alu | 5 Alu | 5 | 71 | 100 |
We show the precision and recall rates of our mobile element (Alu, NCAI and SVA) insertion discovery. We compare our mobile element insertion predictions with both (Xing et al., 2009) and the HuRef genome assembly (Levy et al., 2007). The results demonstrate that our algorithm has a high recall and precision rate.
As shown in Table 1, our method was able to find most of the known/validated mobile element insertions (recall rate is >85%), while the number of invalidated predictions is very low (precision rate ∼90%).
Interestingly, most of the Alu insertions missed by our algorithm were truncated insertions (significantly smaller than the consensus sequences). Figure 5 summarizes the true positive/false negative results with respect to the length of Alu insertion.
Fig. 5.

The length distribution of true positive and false negative mobile element insertion predictions for Alu elements. Note that all of the Alu consensus sequences used in creating chrN are longer than 250 bp.
5.3 Structural Variation Prediction with VariationHunter-CR
In this section, we show that the call set predicted by VariationHunter-CR has a lower false positive rate than the original VariationHunter (Hormozdiari et al., 2009) while retaining the same true positive rate (Supplementary Material includes simulations supporting this claim). As an experiment we compare the deletion predictions using different algorithms including VariationHunter-CR in the genome of a Yoruba African donor (NA18507) sequenced with the Illumina Genome Analyzer platform (Bentley et al., 2008). We include the validated deletions detected in the genome of the same individual using fosmid clone-end sequencing (Kidd et al., 2008) in our comparisons.
We downloaded ∼3.5 billion end sequences (∼1.7 billion pairs) of length 36–41 bp and insert size ∼200 bp from the NCBI Short Read Archive (ftp://ftp.ncbi.nih.gov/pub/TraceDB/ShortRead/SRA000271). Similar to the prescreening methodology used in Hormozdiari et al. (2009), we removed any pairs of reads from consideration if either (or both) end sequences has average phred (Ewing and Green, 1998) quality score <20, or if either (or both) sequences contain more than 2N characters. In total, ∼1.3 billion reads were removed from this dataset. We then mapped all the remaining ∼2.2 billion end sequences to the human reference genome using mrFAST (Alkan et al., 2009). The average insert size was determined to be 209 bp, where the SD was 13.4 bp.
Figure 6 shows the comparison of our new VariationHunter-CR algorithm with the original VariationHunter, the curated (post-processed to remove conflicting predictions) result published in Hormozdiari et al. (2009) and BreakDancer (Chen et al., 2009).
Fig. 6.

Prediction performance comparisons of VariationHunter (black), VariationHunter-CR (red) and BreakDancer (blue). We also show the curated (post-processed) results of VariationHunter in this figure (green). The x-axis represents the number of deletions predicted by each method, and y-axis is the number of validated deletions from (Kidd et al., 2008) that overlaps (>50% reciprocal overlap) with a prediction. It is desirable to obtain a prediction set that is able to find more validated calls with less number of total calls. For VariationHunter and VariationHunter-CR, we give the number of calls and number of validated deletions found for different support levels (number of paired-end read supporting them); less support level results in more predicted deletion intervals. This figure shows that VariationHunter-CR has a better performance than VariationHunter for all support levels, and VariationHunter-CR outperform the BreakDancer algorithm (Chen et al., 2009).
6 CONCLUSIONS
Despite recent advances in algorithm design for the detection of structural variation, due to the difficulty in discovering complex variants in regions of the genome with high plasticity the existing algorithms are still at their infancy. In this article, we presented the ‘Next-Generation’ of our structural variation discovery algorithm VariationHunter that aims to improve both the sensitivity and specificity of SV detection. VariationHunter is now capable of resolving incompatible SV calls through a conflict resolution mechanism that no longer requires post-processing heuristics. Furthermore, we described additional algorithms to discover mobile element insertions that are of known importance to genome evolution and genomic variation. These enhancements provide a much needed step toward a highly reliable and comprehensive structural variation discovery algorithm, which, in turn will enable genomics researchers to better understand the variations in the genomes of newly sequenced human individuals, as well as the genome structures of non-human species.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank T. Brown, L. Brunner and H. Yaghoubi for their help in manuscript preparation. E.E.E. is an investigator of the Howard Hughes Medical Institute.
Funding: Natural Sciences and Engineering Research Council of Canada (NSERC); Genome BC grants (to S.C.S); US National Institutes of Health (grants HG004120 and U01HG005209-02 to E.E.E).
Conflict of Interest: EEE is a SAB member of Pacific Biosciences.
Footnotes
1The two ends of a read pair is referred as end reads.
2Referring to an insertion, deletion and inversion event, respectively.
3In fact the algorithm is optimal with respect to the running time.
4According to our definition of conflicts in a haploid genomes.
5We count the paired-end reads that were not previously covered by any SVs in MS.
6Alu insertions that only contain the internal fragment of Alu are called non-classical Alu insertions (NCAI; Xing et al., 2009).
REFERENCES
- Alkan C, et al. Personalized copy number and segmental duplication maps using next-generation sequencing: Nat. Genet. 2009;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, et al. An Alu transposition model for the origin and expansion of human segmental duplications. Am. J. Hum. Genet. 2003;73:823–834. doi: 10.1086/378594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir A, et al. Evaluation of paired-end sequencing strategies for detection of genome rearrangements in cancer. PLoS Comput. Biol. 2008;4:e1000051. doi: 10.1371/journal.pcbi.1000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekpen C, et al. Death and resurrection of the human IRGM gene. PLoS Genet. 2009;5:e1000403. doi: 10.1371/journal.pgen.1000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, et al. Breakdancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, et al. Integrating sequencing technologies in personal genomics: optimal low cost reconstruction of structural variants. PLoS Comput. Biol. 2009;5:e1000432. doi: 10.1371/journal.pcbi.1000432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. error probabilities. Genome Res. 1998;8:186–94. [PubMed] [Google Scholar]
- Feuk L, et al. Structural variation in the human genome. Nat. Rev. Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- Gupta UI, et al. Efficient algorithms for interval graphs and circular-arc graphs. Networks. 1982;12:459–467. [Google Scholar]
- Hancks DC, et al. Exon-trapping mediated by the human retrotransposon SVA. Genome Res. 2009;19:1983–1991. doi: 10.1101/gr.093153.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollox E, et al. Psoriasis is associated with increased bold beta-defensin genomic copy number. Nat. Genet. 2008;40:23–25. doi: 10.1038/ng.2007.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F, et al. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res. 2009;19:1270–1278. doi: 10.1101/gr.088633.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, et al. Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Kidd JM, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, et al. PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol. 2009;10:R23. doi: 10.1186/gb-2009-10-2-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, et al. A robust framework for detecting structural variations in a genome. Bioinformatics. 2008;24:i59–i67. doi: 10.1093/bioinformatics/btn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, et al. Modil: detecting small indels from clone-end sequencing with mixtures of distributions. Nat. Methods. 2009;6:473–474. doi: 10.1038/nmeth.f.256. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nat. Genet. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev P, et al. Computational methods for discovering structural variation with next-generation sequencing. Nat. Methods. 2009;6:13–20. doi: 10.1038/nmeth.1374. [DOI] [PubMed] [Google Scholar]
- Mills RE, et al. Which transposable elements are active in the human genome? Trends Genet. 2007;23:183–191. doi: 10.1016/j.tig.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, et al. Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Res. 2010 doi: 10.1101/gr.102970.109. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindi S, et al. A geometric approach for classification and comparison of structural variants. Bioinformatics. 2009;25:i222–i230. doi: 10.1093/bioinformatics/btp208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzun E, et al. Fine-scale structural variation of the human genome. Nat. Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- Volik S, et al. End-sequence profiling: sequence-based analysis of aberrant genomes. Proc. Natl Acad. Sci. USA. 2003;100:7696–701. doi: 10.1073/pnas.1232418100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, et al. Mobile elements create structural variation: analysis of a complete human genome. Genome Res. 2009;19:1516–1526. doi: 10.1101/gr.091827.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.