

Abstract
Pancreatic cancer is the fourth leading cause of cancer deaths in the United States with an overall 5-year survival rate of <5%. Pancreatic ductal adenocarcinoma (PDAC), the most common form of pancreatic cancer, is highly resistant to conventional chemotherapies underscoring the critical need for new molecular targets for pancreatic cancer chemotherapy. The KRAS proto-oncogene is mutated in >90% of PDAC. Protein kinase C iota (PKCι) is required for oncogenic Ras-mediated transformed growth in lung cancer and intestinal epithelial cells. However, little is known about the role of PKCι in pancreatic cancer. In this study, we evaluated the expression of PKCι in human pancreatic cancer and the requirement for PKCι for the transformed growth and tumorigenicity of PDAC cells. We find that PKCι is significantly over-expressed in human pancreatic cancer and high PKCι expression correlates with poor patient survival. Inhibition of PKCι expression blocks PDAC cell transformed growth in vitro and tumorigenicity in vivo. Inhibition of PKCι expression in pancreatic tumors also significantly reduces tumor angiogenesis and metastasis. Analysis of downstream PKCι effectors implicates the Rac1-MEK/ERK1/2 signaling axis in PKCι-mediated transformed growth and cellular invasion. Taken together, our data demonstrate a required role for PKCι in the transformed growth of pancreatic cancer cells and reveal a novel role for PKCι in pancreatic cancer cell metastasis and angiogenesis in vivo. Our results strongly indicate that PKCι will be an effective target for pancreatic cancer therapy.
Keywords: pancreatic cancer, transformed growth, invasion, metastasis, protein kinase C iota, Rac1, ERK1/2, VEGF
Introduction
Pancreatic cancer is highly lethal, with patients having a median survival time of <6 months and an overall 5-year survival rate of <5%. The deadly nature of pancreatic cancer is attributed to late detection, rapid growth, a propensity to invade and metastasize, and resistance to conventional chemotherapy. Even patients that undergo “curative” surgery have only a 20% 5-year survival rate (1). Due to the frequent failure of conventional therapies, there is an urgent need for new molecularly-targeted therapies that can improve the outcome for those diagnosed with pancreatic cancer.
Oncogenic KRAS mutations are found in >90% of all advanced pancreatic cancers (2). Anti-sense inhibition of oncogenic K-ras expression in PDAC cell lines blocks cellular transformation, demonstrating a continued requirement for oncogenic K-ras-mediated signaling to maintain the transformed phenotype (3). Currently, there are no clinically effective therapeutic agents that inhibit oncogenic K-ras activity. Farnesyl transferase inhibitors (FTIs) were introduced into the clinic to target mutant Ras, but have not proven to be therapeutically effective in pancreatic cancer (reviewed in (4)). Thus, oncogenic K-ras signaling is critical to pancreatic cancer, but downstream K-ras effector pathways may be better targets for molecularly-targeted therapy in pancreatic cancer.
Our lab and others have identified PKCι as an important effector of oncogenic K-ras in vitro and in vivo (5-11). Here, we tested the hypothesis that PKCι plays a requisite role in pancreatic cancer cell transformed growth and tumorigenesis. We find that PKCι is highly expressed in human pancreatic cancers and that high PKCι expression predicts poor survival. We show that PKCι is required for transformed growth of pancreatic cancer cells in vitro and tumorigenesis in vivo. PKCι promotes transformed growth of pancreatic cancer cells through activation of a proliferative Rac1-MEK/ERK1/2 signaling pathway. Finally, we make the novel observation that inhibition of PKCι expression blocks PDAC tumor angiogenesis and metastasis in vivo. Taken together, these results strongly suggest that PKCι will be an effective target for pancreatic cancer chemotherapy.
Materials and Methods
Reagents and cell culture
Antibodies were obtained from the following sources: PKCι and Rac1 (BD Transduction Laboratories), PKCζ, β-actin, phospho-ERK1/2 Thr202/Tyr204 (p-ERK) and p44/42 ERK (Cell Signaling Technologies), PAK-1 PBD agarose conjugate (Rac/cdc42) (Millipore), 5-bromo-2′-deoxyuridine (BrdUrd) and VEGF (DakoCytomation) and CD31 (PECAM-1) (Santa Cruz Biotechnology, Inc.). U0126 was obtained from Sigma and NSC23766 from Tocris. Human pancreatic cancer cell lines were obtained from ATCC and maintained in a 5% CO2 humidified tissue culture incubator as recommended by ATCC. Retroviral vector encoding firefly luciferase (pSIN-Fluc) was described previously (12).
Patient samples
Biospecimens were obtained from the Mayo Clinic Tissue Registry under an approved Institutional review board protocol. RNA was isolated from a set of pancreatic adenocarcinoma patient samples for which frozen, paired tumor and non-tumor pancreas tissue was available. A second set of formalin-fixed pancreatic adenocarcinoma biospecimens were selected as described previously (13).
RNA isolation, quantitative real-time PCR and analysis
Hematoxylin and eosin (H&E)-stained sections of matched normal and pancreatic tumor tissues were analyzed to confirm the presence of tumor or normal pancreas and overall integrity of the frozen tissue samples. Total RNA was isolated using RNAqueous Isolation Kit (Ambion) according to the manufacturer's protocols. TaqMan® Gene Expression Assay primer and probe sets (Applied Biosystems) were used for real-time, quantitative PCR (qPCR) analysis of hGAPDH (Hs99999905_m1), hPKCζ (Hs00177051_m1) and 18S (Hs99999901_s1). Forward and reverse primer and probe sequences were designed and synthesized for hPKCι (forward-5′-CGTTCTTCCGAAATGTTGATTG-3′, reverse-5′-TCCCCAGAAATATTTGGTTTAAAGG-3′, probe-5′-6FAMTTGCTCCATCATATCC-3′). qPCR analysis was carried out using 10 ng of cDNA or 2 ng cDNA (18S) on an Applied Biosystems 7900 thermal cycler. Data was evaluated using the SDS 2.3 software package. Gene expression in primary pancreatic cancers and in pancreatic cancer cell lines was normalized to 18S and GAPDH, respectively. All data is expressed as 2-(CT(target)-CT(endogenous reference)).
Immunohistochemistry and expression analysis
A second set of formalin-fixed pancreatic cancers (13) were analyzed by immunohistochemistry (IHC) for PKCι expression. A few of the matched normal and pancreatic tumor tissue pairs were also available as formalin-fixed tissues and were analyzed by IHC for PKCι expression. Tissues were processed for IHC as described previously (14). PKCι staining was visualized using the Envision Plus Anti-Mouse Labeled Polymer-HRP (Dako). p-ERK1/2 staining was visualized using the Envision Plus Anti-Rabbit Labeled Polymer-HRP (Dako). Images were captured and analyzed using Aperio and Spectrum software. PKCι expression was scored by a pathologist blinded to patient clinical parameters (TCS). Nuclear and cytoplasmic PKCι levels were scored on a scale of 0-3 and combined for a total cellular expression score of 0-6. Low PKCι was defined as a total expression score of ≤3 and high PKCι as a total expression score >3, yielding two groups consisting of approximately half of the evaluable cases (45 and 40, respectively). Staining with only secondary antibody served as negative controls.
Knock down and re-expression of human PKCι gene expression and immunoblot analysis
Lentiviral vectors carrying short hairpin RNA interference (RNAi) targeting human PKCι were generated and used to obtain stable transfectants as described previously (15). PKCι RNAi #1 construct targets a sequence in the 3′ untranslated region of PKCι (GCCTGGATACAATTAACCATT) and PKCι RNAi #2 construct targets a sequence in the coding region of PKCι (CCTGAAGAACATGCCAGATTT). Cells were stably transfected with pBabe and pBabe-PKCι as described previously (16). PKCι and PKCζ protein expression was determined by immunoblot analysis of total cell lysates.
Cell viability assay
Cell viability was assessed by MTT assay (CellTiter 96 AQueous One Solution, Promega), as recommended by the manufacturer. Pancreatic cancer cells (3×103 cells) were cultured for 1, 3, 5 and 7 days prior to viability assay.
Anchorage-independent growth assays
Panc-1 and MiaPaCa-2 cells (5×103) were plated in soft agar and assessed for anchorage-independent growth as described previously (17).
Rac1 activity assay and signaling analysis
Rac1 activity was assayed as described previously (5, 18). Cells stably expressing PKCι RNAi constructs were co-transfected with LZRS vector or LZRS carrying myc-tagged, constitutively active Rac1 (RacV12) as described previously (15, 16). Transfectants were harvested and subjected to immunoblot analysis as previously described (17).
Orthotopic tumor model
Panc-1 human pancreatic cancer cells carrying pSIN-Fluc and expressing NT or PKCι RNAi (1×106) were mixed with growth factor reduced Matrigel (Becton Dickinson) and injected into the proximal pancreas (n=15 and 16 mice/group respectively) of 4-6 week old male athymic nude mice. For weekly imaging, mice were injected intraperitoneally (IP) with 150 mg/kg body weight D-Luciferin solution (Xenogen), anesthetized with isoflourane and imaged using a bioluminescence imaging system (IVIS Imaging Spectrum System). Bioluminescence was calculated using IVIS Imaging Spectrum software. One hour prior to sacrifice, mice were injected IP with 100μg/g BrdUrd. All of the animal experiments were approved by the Mayo Clinic Institutional Animal Care and Use Committee.
Orthotopic tumor analysis
Formalin-fixed pancreatic tumors were analyzed for proliferation using BrdUrd incorporation as described previously (14, 19, 20). Orthotopic pancreatic tumors were evaluated for apoptosis by TdT-mediated dUTP-biotin nick end labeling (TUNEL) of fragmented DNA as described (20). Angiogenesis was characterized by quantitative analysis of IHC detection of CD31 (PECAM-1) expression as described (17, 20, 21). Expression of p-ERK1/2, ERK1/2, PKCι, VEGF and β-actin was evaluated by immunoblot analysis of total cell lysates from orthotopic tumors.
Cellular invasion assay
Cellular invasion was measured as described (6, 15). The selective MEK inhibitor, U0126 (10 μM), was included in the medium in the upper and lower chambers in some experiments, as noted.
Statistical analysis
Survival rates were calculated using Kaplan-Meier analysis. Differences in survival were analyzed by log-rank test, Fisher Exact test, univariate and multivariate Cox proportional hazard models using SAS 9.1.3 software. All tests were two-sided. One-way Analysis of Variance (ANOVA) and the Pairwise Multiple Comparison Procedures were used to evaluate the statistical significance of the results. p values <0.05 were considered statistically significant.
Results and Discussion
PKCι is highly expressed in human pancreatic cancer
To investigate the role of PKCι in pancreatic cancer, we first evaluated PKCι expression in a panel of 28 human pancreatic tumors and adjacent non-tumor (normal) pancreas mRNA samples (Figure 1A). PKCι mRNA was detected in all 28 primary pancreatic tumors analyzed (Figure 1A). PKCι overexpression, defined as tumor mRNA abundance greater than two standard deviations above the average PKCι mRNA abundance in adjacent non-tumor pancreas, was observed in 27/28 pancreatic tumors analyzed (Figure 1A). Pancreatic tumors exhibited an average 9 ± 2 fold increase in PKCι mRNA expression relative to matched non-tumor pancreas (inset, p<0.001 for paired samples). IHC analysis of PKCι expression was conducted on two matched pancreatic tumor/non-tumor pancreas pairs from this set for which formalin-fixed tissues were available. IHC confirmed increased expression of PKCι in both tumors compared to the matched non-tumor pancreas (Supplemental Figure 1). PKCι localized to both the nucleus and cytoplasm of the tumor cells, with little or no expression of PKCι in the surrounding stromal components (Figure 1B and Supplemental Figure 1). We next validated our analysis of PKCι expression in a second group of 85 pancreatic tumor tissues (13) for which formalin-fixed tissue, clinical follow-up and pathological features were available (Supplemental Table 1). Cases were segregated into high and low PKCι expression groups based on IHC staining intensity and subjected to survival analysis, as described in Materials and Methods, (Figure 1C and Supplemental Table 2). Patients whose tumors exhibit high PKCι expression had significantly shorter survival time (median survival time 492 days for high PKCι expression, vs 681 days for low PKCι expression, p=0.033) and a reduced 5 year survival rate (10% vs 29.5% for low PKCι expression, p=0.032). Multivariate analysis adjusting for age, sex and tumor stage, demonstrates a significant association between high PKCι expression and poor survival of PDAC patients (Hazards Ratio, 1.670; 95% CI, 1.037-2.688, p=0.035).
Figure 1. PKCι is highly expressed in human pancreatic cancer and correlates with poor survival in PDAC patients.
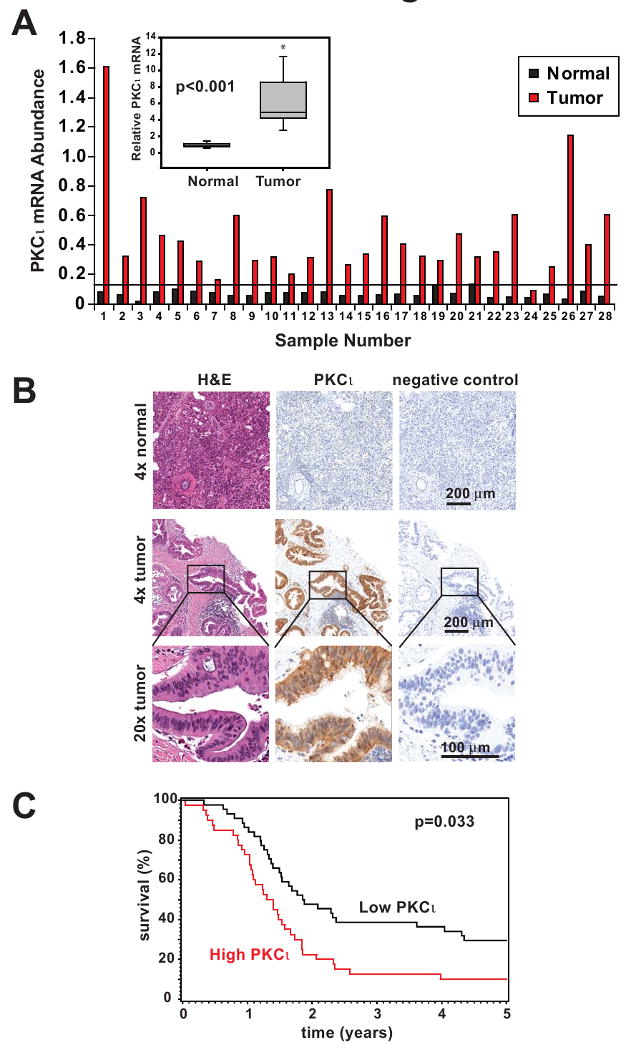
A) qPCR analysis of PKCι mRNA expression in 28 matched human pancreatic tumor and adjacent non-tumor pancreas. Data were normalized to 18S RNA abundance (× 104) to control for RNA concentration. Horizontal line indicates 2 standard deviations above the mean PKCι mRNA abundance in adjacent non-tumor pancreas samples. Inset: PKCι mRNA expression is significantly increased in tumors compared to matched non-tumor pancreas tissue. Average fold increase in PKCι mRNA abundance in tumor/matched non-tumor is plotted. B) Representative images of IHC detection of PKCι expression in formalin-fixed human pancreatic adenocarcinoma and normal pancreas. H&E staining and negative control secondary antibody staining are also shown in serial sections. C) Kaplan-Meier survival curves. PDAC patient tumors were analyzed by IHC for PKCι expression and divided into high (red line) and low (black line) expression groups as described in Materials and Methods.
PKCι is required for transformed growth of PDAC cells in vitro
The prevalence of PKCι overexpression in primary pancreatic tumors, and the association of high PKCι with poor clinical outcome strongly suggests a role for PKCι in the pathology of pancreatic cancer. To evaluate this possibility, we first assessed PKCι mRNA and protein expression in a panel of PDAC cell lines (Figure 2A). Whereas PKCι was abundantly expressed in all PDAC cell lines evaluated, PKCι expression did not directly correlate with known characteristics of the PDAC cell lines including differentiation status, or biological behavior such as soft agar growth, migration or invasion rate, or sensitivity to gemcitabine (22-24).
Figure 2. PKCι is highly expressed in PDAC cell lines and is not required for anchorage-dependent (non-transformed) growth of PDAC cells.
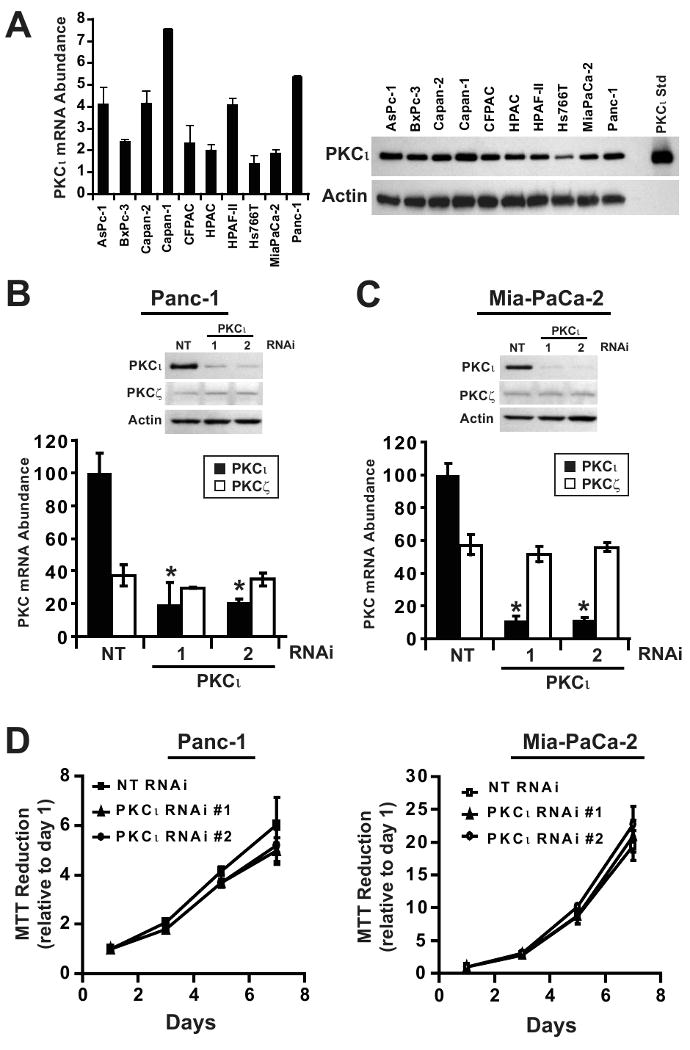
A) Left, qPCR analysis of PKCι mRNA expression in ten human pancreatic cancer cell lines. mRNA abundance is normalized to GAPDH (× 102), n=3. Right, Immunoblot analysis of ten human pancreatic cancer cell lines for expression of PKCι and β-actin. qPCR analysis of PKCι and PKCζ mRNA expression in B) Panc-1 and C) MiaPaCa-2 stably carrying either non target (NT), PKCι-specific RNAi constructs (PKCι #1) or (PKCι #2). PKC mRNA abundance is normalized to GAPDH and presented relative to PKCι in NT RNAi cells. Insets, Immunoblot analysis of PKCι, PKCζ and β-actin protein expression in B) Panc-1 and C) MiaPaCa-2 NT or PKCι-RNAi (PKCι#1 and PKCι#2) constructs. D) Anchorage-dependent growth in Panc-1 (left) and MiaPaCa-2 (right) stably carrying either NT or PKCι-RNAi (PKCι#1 and PKCι#2) was determined by MTT colorimetric assay. Analysis was performed in triplicate and represents two independent experiments.
To directly assess the role of PKCι in the PDAC phenotype, we used lentiviral-mediated RNAi knock down (KD) to inhibit PKCι expression in two widely-utilized human PDAC cell lines, Panc-1 and MiaPaCa-2 (Figure 2B and C). Two PKCι-targeted RNAi constructs significantly inhibited PKCι mRNA and protein expression in both PDAC cell lines. These RNAi constructs had no effect on expression of the closely-related atypical PKC zeta (PKCζ) isozyme, demonstrating their specificity for PKCι (Figure 2B and C). Whereas PKCι KD had no significant effect on log-phase, adherent (non-transformed) cellular growth (Figure 2D) or cellular proliferation (Supplemental Figure 2) of Panc-1 and MiaPaCa-2 cells, PKCι KD significantly inhibited anchorage-independent growth in both PDAC cell lines (Figure 3A and B). While PKCι has been implicated in both pro- and anti-apoptotic signaling (25, 26), we did not observe any effect of PKCι KD on PDAC cell apoptosis (Supplemental Figure 3). These results are consistent with previous findings in intestinal epithelial, NSCLC and ovarian cancer cells (5, 15, 17, 27).
Figure 3. PKCι is required for anchorage-independent growth of PDAC cells.
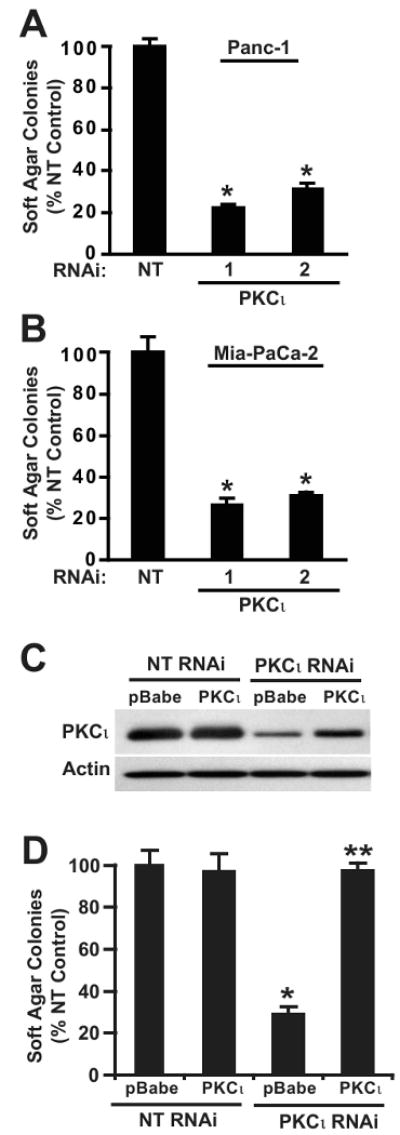
Soft agar colony formation of A) Panc-1 and B) MiaPaCa-2 cells with NT or PKCι-RNAi (PKCι#1 and PKCι#2) constructs. *= significantly different than NT. C) Immunoblot analysis of PKCι expression in Panc-1 cells co-transfected with RNAi (NT or PKCι) and control vector (pBabe) or vector expressing wild type PKCι (PKCι). D) Re-expression of PKCι overcomes the inhibitory effect of PKCι RNAi on soft agar colony formation. *= significantly different than control (NT & pBabe), **= significantly different than PKCι RNAi & pBabe. Mean +/-SEM is plotted and represents two independent experiments.
To confirm that PKCι RNAi-mediated inhibition of transformed growth was due to inhibition of PKCι expression, we expressed human PKCι as a transgene in Panc-1 NT and PKCι RNAi cells (Figure 3C). In this and all further experiments, the PKCι #1 RNAi construct was used to knock down PKCι expression because it targets the 3′UTR of the endogenous human PKCι mRNA, making it possible to reconstitute PKCι expression in PKCι RNAi cells using an exogenous human PKCι cDNA construct lacking the 3′UTR target sequence (15). Re-expression of PKCι reconstituted the anchorage-independent growth of Panc-1 PKCι RNAi cells demonstrating the specific requirement for PKCι in the transformed growth of PDAC cells (Figure 3D).
Rac1 is a critical effector of PKCι-mediated transformed growth in pancreatic cancer cells
We previously demonstrated that PKCι drives transformed growth of intestinal epithelial cells and NSCLC cells by activating a Rac1-MEK/ERK signaling pathway (15, 17, 28). Having established a requirement for PKCι for PDAC cell transformed growth, we evaluated the required signaling downstream of PKCι. We first assessed the requirement for Rac1 and MEK in Panc-1 cell transformed growth. Pharmacological inhibition of either Rac1 (NSC23766) or MEK (U0126) significantly inhibited Panc-1 cell soft agar colony formation (29) (Supplemental Figure 4), demonstrating the requirement for both Rac1 and MEK activity for Panc-1 cell transformed growth. We next assessed whether PKCι regulates Rac1 and MEK activity in PDAC cells. PKCι RNAi significantly reduced Rac1 activity (Figure 4A) and ERK phosphorylation in Panc-1 cells (Figure 4B and 4C). Similar results were observed in MiaPaCa-2 cells (data not shown). To determine whether Rac1 and MEK/ERK activity are required downstream of PKCι in the transformed growth of PDAC cells, we evaluated the ability of myc-tagged constitutively active Rac1 (RacV12) to reconstitute transformed growth in PKCι RNAi Panc-1 cells. Expression of RacV12 restored the transformed growth of PKCι RNAi cells (Figure 4D) and ERK1/2 phosphorylation (Figure 4B and 4C), without affecting PKCι or ERK1/2 expression (Figure 4B). Furthermore, inhibition of MEK blocks RacV12-mediated reconstitution of transformed growth in PKCι KD cells (Figure 4D). Taken together, these data demonstrate that a PKCι-Rac1-MEK/ERK signaling pathway is required for transformed growth of Panc-1 cells.
Figure 4. Constitutively active Rac1 (RacV12) recovers transformed growth of PKCι RNAi PDAC cells in a MEK-dependent manner.
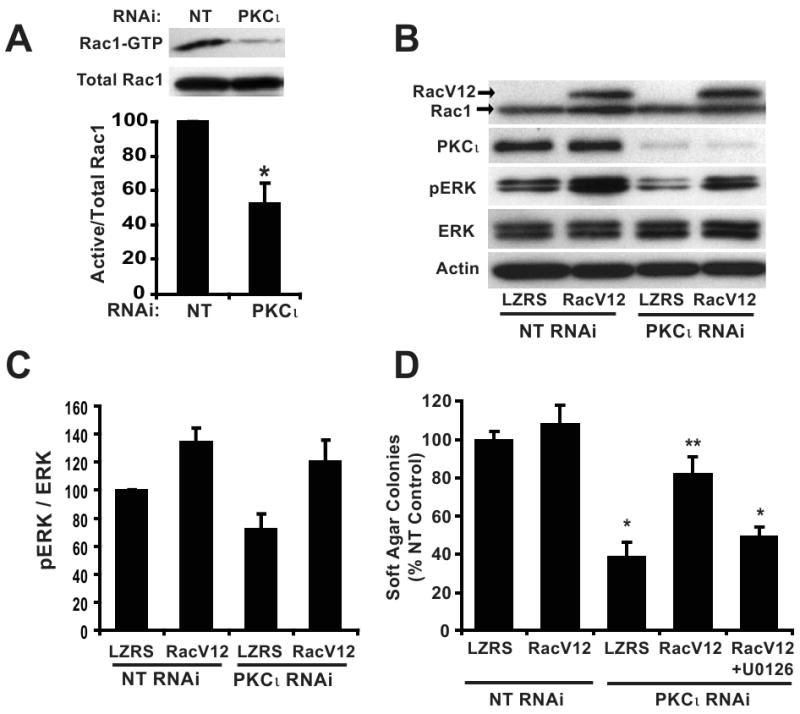
A) Panc-1 cells stably expressing NT or PKCι RNAi were assayed for Rac1 activity. Top panel, (Active) Rac1-GTP was precipitated from cell extracts with PAK-1 PBD agarose. Immunoblot analysis of precipitates and total cellular extracts (total Rac1) was performed using an anti-Rac1 antibody. Bottom panel, Quantitative, densitometric analysis of relative Rac1 activity (active Rac1/total Rac1). Mean +/- SEM is plotted, n=3. B) Panc-1 cells co-transfected with RNAi (NT or PKCι) and control vector (LZRS) or vector expressing RacV12 were subject to immunoblot analysis for expression of Rac1, PKCι, p-ERK1/2 (Thr202/Tyr204), ERK1/2 and actin as a loading control. Arrows indicate migration of endogenous Rac1 and slower migrating myc-tagged RacV12. C) Quantitation of densitometric analysis of relative p-ERK1/2 to ERK1/2 expression. Mean +/- SEM is plotted, n=3. D) Expression of RacV12 recovers the inhibitory effect of PKCι RNAi on soft agar colony formation and requires MEK activity. +U0126= 10 μM U0126 was included in the assay media and agar; *= significantly different than control (NT & LZRS) and RacV12 reconstituted (PKCι RNAi & RacV12); **= significantly different than PKCι KD (PKCι RNAi & LZRS) and MEK-inhibited (PKCι RNAi & RacV12+U0126). Mean +/-SEM is plotted and represents two independent experiments.
PKCι plays a critical role in PDAC cell tumorigenesis
We next utilized an orthotopic pancreatic tumor model to evaluate the role of PKCι in PDAC tumor growth and metastasis in vivo (30). Panc-1 cells expressing the firefly luciferase gene (pSIN-Fluc) and either NT or PKCι RNAi were injected orthotopically into the pancreas of nude mice (31). Tumor growth was monitored by bioluminescence weekly over a 5 week time course (Figures 5A). Tumor formation was observed in all mice injected with either NT or PKCι RNAi-expressing Panc-1 cells. However, PKCι RNAi tumors grew at a slower rate than NT RNAi tumors, resulting in significantly smaller tumors (Figure 5A). We hypothesized that the smaller size of PKCι RNAi tumors was due to reduced proliferation of the tumor cells. As predicted, tumor cell proliferation, as detected by BrdUrd incorporation, was significantly inhibited in PKCι RNAi tumors when compared to NT RNAi tumors (Figure 5B). In contrast, PKCι KD had no effect on tumor apoptosis (Figure 5C). Thus, the reduced tumor volume of PKCι RNAi pancreatic tumors is due to decreased cellular proliferation of the tumor cells.
Figure 5. Inhibition of PKCι blocks orthotopic pancreatic tumor proliferation and proliferative signaling.
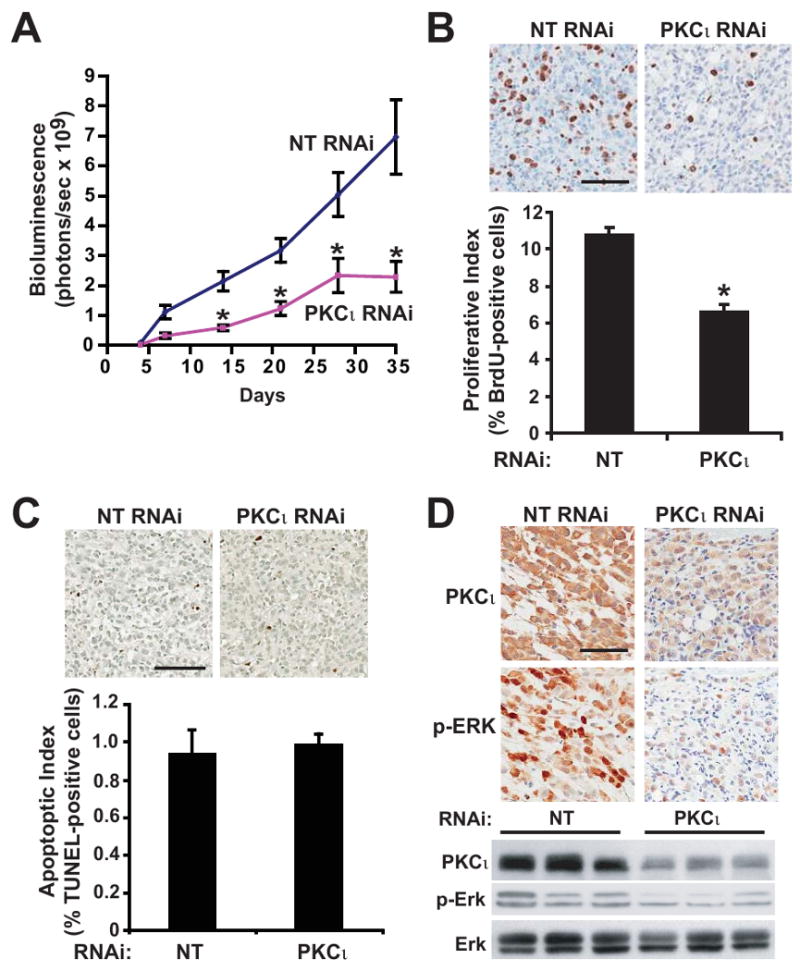
A) Tumor growth was monitored by bioluminescence (total flux, photons/sec) detected by IVIS imaging of orthotopic Panc-1 NT versus Panc-1 PKCι RNAi pancreatic tumors in live, anesthetized mice at weekly intervals after tumor implantation. n=15, 16/group. *= significantly different than NT RNAi tumors. B) Top: Immunohistochemical analysis of BrdUrd incorporation. Bar=100μm. Bottom: Quantitative analysis of BrdUrd incorporation into Panc-1 tumors. Mean+/-SEM is plotted. C) Top: Immunohistochemical detection of TUNEL staining in representative tumors. Bar=100μm. Bottom: Quantitative analysis of TUNEL staining. Mean+/-SEM is plotted. D) Representative images of IHC detection of PKCι and p-ERK1/2 (Thr 202/Tyr 204) in NT and PKCι RNAi tumors. Bar=100μm. Representative immunoblot analysis of PKCι, p-ERK1/2 (Thr 202/Tyr 204) and ERK1/2 in Panc-1 NT and PKCι RNAi orthotopic pancreatic tumors. Equivalent amounts of protein from each tumor sample were analyzed.
To investigate whether MEK/ERK1/2 activity is regulated by PKCι expression in Panc-1 cells in vivo, as observed in vitro (Figure 4), we evaluated the status of ERK1/2 phosphorylation in NT and PKCι RNAi tumors. IHC analysis revealed a dramatic decrease in p-ERK1/2 and PKCι in PKCι RNAi tumors when compared to NT RNAi tumors (Figure 5D). Immunoblot analysis confirmed reduced PKCι expression and reduced p-ERK1/2 in PKCι RNAi tumors compared to NT RNAi tumors (Figure 5D). These data strongly implicate PKCι-mediated activation of a Rac1-MEK/ERK1/2 proliferative signaling pathway in PDAC tumorigenesis in vivo. In this regard, we detect elevated ERK phosphorylation in our panel of human pancreatic tumors (Supplemental Figure 5) as previously described (32, 33).
PKCι expression regulates angiogenesis, invasion and metastasis of PDAC orthotopic tumors
Angiogenesis plays an important role in tumor cell proliferation. We therefore evaluated the effect of PKCι KD on angiogenesis in orthotopic PDAC tumors by IHC detection of the endothelial cell marker CD31 in NT and PKCι RNAi tumors (Figure 6A). CD31 expression was significantly decreased in PKCι RNAi tumors (Figure 6A), indicating that PKCι in tumor cells regulates tumor angiogenesis. Vascular endothelial cell growth factor (VEGF) is a major pro-angiogenic factor expressed in tumor tissue. VEGF expression was considerably decreased in PKCι RNAi tumors (Figure 6A); supporting the conclusion that PKCι drives tumor angiogenesis by regulating VEGF expression in pancreatic tumors. Since ERK activity has been shown to regulate VEGF expression in PDAC cells (34), it is possible that PKCι regulates VEGF expression, at least in part, via regulation of ERK activity. The significantly higher level of tumor angiogenesis in NT RNAi tumors may contribute to the increased tumor proliferation observed in NT RNAi tumors compared to PKCι RNAi tumors.
Figure 6. Inhibition of PKCι blocks PDAC angiogenesis and metastasis.
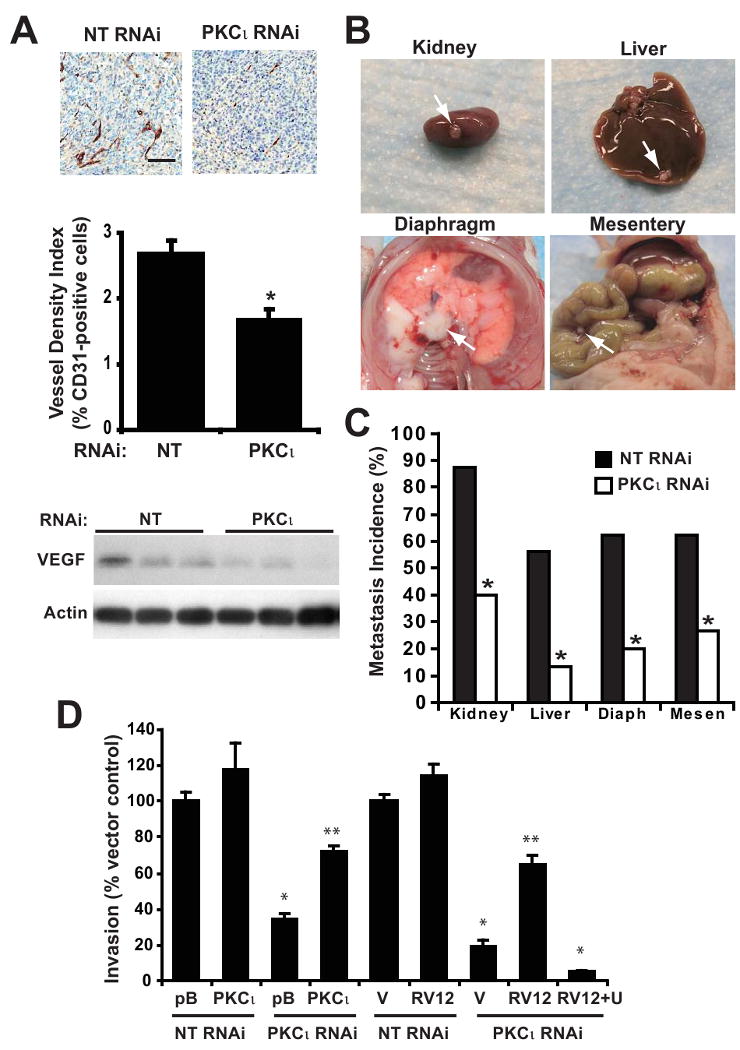
A) Top: Immunohistochemical detection of CD31 staining. Bar=100μm. Middle: Quantitative analysis of CD31 positive staining in Panc-1 tumors, calculated as the ratio of CD31-positive pixels to the sum of all pixels. Mean+/-SEM is plotted. Bottom: Representative immunoblot analysis of VEGF and actin in Panc-1 NT and PKCι RNAi orthotopic pancreatic tumors. B) Representative images of tumor metastases to various organs. C) Percent of orthotopic Panc-1 NT and PKCι RNAi pancreatic tumors that metastasized to various organs is plotted. *= significantly different than NT RNAi tumors. D) Panc-1 cells were assayed for cellular invasion through Matrigel-coated chambers as described in Materials and Methods. pB=pBABE control vector; V=LZRS control vector; RV12= RacV12; RV12+U= RacV12 treated with 10 μM U1026; *= significantly different than control (NT & pB) or (NT & V), **= significantly different than (PKCι RNAi & pB) or (PKCι RNAi & V) or (PKCι RNAi & RV12 +U). Mean +/-SEM is plotted and represents two independent experiments.
Since tumor angiogenesis can be permissive for tumor metastasis, we determined the effect of PKCι RNAi on the metastatic capacity of Panc-1 orthotopic tumors in vivo (Figure 6B and 6C). As described previously (35, 36), Panc-1 cells not only form orthotopic tumors in the pancreas but also develop metastases in other organs. Metastases to the kidney, liver, diaphragm and mesentery were observed in more than 50% of the mice harboring NT RNAi tumors (Figure 6B and 6C). In contrast, PKCι RNAi tumors exhibited significantly reduced metastases to all of these organ sites (Figure 6C). To investigate the mechanism by which PKCι regulates tumor metastasis, we evaluated the requirement for PKCι for cellular invasion (Figure 6D). Cellular invasion is significantly inhibited in PKCι RNAi Panc-1 cells (Figure 6D). Expression of exogenous PKCι reconstituted the PKCι KD phenotype, demonstrating the specific requirement for PKCι for invasion (Figure 6D). Since Rac1 and MEK activity are also required for Panc-1 cellular invasion (Supplemental Figure 6), we evaluated whether Rac1/MEK signaling is downstream of PKCι in cellular invasion (Figure 6D). Expression of Racv12 reconstituted cellular invasion in PKCι KD Panc-1 cells in a MEK-dependent manner (Figure 6D). These data reveal a novel required role for PKCι-Rac1-MEK/ERK signaling in cellular invasion in vitro and taken together with our in vivo data, suggest a role for PKCι-Rac1-MEK/ERK signaling in PDAC tumor cell invasion and metastasis.
Pancreatic cancer is a highly lethal disease with no effective therapeutic options. The overall goal of our research is to reduce this statistic by identifying and characterizing new molecular targets for more effective pancreatic cancer therapy. Our results demonstrate that PKCι is dispensable for adherent pancreatic cell growth, but is required for transformed growth of PDAC cells in vitro and tumorigenicity in vivo. This observation suggests that chemotherapeutic interventions targeting PKCι will specifically inhibit the growth of transformed pancreatic tumor cells while having little effect on non-transformed pancreatic epithelial cells. In this regard, we have identified and characterized a molecularly-targeted inhibitor of PKCι, aurothiomalate (18, 21, 37), which is currently in clinical trials for the treatment of advanced lung and ovarian cancers. Future studies will evaluate the ability of aurothiomalate to inhibit pancreatic tumor growth and metastasis in vivo. Interestingly, a small molecule (oncrasin-1) identified for its ability to selectively induce apoptosis in mutant K-ras-expressing cells requires not only mutant K-ras, but also expression of PKCι, for its cytotoxic effects (38). While the target of oncrasin-1 remains to be elucidated (it does not directly inhibit PKCι activity in vitro)(38), this study provides further support for the conclusion that PKCι plays a critical role in the transformed phenotype of oncogenic K-ras-mediated cancer.
Our study elucidates a critical molecular mechanism by which PKCι promotes transformed growth and cellular invasion of PDAC cells. PKCι regulated Rac1 and ERK1/2 activity, both of which are required for PDAC cell transformed growth and invasion. Specifically, PKCι KD in Panc-1 cells inhibits in vitro transformed growth and invasion which can be reconstituted by RacV12 in a MEK-dependent manner. These results are consistent with the role of PKCι in NSCLC cells (17). We further demonstrate a required role for PKCι in PDAC tumorigenesis in vivo and a novel role for PKCι in PDAC tumor metastasis. Since PKCι also regulates ERK activation in vivo, these data suggests that a Rac1-MEK/ERK1/2 signaling pathway plays a role in PKCι-dependent PDAC tumor cell proliferation and metastasis in vivo. Interestingly, similar to high PKCι expression, ERK phosphorylation predicts poor survival of pancreatic cancer patients (32, 33).
We have recently demonstrated that PKCι is overexpressed in a majority of primary lung cancers, and that high PKCι expression predicts poor survival in NSCLC patients (39). In lung squamous cell cancers, as well as ovarian cancer, elevated PKCι expression is the result of tumor-specific amplification of the PKCι gene (27, 39). As gene amplification frequently occurs in pancreatic cancer, this is a possible mechanism for increased PKCι in human pancreatic cancers. Future studies will address this possibility.
In summary, PKCι is highly overexpressed in the majority of primary pancreatic cancers and elevated PKCι expression correlates with decreased survival time. PKCι and its downstream effectors Rac1 and MEK/ERK1/2 are required for PDAC transformed growth and cellular invasion in vitro and PKCι is required for PDAC tumorigenicity and tumor cell proliferation in vivo. Finally, we describe a previously unappreciated role for PKCι in PDAC tumor angiogenesis and metastasis. These data identify PKCι as an attractive therapeutic target for the treatment of pancreatic cancer.
Supplementary Material
Acknowledgments
Grant Support: NIH grants CA128661 (N.R. Murray) and CA081436-12 (A.P. Fields), Mayo Clinic SPORE in Pancreatic Cancer Career Development award P50 CA102701 (N.R. Murray), Daniel Foundation of Alabama Postdoctoral Fellowship (M.L. Scotti) and The Mayo Clinic Foundation. We thank Amanda Butler, Shelly R. Calcagno, Brandy Edenfield, Dr. Lee Jamieson, Alyssa Kunz and Dr. Shuhua Li of the Mayo Clinic, for excellent technical support, Dr. Yasurhiro Ikeda of the Mayo Clinic for providing the pSIN-Fluc vector and the Mayo Clinic RNA Interference Technology Resource for RNAi reagents. We thank Dr. Lizhi Zhang and Kari Rabe of the Mayo Clinic for assistance with acquisition of biospecimens. We acknowledge the Mayo Clinic SPORE in Pancreatic Cancer Patient Registry and Tissue Core (supported by P50 CA102701 (Gloria Petersen, PI) and the Lustgarten Foundation for Pancreatic Cancer Research) for providing the biospecimens used in this study.
Abbreviations used
- PKC
protein kinase C
- qPCR
quantitative real time polymerase chain reaction
- BrdUrd
5-bromo-2′-deoxyuridine
- H&E
hematoxylin and eosin
- RNAi
RNA interference
- KD
knock down
References
- 1.Lebedeva IV, Sarkar D, Su ZZ, et al. Molecular target-based therapy of pancreatic cancer. Cancer Res. 2006;66:2403–13. doi: 10.1158/0008-5472.CAN-05-3510. [DOI] [PubMed] [Google Scholar]
- 2.Klimstra DS, Longnecker DS. K-ras mutations in pancreatic ductal proliferative lesions. Am J Pathol. 1994;145:1547–50. [PMC free article] [PubMed] [Google Scholar]
- 3.Aoki K, Yoshida T, Matsumoto N, Ide H, Sugimura T, Terada M. Suppression of Ki-ras p21 levels leading to growth inhibition of pancreatic cancer cell lines with Ki-ras mutation but not those without Ki-ras mutation. Mol Carcinog. 1997;20:251–8. [PubMed] [Google Scholar]
- 4.Saad ED, Hoff PM. Molecular-targeted agents in pancreatic cancer. Cancer Control. 2004;11:32–8. doi: 10.1177/107327480401100105. [DOI] [PubMed] [Google Scholar]
- 5.Murray NR, Jamieson L, Yu W, et al. Protein kinase C{iota} is required for Ras transformation and colon carcinogenesis in vivo. J Cell Biol. 2004;164:797–802. doi: 10.1083/jcb.200311011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J, Anastasiadis PZ, Liu Y, Thompson EA, Fields AP. Protein kinase C (PKC) betaII induces cell invasion through a Ras/Mek-, PKC iota/Rac 1-dependent signaling pathway. J Biol Chem. 2004;279:22118–23. doi: 10.1074/jbc.M400774200. [DOI] [PubMed] [Google Scholar]
- 7.Bjorkoy G, Perander M, Overvatn A, Johansen T. Reversion of Ras- and phosphatidylcholine-hydrolyzing phospholipase C-mediated transformation of NIH 3T3 cells by a dominant interfering mutant of protein kinase C lambda is accompanied by the loss of constitutive nuclear mitogen-activated protein kinase/extracellular signal-regulated kinase activity. J Biol Chem. 1997;272:11557–65. doi: 10.1074/jbc.272.17.11557. [DOI] [PubMed] [Google Scholar]
- 8.Kampfer S, Windegger M, Hochholdinger F, et al. Protein kinase C isoforms involved in the transcriptional activation of cyclin D1 by transforming Ha-Ras. J Biol Chem. 2001;276:42834–42. doi: 10.1074/jbc.M102047200. [DOI] [PubMed] [Google Scholar]
- 9.Uberall F, Hellbert K, Kampfer S, et al. Evidence that atypical protein kinase C-lambda and atypical protein kinase C-zeta participate in Ras-mediated reorganization of the F-actin cytoskeleton. J Cell Biol. 1999;144:413–25. doi: 10.1083/jcb.144.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coghlan MP, Chou MM, Carpenter CL. Atypical protein kinases Clambda and -zeta associate with the GTP-binding protein Cdc42 and mediate stress fiber loss. Mol Cell Biol. 2000;20:2880–9. doi: 10.1128/mcb.20.8.2880-2889.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Regala RP, Davis RK, Kunz A, Khoor A, Leitges M, Fields AP. Atypical Protein Kinase C{iota} Is Required for Bronchioalveolar Stem Cell Expansion and Lung Tumorigenesis. Cancer Res. 2009 doi: 10.1158/0008-5472.CAN-09-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hasegawa K, Nakamura T, Harvey M, et al. The use of a tropism-modified measles virus in folate receptor-targeted virotherapy of ovarian cancer. Clin Cancer Res. 2006;12:6170–8. doi: 10.1158/1078-0432.CCR-06-0992. [DOI] [PubMed] [Google Scholar]
- 13.Pongprasobchai S, Pannala R, Smyrk TC, et al. Long-term survival and prognostic indicators in small (<or=2 cm) pancreatic cancer. Pancreatology. 2008;8:587–92. doi: 10.1159/000161009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calcagno SR, Li S, Colon M, et al. Oncogenic K-ras promotes early carcinogenesis in the mouse proximal colon. Int J Cancer. 2008;122:2462–70. doi: 10.1002/ijc.23383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frederick LA, Matthews JA, Jamieson L, et al. Matrix metalloproteinase-10 is a critical effector of protein kinase Ciota-Par6alpha-mediated lung cancer. Oncogene. 2008 doi: 10.1038/onc.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murray NR, Jamieson L, Yu W, Zhang J, Gokmen-Polar Y, Sier D, Anastasiadis P, Gatalica Z, Thompson EA, Fields AP. Protein kinase Ci is required for ras transformation and colon carcinogenesis in vivo. Journal of Cell Biology. 2004;164:797–802. doi: 10.1083/jcb.200311011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Regala RP, Weems C, Jamieson L, Copland JA, Thompson EA, Fields AP. Atypical protein kinase Ciota plays a critical role in human lung cancer cell growth and tumorigenicity. J Biol Chem. 2005;280:31109–15. doi: 10.1074/jbc.M505402200. [DOI] [PubMed] [Google Scholar]
- 18.Stallings-Mann M, Jamieson L, Regala RP, Weems C, Murray NR, Fields AP. A novel small-molecule inhibitor of protein kinase Ciota blocks transformed growth of non-small-cell lung cancer cells. Cancer Res. 2006;66:1767–74. doi: 10.1158/0008-5472.CAN-05-3405. [DOI] [PubMed] [Google Scholar]
- 19.Murray NR, Weems C, Chen L, et al. Protein kinase C betaII and TGFbetaRII in omega-3 fatty acid-mediated inhibition of colon carcinogenesis. J Cell Biol. 2002;157:915–20. doi: 10.1083/jcb.200201127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fields AP, Calcagno SR, Krishna M, Rak S, Leitges M, Murray NR. Protein kinase Cbeta is an effective target for chemoprevention of colon cancer. Cancer Res. 2009;69:1643–50. doi: 10.1158/0008-5472.CAN-08-3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Regala RP, Thompson EA, Fields AP. Atypical protein kinase C iota expression and aurothiomalate sensitivity in human lung cancer cells. Cancer Res. 2008;68:5888–95. doi: 10.1158/0008-5472.CAN-08-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Missiaglia E, Blaveri E, Terris B, et al. Analysis of gene expression in cancer cell lines identifies candidate markers for pancreatic tumorigenesis and metastasis. Int J Cancer. 2004;112:100–12. doi: 10.1002/ijc.20376. [DOI] [PubMed] [Google Scholar]
- 23.Giroux V, Malicet C, Barthet M, et al. p8 is a new target of gemcitabine in pancreatic cancer cells. Clin Cancer Res. 2006;12:235–41. doi: 10.1158/1078-0432.CCR-05-1700. [DOI] [PubMed] [Google Scholar]
- 24.Sato N, Maehara N, Mizumoto K, et al. Telomerase activity of cultured human pancreatic carcinoma cell lines correlates with their potential for migration and invasion. Cancer. 2001;91:496–504. doi: 10.1002/1097-0142(20010201)91:3<496::aid-cncr1028>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 25.Win HY, Acevedo-Duncan M. Role of protein kinase C-iota in transformed non-malignant RWPE-1 cells and androgen-independent prostate carcinoma DU-145 cells. Cell Prolif. 2009;42:182–94. doi: 10.1111/j.1365-2184.2009.00582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Staiger K, Schatz U, Staiger H, et al. Protein kinase C iota mediates lipid-induced apoptosis of human coronary artery endothelial cells. Microvasc Res. 2009;78:40–4. doi: 10.1016/j.mvr.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 27.Zhang L, Huang J, Yang N, et al. Integrative genomic analysis of protein kinase C (PKC) family identifies PKCiota as a biomarker and potential oncogene in ovarian carcinoma. Cancer Res. 2006;66:4627–35. doi: 10.1158/0008-5472.CAN-05-4527. [DOI] [PubMed] [Google Scholar]
- 28.Ray S, Lu Y, Kaufmann SH, Gustafson WC, Karp JE, Boldogh I, Fields AP, Brasier AR. Genomic mechanisms of p210BCR-ABL signaling: induction of heat shock protein 70 through the GATA response element confers resistance to paclitaxel-induced apoptosis. JBiolChem. 2004;279 doi: 10.1074/jbc.M401851200. [DOI] [PubMed] [Google Scholar]
- 29.Guha S, Lunn JA, Santiskulvong C, Rozengurt E. Neurotensin stimulates protein kinase C-dependent mitogenic signaling in human pancreatic carcinoma cell line PANC-1. Cancer Res. 2003;63:2379–87. [PubMed] [Google Scholar]
- 30.Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia. 1999;1:50–62. doi: 10.1038/sj.neo.7900005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kunnumakkara AB, Guha S, Krishnan S, Diagaradjane P, Gelovani J, Aggarwal BB. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-kappaB-regulated gene products. Cancer Research. 2007;67:3853–61. doi: 10.1158/0008-5472.CAN-06-4257. [DOI] [PubMed] [Google Scholar]
- 32.Chadha KS, Khoury T, Yu J, et al. Activated Akt and Erk expression and survival after surgery in pancreatic carcinoma. Ann Surg Oncol. 2006;13:933–9. doi: 10.1245/ASO.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 33.Javle MM, Gibbs JF, Iwata KK, et al. Epithelial-mesenchymal transition (EMT) and activated extracellular signal-regulated kinase (p-Erk) in surgically resected pancreatic cancer. Ann Surg Oncol. 2007;14:3527–33. doi: 10.1245/s10434-007-9540-3. [DOI] [PubMed] [Google Scholar]
- 34.Billadeau DD, Chatterjee S, Bramati P, et al. Characterization of the CXCR4 signaling in pancreatic cancer cells. Int J Gastrointest Cancer. 2006;37:110–9. doi: 10.1007/s12029-007-0011-7. [DOI] [PubMed] [Google Scholar]
- 35.Loukopoulos P, Kanetaka K, Takamura M, Shibata T, Sakamoto M, Hirohashi S. Orthotopic transplantation models of pancreatic adenocarcinoma derived from cell lines and primary tumors and displaying varying metastatic activity. Pancreas. 2004;29:193–203. doi: 10.1097/00006676-200410000-00004. [DOI] [PubMed] [Google Scholar]
- 36.Fukasawa M, Korc M. Vascular endothelial growth factor-trap suppresses tumorigenicity of multiple pancreatic cancer cell lines. Clin Cancer Res. 2004;10:3327–32. doi: 10.1158/1078-0432.CCR-03-0820. [DOI] [PubMed] [Google Scholar]
- 37.Erdogan E, Lamark T, Stallings-Mann M, et al. Aurothiomalate inhibits transformed growth by targeting the PB1 domain of protein kinase Ciota. J Biol Chem. 2006;281:28450–9. doi: 10.1074/jbc.M606054200. [DOI] [PubMed] [Google Scholar]
- 38.Guo W, Wu S, Liu J, Fang B. Identification of a small molecule with synthetic lethality for K-ras and protein kinase C iota. Cancer Res. 2008;68:7403–8. doi: 10.1158/0008-5472.CAN-08-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Regala RP, Weems C, Jamieson L, et al. Atypical protein kinase C iota is an oncogene in human non-small cell lung cancer. Cancer Res. 2005;65:8905–11. doi: 10.1158/0008-5472.CAN-05-2372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.