

Abstract
Motivation: Finding novel or non-standard metabolic pathways, possibly spanning multiple species, has important applications in fields such as metabolic engineering, metabolic network analysis and metabolic network reconstruction. Traditionally, this has been a manual process, but the large volume of metabolic data now available has created a need for computational tools to automatically identify biologically relevant pathways.
Results: We present new algorithms for finding metabolic pathways, given a desired start and target compound, that conserve a given number of atoms by tracking the movement of atoms through metabolic networks containing thousands of compounds and reactions. First, we describe an algorithm that identifies linear pathways. We then present a new algorithm for finding branched metabolic pathways. Comparisons to known metabolic pathways demonstrate that atom tracking enables our algorithms to avoid many unrealistic connections, often found in previous approaches, and return biologically meaningful pathways. Our results also demonstrate the potential of the algorithms to find novel or non-standard pathways that may span multiple organisms.
Availability: The software is freely available for academic use at: http://www.kavrakilab.org/atommetanet
Contact: kavraki@rice.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Over the last few decades, experimental studies on metabolic networks, coupled with computational methods, have generated increasingly large amounts of data. In turn, many specialized databases have been created to store and organize information about metabolic networks (Caspi et al., 2008; Kanehisa et al., 2008). These networks are usually presented as many small subpathways that are manually divided based on function. However, it is often difficult to navigate these subpathways to find connections between compounds, especially for novel or non-standard routes used in applications such as metabolic engineering. Furthermore, as more information is accumulated from metagenome work on multi-species communities, it is of interest to find pathways that are a composition of the metabolic pathways from multiple organisms. Combining parts of pathways existing in different organisms can lead to new ways of considering how metabolism works in complex communities and provide novel routes to form important and useful compounds. Consequently, computational identification of biologically relevant pathways in large metabolic networks is required for applications such as metabolic engineering, metabolic network analysis and metabolic network reconstruction (Faust et al., 2009; Planes and Beasley, 2008).
The main contribution of this article is a pair of algorithms that find metabolic pathways by using atom mapping data to track the movement of atoms through metabolic networks. One algorithm finds linear pathways and the other algorithm finds branched pathways. They both take as input atom mapping data, a start compound, a target compound, a minimum number of atoms to conserve and a maximum number of pathways to return. A set of metabolic pathways, which conserve at least given number of atoms from the start compound to the target compound, are returned.
Atom tracking is a crucial feature in finding meaningful metabolic pathways because it essentially eliminates spurious connections and reactions that do not correspond to useful or real biochemical pathways or reactions, which are present in earlier work (Arita, 2004; Faust et al., 2009). Furthermore, we are able to harness atom tracking in order to find branched pathways. While atom tracking does increase the complexity of finding pathways, we demonstrate that our algorithms can efficiently identify both linear and branched metabolic pathways, in which a certain threshold of atoms are conserved. The resulting metabolic pathways are validated on known functional pathways and reveal the potential of our algorithms to find novel or alternative pathways that may span multiple organisms.
2 PREVIOUS WORK
Prior work on path finding in metabolic networks has focused on finding realistic linear pathways and mostly avoided using atom tracking, perhaps due to the increase of complexity and the previous unavailability of data. Initial analysis of metabolic networks was based on the shortest paths in directed graph representations of metabolic networks (Jeong et al., 2000; Ravasz et al., 2002). However, it was revealed that many of the shortest pathways in the directed graph may be biologically meaningless because they route through highly connected cofactors or pool metabolites (Arita, 2003, 2004; Ma and Zeng, 2003). Several approaches have been developed to overcome the problem of meaningless connections, such as removing these compounds from the graph (Gerlee et al., 2009; Wagner and Fell, 2001) or adding weights based on the degree of the nodes (Croes et al., 2006; Faust et al., 2009). Other approaches use measures of structural similarity between compounds as a heuristic to avoid spurious connections when finding metabolic pathways (McShan et al., 2003; Rahman et al., 2005).
Earlier work has also identified pathways by building stoichiometric models of metabolic networks (Planes and Beasley, 2008). These stoichiometric approaches typically focus on one organism or system, and require the user to define which compounds are present or available to the cell. In contrast, our approach strives to find interesting metabolic systems without information such as what organisms or what environments the organisms function in.
Our approach is more closely related to approaches that explicitly use atom mapping data, which identifies exactly where each atom in each input compound ends up in the output compounds of a reaction (Boyer and Viari, 2003; Pitkänen et al., 2009). Atom mapping data has primarily been used in earlier work as a filter to remove pathways that do not conserve at least one atom, usually a carbon, from the start to the end compound (Arita, 2003, 2004; Blum and Kohlbacher, 2008a, b). It also improved pathfinding results when used to construct a graph where edges are only drawn between compounds that share an atom mapping (Faust et al., 2009; Mithani et al., 2009).
Biochemical intuition says that pathways that move a high percentage of atoms from start to finish compounds will be biologically relevant. It has been shown previously that this problem is PSPACE-complete; when a compound can only be used once in a pathway, the problem is NP-complete (Boyer and Viari, 2003). Despite the complexity, this previous work provides inspiration that linear atom conserving pathways can be found efficiently in practice. The linear pathfinding algorithm in our article is able to run efficiently on a dataset containing about twice as many compounds and three and half times more atom mappings than the dataset used in Boyer and Viari (2003). Furthermore, explicit tracking of the atom enables the identification of branched pathways.
The first algorithm to use atom mapping information to find branched pathways, called ReTrace, was recently introduced and successfully used to reconstruct the metabolic network of Trichoderma reesei (Jouhten et al., 2009; Pitkänen et al., 2009). Our branched pathway algorithm, developed independently, also augments linear pathways to find branched pathways that maximize the number of atoms conserved from the start to the target compound. One key difference is that our method explicitly finds linear pathways that conserve at least a given number of atoms. In contrast, ReTrace finds pathways that conserve one atom and requires weighting heuristics to help find paths that conserve a larger number of atoms. Since both methods use various heuristics and cutoffs to overcome the high complexity of finding branched pathways, the selection of which method to use may be dependent on the specific application or compounds being studied. As these methods are adopted, a better understanding and comparison of the practical performance of these methods will become possible and help identify areas for future improvement.
This article describes how a graph containing atom mapping information (Section 3) can be used to find atom conserving linear pathways (Section 4) that give rise to complex branched pathways (Section 5). Our results are demonstrated through representative examples (Section 6).
3 ATOM MAPPING GRAPH CONSTRUCTION
Until recently, the development of automated ways to track atoms through large metabolic networks has been hindered by a lack of atom mapping data. Fortunately, large scale curation efforts have resulted in the increased availability of atom mapping data for chemical reactions. Progress has also been made in computational tools for automatically generating correct atom mappings, which can be used to fill in the gaps of the manual curation process (Akutsu, 2004; Blum and Kohlbacher, 2008a, b). In this work, we use data from the Kyoto Encyclopedia of Genes and Genomes (KEGG) due to the existence of the curated KEGG RPAIR database (Kanehisa et al., 2008). Each KEGG RPAIR entry contains structural information for each compound, an alignment mapping atoms between the two compounds and a list of associated reactions (Kanehisa et al., 2006). We process the KEGG RPAIR data to create a universal index for each atom in each compound as well as remove entries where the atom types mapped are different. The data also contains generic compounds such as ‘alcohol’ where the structure contains a ‘R’. We include mappings with generic mappings where the ‘R’ is not mapped to a specific atom, otherwise it is discarded. This processing of the data results in discarding about one percent of the KEGG RPAIR entries.
A frequently used representation of metabolic networks is a directed graph where there are reaction nodes and compound nodes, and edges are drawn between the compounds and the reactions they participate in. We found that tracking atoms through this representation typically results in unreasonably high computational cost, because of compounds, such as cofactors, participating in a large number of reactions. Therefore, we create an atom mapping graph, Gam, built upon the observation that the same atom mapping pattern between two compounds often appears in multiple reactions (Arita, 2003). For example, adenosine triphosphate (ATP) to adenosine diphosphate (ADP) occurs in many reactions, but the atom mapping remains the same between the two compounds. When searching Gam, only one node representing the ATP to ADP atom mapping needs to be explored. This is more efficient than explicitly exploring all of the reactions containing the atom mapping. In our experiments, this representation is important to help reduce the computational cost required to find atom conserving pathways.
Gam is a directed bipartite graph containing compound nodes and mapping nodes. Building Gam starts by adding a compound node for each compound in the RPAIR database. Each compound node has a unique identifier as well as a unique identifier for each of the atoms in the compound. We add a mapping node for each RPAIR atom mapping entry and create two directed edges, one from the first compound to this node and one from this node to the second compound. The mapping nodes contain atom mapping information, such that the atom identifiers from the reactant compounds are associated with the atom indices in the output compounds. For each mapping node, another mapping node is added to enable the reverse direction; it has the same edges created but in the reverse direction.
We currently make all mappings reversible due to the lack of readily available reversibility information of reactions. As this information becomes more readily available, it could be incorporated into the graph easily by only allowing the proper direction to be added to the graph. The KEGG RPAIR data also does not typically account for molecular symmetry. If we know a compound is symmetric, we can then create additional mapping nodes to account for the symmetry of the molecule. However, automatically identifying symmetric molecules can be complicated by stereochemistry, and therefore in this article we only add nodes explicitly represented in the KEGG RPAIR data.
Figure 1 shows a small subgraph of Gam. In this subgraph, a linear path from C00231 to C05345 through RP00080 and RP13340 conserves three carbon atoms. All mapping nodes only have one input edge and one output edge connected to two different compound nodes. The compound nodes have the same number of outgoing and incoming edges equal to the number of atom mapping entries they participate in. Therefore, the degree of the nodes of Gam is less than the more traditional compound and reaction directed graph, which in turn contributes to the efficiency of the pathfinding methods.
Fig. 1.

A small subgraph of Gam containing three compound nodes and two mapping nodes representing two atom mappings: RP00080 between C00231 and C00118, and RP13340 between C00118 and C05345. The KEGG RPAIR database contains information for all non-hydrogen atoms, but only carbons are depicted for clarity.
4 LINEAR ATOM CONSERVING PATHWAYS
The linear pathway search guarantees finding the k-shortest pathways that conserve at least a given number of atoms. The linear pathway search takes as input Gam, a start compound, a target compound and the minimum number of atoms to be conserved. Maximal atom conserving linear pathways are found by starting with the number of atoms in the smaller of the start and target compounds and decrementing by one until pathways are found or a minimal number of atoms is reached.
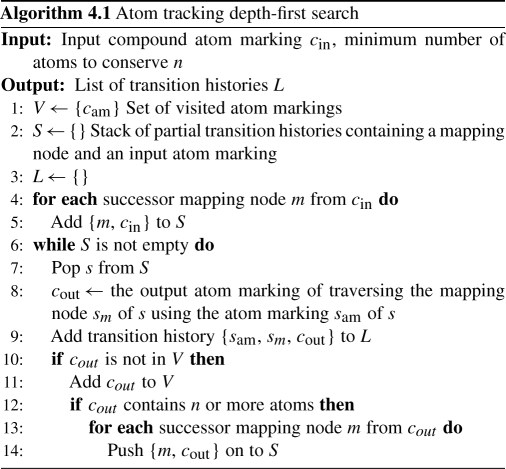
Finding linear pathways starts with an exploration step that traverses Gam in a depth-first manner while explicitly tracking, where each atom from the starting compound goes along the way. The exploration is a modified version of a standard depth first search because the semantics of Gam are different. These semantics require that the exact set of atoms visited in each compound are recorded, and not just the fact that the node itself is visited as in traditional graph traversals. Therefore, we term a string containing the compound identifier along with an ordered list of atom identifiers an atom marking. When the traversal moves through a mapping node, we use the input atom marking to compute the output atom marking based upon the mapping contained in the mapping node. For example, in Figure 1 if we started with all carbon atoms in d-xylulose 5-phosphate the atom marking would be ‘C00231 C0 C1 C2 C3 C4’, then taking the mapping node RP00080 would result in the atom marking ‘C00118 C0 C1 C2’. An object containing the input atom marking, the identifier of the mapping node taken and the resulting output atom marking is termed a transition history. Using the atom markings and transition histories, we can now introduce atom tracking depth-first search in algorithm 4.1. This search starts from the starting compound and explores Gam to find all reachable states that conserve the given number of atoms. The result of the search is a list of transition histories, L, which is then used to build an auxiliary graph.
The auxiliary graph has the important property that it contains all paths from the starting compound's atom marking that conserve at least the given number of atoms. This allows us to use it as input to standard algorithms for finding the k-shortest paths in a graph. The auxiliary graph contains a node labeled with each atom marking found in L. For each transition history, a new node containing the mapping node identifier is added. Due to the fact that the same mapping node can be traversed using different atom markings, the mapping node identifier is appended with a counter that is incremented every time the same mapping node is added. Then an edge is drawn from the input atom marking node to the node representing the mapping node, and then from the node representing the mapping node to the output atom marking node. The auxiliary graph has the important property that it contains all paths from the starting compound's atom marking that conserve at least the given number of atoms. Finally, Eppstein's k-shortest path algorithm is then run on this auxiliary graph with the appropriate start and target nodes as input and the resulting set of pathways are linear atom conserving pathways (Eppstein, 1998). Depictions of the auxiliary graph construction can be found in Supplementary Figures 2–4.
5 BRANCHED ATOM CONSERVING PATHWAYS
In addition to eliminating inappropriate transitions, atom tracking identifies where atoms are lost and gained along a linear pathway and enables finding branched pathways. Branched pathway finding starts by obtaining a set of linear pathways between the desired start and target compounds by using the previously described algorithm. In this section, we first describe how the linear pathways give rise to seed pathways, where transitions that lose or gain atoms are replaced by specific reactions. Then, we describe how seed pathways are used to obtain the resulting set of branched pathways.
5.1 Seed pathways
Let Pl be the set of linear atom conserving pathways found in Section 4. Each pathway in Pl will potentially give rise to a number of seed pathways, in which loss or gain atom mappings are replaced by reactions. While Gam contains enough information to find linear pathways, it is missing information about which compounds the atoms are lost or gained through. This information is found in the reactions in the KEGG REACTION database that correspond to the atom mappings. Therefore, we store a correspondence, provided by KEGG, between mapping nodes and reactions. For example, RP00080 in Figure 1 is associated with six different reactions in KEGG, which use and produce different compounds in addition to d-xylulose 5-phosphate and d-glyceraldehyde 3-phosphate. Two of the reactions are illustrated in Supplementary Figure 1. In one reaction, d-xylulose 5-phosphate reacts with formaldehyde to produce d-glyceraldehyde 3-phosphate and glycerone. In the other, d-xylulose 5-phosphate reacts with orthophosphate to produce d-glyceraldehyde 3-phosphate and acetyl phosphate. The important difference is that in the first reaction the C0 and C1 carbons of d-xylulose 5-phosphate end up in glycerone and in the second reaction they end up in acetyl phosphate. Hence, the starting compound of the branch is different depending upon which reaction is used.
For each p ∈ Pl, two types of mapping nodes are identified: loss mapping nodes (LMNs) and gain mapping nodes (GMNs). LMNs are mapping nodes where the atom mapping does not map all of the atoms in the input compound and GMNs are mapping nodes where the atom mapping does not map all of the atoms in the output compound. For example, RP00080 in Figure 1 would be considered an LMN. To create the seed pathways, we need to (i) replace each LMN in p with all possible corresponding reactions—called loss reactions nodes (LRNs) and (ii) replace each GMN in p with all possible corresponding reactions—called gain reaction nodes (GRNs). All possible combinations should be produced to obtain the seed pathways. For example, since RP00080 in Figure 1 is found in six reactions in KEGG at least six seed pathways would be created, each one containing one of the reactions. Depending on the reactions corresponding to GMN RP13340 of Figure 1 more seed pathways may be generated until we have pathways containing all possible combinations of LRNs and GRNs. Again, we face the possibility of theoretical combinatorial explosion as the number of seed pathways is equal to the number of possible combinations of reactions along the pathway. However, in practice, we observed most pathways have few LMNs and GMNs, which also typically correspond to only a few reactions.
5.2 Finding branched pathways from seed pathways
To attach branches properly to seed pathways, we must now add compounds nodes involved in the reactions represented by LRNs and GRNs through which atoms can be lost and gained. These ‘new’ compound nodes will be the starts and ends of branches. For each seed pathway, we examine its LRNs and add the output compound nodes from the corresponding reaction through which atoms may be lost; a directed edge is created from the LRN to the node. We then examine the seed pathways' GRNs and add input compound nodes from the corresponding reaction through which atoms may be gained; a directed edge is created from the node to the GRN. This new construction containing compound nodes through which atoms can be lost and gained, is termed an augmented seed pathway.

We now present Algorithm 5.1, which takes as input augmented seed pathways, and finds possible linear branches searching Gam (lines 7–10). Then, all combinations of possible branches must be tried systematically for attachment to the augmented seed pathway, because adding a branch to an augmented seed pathway effects the atom tracking down the pathway (lines 11–15). Our experimentation showed that trying all combinations can lead to long run times without substantially improving biological value. Therefore, we provide the option to limit the maximum number of branches that can be added by b. Extra care is taken to maintain any computed branches in a global data structure, Mb, so that branches can be reused to reduce computation time. The result is an ordered list of branched pathways sorted first by the number of atoms conserved and second by the total number of nodes in the pathway. A slightly modified version of Algorithm 5.1 that accepts branched pathways has also been implemented but not described here due to space limitations. That version can be repeatedly applied to our branched pathways until either the maximal number of atoms are conserved from the start or feasible linear pathways cannot be found. However, we observed that even for our most complex pathways this only needed to be done once.
6 RESULTS
In this section, we first provide a brief evaluation of our approach on linear pathways. We then present three biologically motivated examples of branched pathways found by our algorithms that cannot be found using pathfinding methods available in the literature. The atom mapping graph used in all of the experiments contained 5844 compound nodes and 22 920 mapping nodes, built from KEGG RPAIR data. From the KEGG REACTION database, we obtain 7340 reactions from over 1000 organisms which have corresponding KEGG RPAIR entries. The KEGG data was acquired in July 2009. For the purposes of the results in this article, only carbon atoms were tracked, but the methods described can be used to track atoms of interest as long as proper atom mapping data is provided. The resulting pathways presented are ranked first by the number of carbon atoms they conserve and then by the number of reactions they contain.
The implementation was done in Java using the Chemical Development Kit (Steinbeck et al., 2006) and the Java Universal Network/Graph Framework (http://jung.sourceforge.net/). All result figures are drawn using Graphviz (http://www.research.att.com/sw/tools/graphviz/). All experiments were run on a single core from a 2.83 GHz Intel Xeon E5440 with access to 16 GB of RAM. For the branched pathways, the input k, for Eppstein's k-shortest paths algorithm, was set to one million and the seed pathways were chosen as all paths, without cycles, containing less mapping nodes than the shortest path plus four.
6.1 Evaluation on linear pathways
Evaluating metabolic pathfinding methods can be difficult, even for linear pathways, because there is no standard test set. To provide a base line comparison for our methods, we downloaded known metabolic pathways from a recent evaluation on linear pathways (Faust et al., 2009). We removed pathways where either atom mapping data was missing or no carbons made it from the start to target compounds, resulting in a set of 48 known pathways. We compared these known pathways to the computed shortest paths using three different types of atom tracking: (i) conserving the maximum number of carbon atoms; (ii) conserving at least one carbon; and (iii) not using atom tracking at all.
The results on comparing the known pathways with the computed pathway are found in Table 1. True positives (TP) are compounds found in both pathways; false negatives (FN) are compounds in the known pathway, which are not in the computed pathway; false positives (FP) are compounds not in the known pathway, which are in the computed pathway. For each pathway, we use measurements found previously in the literature to calculate the sensitivity Sn = TP/(TP+FN), positive predictive value (PPV) = TP/(TP+FP) and accuracy Ac = (Sn+PPV)/2 (Blum and Kohlbacher, 2008a, b; Croes et al., 2006; Faust et al., 2009). Positive predictive value is used instead of specificity because true negatives do not exist in this comparison. The poor performance of (iii) is expected, as this result is found in a number of previous evaluations (Arita, 2004; Croes et al., 2006; Faust et al., 2009). The main result is the level of improvement between (ii) and our approach (i). This indicates that one important characteristic for many metabolic pathways is the movement of carbon atoms from the start compound to the target compound.
Table 1.
Average accuracy, positive predictive value and sensitivity for the 48 pathways tested
| Atom tracking | Top Path | Best of top five paths | ||||
|---|---|---|---|---|---|---|
| Ac | PPV | Sn | Ac | PPV | Sn | |
| (a) Max carbons | 0.64 | 0.70 | 0.58 | 0.85 | 0.89 | 0.80 |
| (b) One carbon | 0.37 | 0.41 | 0.33 | 0.65 | 0.71 | 0.59 |
| (c) No carbon | 0.15 | 0.19 | 0.10 | 0.39 | 0.5 | 0.28 |
6.2 Branched pathway: sn-glycero-3-phosphocholine to l-threonine
Threonine is an essential amino acid primarily manufactured by using engineered strains of bacteria, and therefore there has been a major focus toward improving the yield (Leuchtenberger et al., 2005). A strategy to increase yield may include using pathways that transform degradation products, which otherwise might be lost, into the desired product. In Figure 2, we show results from our branched path finding algorithm for starting with sn-glycero-3-phosphocholine, a common degradation intermediate of triglycerides containing eight carbons, to threonine, which contains four carbons. The seed pathway search conserved at least two carbons from start to finish generating 2155 seed pathways, with the shortest seed pathway containing six reactions. The whole branched search pathfinding process took 24 min.
Fig. 2.

The solid edges shows the known pathway, for sn-glycero-3-phosphocholine to threonine, containing 11 reactions, ranked third in our results. The dashed edges show how the shortest pathway containing 10 reactions differs. Compound nodes are ovals, mapping nodes are boxes and reactions are diamonds.
Many of the resulting branched pathways split sn-glycero-3-phosphocholine into sn-glycerol-3-phosphate and choline, each of which begin paths conserving two carbons and end at acetaldehyde and glycine, which then join to make the four carbon threonine. This general branching scheme is an expected result and no linear pathways were found that conserved all four carbons. The top-ranked result is depicted by the dashed edges in Figure 2. This result takes an unusual, likely infeasible, shortcut through acetyl phosphate. Reversibility information may help improve the results because the reaction from acetyl phosphate to glycine is only observed in the reverse direction. However, the reaction from betaine to acetyl phosphate is a feasible reaction that may not typically be considered and could lead to other interesting pathways. Therefore, interesting paths and reactions may be automatically revealed that might normally not be foremost to those familiar with specific subpathways. Additionally, we observe the known pathway from choline to glycine via demethylation in the next longest set of pathways, with 11 reactions, and it is depicted by the solid edges in Figure 2. The pathway from sn-glyerol-3-phophate to acetaldehyde demonstrates the difficulty in finding the balance between finding unusual but likely shortcuts and very unlikely shortcuts. In this pathway, most likely pyruvate is generated from glycerone phosphate via glycolysis instead of through methylglyoxal. However, the overall scheme returned by our search is correct, and in the last section we discuss potential ways to help address shortcuts around standard pathways such as glycolysis.
6.3 Branched pathway: chorismate to (S)-norcoclaurine
(S)-norcoclaurine is a key intermediate in the formation of benzylisoquinoline alkaloids, leading to more complex molecules such as morphine and codeine (Minami et al., 2008). In this example, we demonstrate how starting from multiple molecules can be incorporated by adding a new compound node representing two molecules of chorismate that are connected to the rest of the network by a reaction that creates two molecules of chorismate. The search proceeds as normal because the seed pathways use one of the chorismate molecules, while the other one is considered as a branch start point. Each molecule of chorismate contains 10 carbons and (S)-norcoclaurine contains 16 carbons. The seed pathways conserve 7 carbons, the shortest contained 5 reactions and 80 seed pathways were used. The overall search took 8 min. The top-three ranked, branched pathways found by our search are known pathways for the synthesis of (S)-norcoclaurine. An illustration of these three pathways can be found in Figure 3.
Fig. 3.

The top three pathways for chorismate to (S)-norcoclaurine, merged together with the solid line indicating one of the results and the dashed lines showing how the other two pathways differ. Compounds nodes are ovals, mapping nodes are boxes and reactions are diamonds.
All of the pathways share the same path from chorismate to (S)-norcoclaurine through 4-hydroxyphenylacetaldehyde, which is the shortest seed pathway. In this case, the variation can be discovered in two ways, one being by returning all branches of equal length. However, in an implementation where only one of the shortest branches was selected, the same paths were still returned as the top results. This is because the paths from chorismate to (S)-norcoclaurine via dopamine are also in the set of seed pathways. Since they are two reactions longer, they are much further down in the list of paths, but by adding the appropriate branch through 4-hydroxyphenylacetaldehyde they rise to the top of the branched pathway results over other unlikely seed pathways. This illustrates that identifying branched pathways may help to avoid undesirable pathways.
6.4 Branched pathway: α-d-glucose 6-phosphate to l-tryptophan
Our final result demonstrates how complex topologies of branched pathways can be revealed by our algorithm. Tryptophan, similar to threonine, is an essential amino acid mainly produced by microbial fermentation (Leuchtenberger et al., 2005). The trypthophan pathway is relatively complex, with a number of places where carbons are lost and gained along the way. We search for branched pathways starting with two molecules of α-d-glucose 6-phosphate. The minimum number of carbons for the seed pathways was 4, resulting in 798 seed pathways with the shortest seed path having 12 reactions. The overall walltime for the branched pathway search was 30 min.
Due to the length of the pathway, there were many more shortcuts revealed by the search than in our other results. Many of these shortcuts proved to be unlikely to occur, such as ones through l-formylkynurenine to tryptophan and catechol to anthranilate, both of which normally occur in the other direction. While the top results using our current ranking are unlikely to model real metabolic systems, an important aspect of our algorithm is that it returns a large number of potential pathway results. There are a number of simple ways to filter the results that enable a user to discover other interesting pathways. One way is for the user to identify undesirable reactions, another is to ask for only pathways that go through specific intermediates. For example, Figure 4 displays a pathway that is far down in the overall ranking, but is the fourth-ranked result from the subset of results that go through shikimate, a known intermediate for tryptophan. This result illuminates a number of interesting properties of the tryptophan pathway, e.g. that serine and 5-phospho-α-d-ribose 1-diphosphate both can be made from compounds further upstream. These complex relationships, automatically discovered by our algorithm, are of importance for metabolic pathway analysis and design. We also observe that the second α-d-glucose 6-phosphate is ultimately used to create two molecules of PEP. As with the threonine pathway, these molecules would typically be created via glycolysis, but here they are created by a different scheme because we favor pathways utilizing fewer reactions.
Fig. 4.

A single branched pathway result for tryptophan, demonstrates how branching schemes can become quite complicated. The bold edges indicate the initial seed pathway, the normal solid lines are the branches attached in the first stage of the algorithm and the dotted line is a final branch added after the initial branches were attached. Compound nodes are ovals, mapping nodes are boxes and reactions are diamonds. The numbers are to assist the reader in following the pathway, as the figure is split into half to fit on the page.
7 DISCUSSION AND CONCLUSIONS
The metabolic pathfinding algorithms we have presented are part of a growing set of analysis tools that will assist in understanding metabolic networks and designing of novel pathways for applications such as metabolic engineering and synthetic biology. Atom tracking methods, such as ours and ReTrace (Pitkänen et al., 2009), enable graph-theoretical methods to find biologically meaningful linear and branched metabolic pathways in genome-scale metabolic networks. While the theoretical complexity of finding even linear atom conserving pathways is high, by choosing the appropriate representations and heuristics, and perhaps due to the structure of the underlying data, these algorithms have reasonable running times in practice.
The pathways found by our algorithms demonstrate that they are able to avoid spurious connections and have the potential to find biologically interesting pathways. Our results also point towards a number of interesting areas for future applications and improvements. For example, in this work we have focused on tracking carbon atoms, but the methods can be applied to other atoms of interest, such as nitrogen or sulfur, to better understand metabolism as a whole. We have also focused on searching across all of the data in KEGG, but for some applications, one may only want to look at the metabolic network of a single organism or a subset of all of the organisms. These organism-specific networks would be smaller than the network used in this work and we would expect similar, possibly faster, performance.
The search algorithms may also be improved by using knowledge about highly conserved metabolic pathways, such as glycolysis. A small number of these pathways could be cataloged and be used in the search or as a post-processing step to guide towards more feasible pathways. Using weighting schemes may provide another way to improve performance. There are several previously proposed weighting schemes based upon compound degree, that could potentially be incorporated with full atom tracking to improve pathway ranking (Blum and Kohlbacher, 2008a; Croes et al., 2006). Other weighting schemes based upon characteristics such as energy consumption, or what organisms can perform the reactions may help find good candidates for in vivo experimentation. Since it is unlikely that there will be a single perfect ranking scheme for all applications of metabolic pathfinding, future studies on the practical performance of such methods will be required.
Funding: Hamill Innovation Award from the Institute of Biosciences and Bioengineering at Rice University and Rice University funds (in parts); Training fellowship from the Biomedical Discovery Training Program of the W. M. Keck Center for Interdisciplinary Bioscience Training of the Gulf Coast Consortia to A.P.H. (National Institutes of Health Grant No. T90 DA022885 and R90 DA023418); Computational resources were provided by the Shared University Grid at Rice, funded by the National Science Foundation under Grant EIA-0216467 and partnership between Rice University, Sun Microsystems, and Sigma Solutions, Inc.
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Akutsu T. Efficient extraction of mapping rules of atoms from enzymatic reaction data. J. Comput. Biol. 2004;11:44962. doi: 10.1089/1066527041410337. [DOI] [PubMed] [Google Scholar]
- Arita M. In silico atomic tracing by substrate-product relationships in Escherichia coli intermediary metabolism. Genome Res. 2003;13:2455–2466. doi: 10.1101/gr.1212003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita M. The metabolic world of Escherichia coli is not small. Proc. Natl Acad. Sci. USA. 2004;101:1543–1547. doi: 10.1073/pnas.0306458101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum T, Kohlbacher O. MetaRoute: fast search for relevant metabolic routes for interactive network navigation and visualization. Bioinformatics. 2008a;24:2108–2109. doi: 10.1093/bioinformatics/btn360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum T, Kohlbacher O. Using atom mapping rules for an improved detection of relevant routes in weighted metabolic networks. J. Comput. Biol. 2008b;15:565–576. doi: 10.1089/cmb.2008.0044. [DOI] [PubMed] [Google Scholar]
- Boyer F, Viari A. Ab initio reconstruction of metabolic pathways. Bioinformatics. 2003;19:26ii–34ii. doi: 10.1093/bioinformatics/btg1055. [DOI] [PubMed] [Google Scholar]
- Caspi R, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2008;36:D623–D631. doi: 10.1093/nar/gkm900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croes D, et al. Inferring meaningful pathways in weighted metabolic networks. J. Mol. Biol. 2006;356:222–236. doi: 10.1016/j.jmb.2005.09.079. [DOI] [PubMed] [Google Scholar]
- Eppstein D. Finding the k shortest paths. SIAM J Comput. 1998;28:652–673. [Google Scholar]
- Faust K, et al. Metabolic pathfinding using RPAIR annotation. J. Mol. Biol. 2009;388:390–414. doi: 10.1016/j.jmb.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Gerlee P, et al. Pathway identification by network pruning in the metabolic network of Escherichia coli. Bioinformatics. 2009;25:3282–3288. doi: 10.1093/bioinformatics/btp575. [DOI] [PubMed] [Google Scholar]
- Jeong H, et al. The large-scale organization of metabolic networks. Nature. 2000;407:651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- Jouhten P, et al. 13C-metabolic flux ratio and novel carbon path analyses confirmed that Trichoderma reesei uses primarily the respirative pathway also on the preferred carbon source glucose. BMC Syst. Biol. 2009;3:104. doi: 10.1186/1752-0509-3-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuchtenberger W, et al. Biotechnological production of amino acids and derivatives: current status and prospects. Appl. Microbiol. Biotechnol. 2005;69:1–8. doi: 10.1007/s00253-005-0155-y. [DOI] [PubMed] [Google Scholar]
- Ma H, Zeng A.-P. Reconstruction of metabolic networks from genome data and analysis of their global structure for various organisms. Bioinformatics. 2003;19:270–277. doi: 10.1093/bioinformatics/19.2.270. [DOI] [PubMed] [Google Scholar]
- McShan DC, et al. PathMiner: predicting metabolic pathways by heuristic search. Bioinformatics. 2003;19:1692–1698. doi: 10.1093/bioinformatics/btg217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami H, et al. Microbial production of plant benzylisoquinoline alkaloids. Proc. Natl Acad. Sci. USA. 2008;105:7393–7398. doi: 10.1073/pnas.0802981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mithani A, et al. Rahnuma: hypergraph-based tool for metabolic pathway prediction and network comparison. Bioinformatics. 2009;25:1831–1832. doi: 10.1093/bioinformatics/btp269. [DOI] [PubMed] [Google Scholar]
- Pitkänen E, et al. Inferring branching pathways in genome-scale metabolic networks. BMC Syst. Biol. 2009;3:103. doi: 10.1186/1752-0509-3-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planes FJ, Beasley JE. A critical examination of stoichiometric and path-finding approaches to metabolic pathways. Brief. Bioinform. 2008;9:422–436. doi: 10.1093/bib/bbn018. [DOI] [PubMed] [Google Scholar]
- Rahman SA, et al. Metabolic pathway analysis web service (Pathway Hunter Tool at CUBIC) Bioinformatics. 2005;21:1189–1193. doi: 10.1093/bioinformatics/bti116. [DOI] [PubMed] [Google Scholar]
- Ravasz E, et al. Hierarchical organization of modularity in metabolic networks. Science. 2002;297:1551–1555. doi: 10.1126/science.1073374. [DOI] [PubMed] [Google Scholar]
- Steinbeck C, et al. Recent developments of the chemistry development kit (CDK) - an open-source Java library for chemo- and bioinformatics. Curr. Pharm. Des. 2006;12:2111–2120. doi: 10.2174/138161206777585274. [DOI] [PubMed] [Google Scholar]
- Wagner A, Fell DA. The small world inside large metabolic networks. Proc. R. Soc. Lond. B. 2001;268:1803–1810. doi: 10.1098/rspb.2001.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.