

Abstract
MHC class II (MHCII) genes have been implicated in the regulation of T lymphocyte responses. However, the mechanism of MHCII-driven regulation remains unknown. Matching for MHCII between donors and recipients of allografts favors regulatory T cell tolerance to transplants and provides a unique opportunity to study this regulation. In this study, we investigated MHCII regulation using transfer of donor MHCII genes in recipients of cardiac allografts. Transfer of MHCII IAb genes in the bone marrow of CBA mice (H-2k) prior to the grafting of IAb+ fully allogeneic C57BL/6 (B6, H-2b) heart transplants resulted in donor-specific tolerance associated with long-term survival of B6, but not third-party, allografts without sustained immunosuppression. Strikingly, the majority of accepted heart transplants (>170 d) were devoid of allograft vasculopathy. Further studies indicated that intracellular IAb initiated the tolerogenic process, which was mediated by regulatory T cells (Tregs) that polarized antigraft responses to Th2 cytokine producers. This mechanism seems to be unique to MHCII genes, because previous MHC class I gene-based therapies failed to produce Tregs. These results demonstrate the key role of MHCII in the induction of Tregs. They also underscore a potential mechanism of specific inactivation of T cells in this model; when activated by IAb+ grafts, IAb-specific Tregs repress the entire alloresponse to C57BL/6 transplants (including MHC I and minor Ags), thus mediating T cell tolerance.
Major histocompatibility complex class II (MHCII) glycoproteins are known to play a deciding role in the selection of helper and regulatory CD4+ T lymphocytes (1). The contribution of MHCII molecules plus self-peptide (pMHCII) complexes to CD4+ T cell differentiation/activation has been well documented (2). Likewise, pMHCII complexes are involved in the thymic differentiation of naturally occurring CD4+ CD25+Foxp3+ regulatory T cells (Tregs) (3, 4). In addition to their prominent role in CD4+ cell differentiation, MHCII molecules have been implicated in the regulation of immune responses, a function that was the cornerstone of their discovery (5) but remains poorly understood. It is widely accepted that the avidity of MHCII heterodimers for foreign peptides and the density of resulting pMHCII complexes on APCs modulate CD4+ T cell activation (6-9). However, there is mounting evidence that the regulatory role of MHCII molecules is not confined to their ability to bind or not bind peptides. Indeed, inverse correlations between peptide avidity for MHCII and the magnitude of T cell responses were reported (10, 11). Other instances in clinical and experimental transplantation showed that matching for MHCII loci between graft donors and hosts downmodulated T cell responses to allografts and improved graft survival (12, 13). These data indicated that immune regulation via MHCII was mostly independent of the avidity of MHCII for graft-derived peptides and that transplantation models may be especially suited to decipher the regulatory function of MHCII genes.
To assess whether MHCII regulatory control on T cell responses was mediated by MHCII genes and not by other genes mapping in the MHCII locus, pilot gene-therapy experiments were carried out in a preclinical transplantation model (14). The results demonstrated that transfer of donor MHCII genes into bone marrow cells (BMCs) of future graft recipients induced immune tolerance to kidney transplants that were fully allogeneic to the hosts (15, 16). They also suggested that the MHCII-induced tolerance was regulatory, because T cell responses to all donor major and minor Ags were repressed. However, the mechanism of downregulation of immune responses by MHCII remains to be demonstrated.
The present study investigated the mechanism of MHCII control over T cell responses to allogeneic cardiac transplants in mice. We demonstrated that transfer of a single donor MHCII gene in recipient BMCs resulted in donor-specific tolerance and long-term survival of fully allogeneic transplants expressing this donor MHCII gene. MHCII-induced tolerance was selectively carried by Tregs and involved cytosolic forms of transferred MHCII molecules expressed in/on APCs. Furthermore, MHCII gene transfer prevented the onset of chronic rejection characterized by cardiac allograft vasculopathy (CAV), the chief cause of graft loss in clinical transplantation (17). These findings provide a novel mechanism to account for the regulatory role of MHCII genes, by showing that MHCII has the unique property of inducing Tregs that ultimately suppress the T cell alloresponses involved in acute and chronic allograft rejection.
Materials and Methods
Animals
Six- to eight-week-old female BALB/c (H-2d), CBA (H-2k), and C57BL/6 (B6: H-2b) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). They were maintained in our pathogen-free facility at the Massachusetts General Hospital animal facility and treated according to institutional guidelines.
Cells
Dendritic cells (DCs) were derived from retrovirally transduced BMCs from CBA/J mice. Nonadherent transduced BMCs (2 × 106 cells/ml) were plated in 4 ml RPMI 1640 medium containing 500 U/ml murine rGM-CSF (BioSource International, Camarillo, CA) in six-well plates. Every other day, one-half of the medium was refreshed. On day 7, 50 U/ml recombinant murine IL-4 (BioSource International) was added in addition to GM-CSF to derive mature DCs (mDCs). Cells were harvested for analyses 4 d later. CD11c+ cells for quantitative RT-PCR (qRT-PCR) analysis were purified over magnetic columns after staining with biotin-conjugated anti-CD11c mAb (clone HL3, BD Pharmingen, San Diego, CA), followed by anti-Biotin MicroBeads (Miltenyi Biotec, Auburn, CA). CD4+ T cell subset enrichment (>90%) was performed on splenocytes using the Regulatory T Cell Isolation Kit (Miltenyi Biotec).
Retroviral vectors
Constructs contain the LN vector backbone (18), modified with a myeloproliferative sarcoma virus enhancer in the 3′ long terminal repeat (19). The IAb.RV construct derived from the plasmid pPBM19 containing the porcine MHCII DRA and DRB cDNA, spaced by the internal ribosome entry site (IRES), from the 5′ untranslated IgH-binding protein (19). The following modifications were made: the EMC-neo(r)-SV40 ori fragment was removed by digestion with BglII and ClaI, and the SLA-DRB and SLA-DRA sequences were replaced, respectively, with murine sequences for I-Aβb (bases −16−822, 900-bp EcoRI × HincII fragment from plasmid pcEXV-Aβb) and I-Aαb (bases −77−901, 985-bp EcoRI fragment from plasmid pcEXV-Aαb), kindly provided by R.N. Germain, National Institutes of Health, National Institute of Allergy and Infectious Diseases (Bethesda, MD).
To construct the IAb-GFP.RV plasmid, the stop codon of the I-Aαb cDNA in IAb.RV was mutated (in bold, underlined), and a 3′ AgeI restriction site was added using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) and primers 5′-CCCAGGGCCTTTAAGAGTCACACCGGTGAAAGGAAGGCG-3′ and5′-CGCCTTCCTTTCACCGGTGTGACTCTTAAAGGCCCTGGG-3′. The enhanced GFP (eGFP) cDNA, isolated from plasmid pEGFP-1 (BD Clontech, Palo Alto, CA), was cloned in-frame with the I-Aαb mutated cDNA. The GFP.RV construct (pPBM25-1) was previously reported, and virus-containing supernatants were prepared as described (20).
Retroviral transduction
CBA/J mice were treated with fluorouracil (150 mg/kg, i.v.), and BMCs were harvested 7 d later for transduction. Transduction was performed as described (20), with some modifications: recombinant mouse stem cell factor was from BioSource International, and retronectin-coated wells were preloaded with retrovirus for 3 h at 37°C. L cell fibroblasts were transduced using supernatants diluted 1:5 in complete DMEM + 16 μg/ml polybrene for 2 h at 37°C.
Conditioning protocols for bone marrow and heart transplantation
Transplantation of transduced BMCs (1 × 106 cells/mouse, i.v.) was performed in CBA mice receiving two injections of anti-CD4 (GK1.5), anti-CD8 (2.43) Abs (1 mg each/mouse, i.p.) at days −3 and −1 relative to bone marrow transplantation. Animals also received 10 mg/kg Busulfex at days −4, −3, −2, and −1 (21). Eight weeks later, animals received B6 or BALB/c heart grafts together with 1 mg anti-CD8 i.p. (22). Transplants were monitored daily by palpation in a blinded fashion. Undetectable heart impulses for two consecutive days was considered rejection. Statistical analysis was performed using the Student t test. The prevalence and severity of CAV were quantified by a morphometric approach (23). Briefly, a minimum of 50 vessels were examined per specimen using elastic-stained tissue sections. Severity of CAV was quantified as the percent stenoses of involved vessels ([intimal area ÷ intimal area + luminal area] × 102) and is reported as the mean (± SE) stenosis of all vessels with CAV. For adoptive transfers, long-term tolerant and naive CBA mice received donor-specific transfusions (DSTs; 250 μl blood from B6 mice /mouse) 3 and 2 wk prior to cell transfer. Purified CD4+CD25+ or CD4+CD25− T cells from tolerant or naive CBA mice were infused i.v. into naive CBA mice at day 0, along with 1 mg/mouse anti-CD8 Ab. Mice were then transplanted with B6 hearts.
Abs
mAbs used in this study included biotin-conjugated anti-IAk (clone 590H-4-2) (24), anti-IAb (clone 34-5-3, Cedarlane Laboratories, Burlington, NC), and anti-CD11c (clone HL3, BD Pharmingen), along with PE-conjugated anti-CD11c (clone HL3, BD Pharmingen).
RT-PCR and qRT-PCR
RNAwas isolated from cells and tissues using the RNeasy Mini Kit (Qiagen, Valencia, CA). First-strand cDNA was prepared from 5 μg DNAse-treated RNA using oligo(dT) primers and the Superscript First Strand Synthesis System (Invitrogen, Carlsbad, CA). One-fourth of the total cDNA was used as template for each PCR reaction. Primers were 5′-AAGATGTTGAGCGGCATCGG (I-Aβb) and 5′-CCAGACAGTCTCCTTCTTATCC (I-Aαb) for IAb proviral sequences; 5′-CACCATCTTCTTCAAGGAG and 5′-TGTTCTGCTGGTAGTGGTCG-3′ for eGFP; and 5′-GGGAATTCGGAGAAAWGRTGARGAGC and 5′-GGGATCCTCTGCTGGTGARCYTGTGTGC for detection of CD2. Reaction conditions for IAb and eGFP sequences were 1× Buffer A (Fisher Scientific), 200 μM each 2′-deoxynucleoside 5′-triphosphate, 0.5 μM primer, and 2.5 U Taq polymerase (Fisher Scientific) in 50 μl. Cycle conditions were 94°C/30 s, 55°C/30 s, and 72°C/1 min × 30; 72°C/10 min; and 4°C soak. For CD2, 1× HotStarTaq Buffer (Qiagen), 200 μM 2′-deoxynucleoside 5′-triphosphate, 0.5 μM each primer, and 2.5 U HotStarTaq (Qiagen) in a 50-μl volume. Conditions were 94°C/15 min; (94°C/30 s; 53°C/30 s, 72°C/1 min) × 30; 72°C/10 min; and 4° C soak. PCR products were analyzed on 3% agarose gel, transferred to Nytran membranes, and hybridized to a [32P]-end–labeled primer specific for I-Aαb, 5′-ACGCGTGACCACCATGCTCAGCCTC. For qRT-PCR of transduced CD11c+ DCs, first-strand cDNA was prepared from 1 μg DNAse-treated RNA, as detailed above. PCR amplifications were done in triplicate in a 25-μl final volume containing 1× SYBR green PCR buffer (Stratagene), 5.5 mM MgCl2, 200 nM each primer, 0.6 U platinum Taq DNA polymerase, and 2 μl cDNA. PCR amplifications were carried out in an Mx3000P QPCR System (Stratagene), with an initial denaturing step at 94°C for 10 min, followed by 40 cycles of 94°C for 30 s, 52°C for 30 s, and 72°C for 1 min. Expression levels were normalized to hypoxanthine phosphoribosyltransferase. The primers for eGFP were 5′-AGCACGACTTCTTCAAGTCCG and 5′-GTGTCGCCCTCGAACTTCAC-3′. For hypoxanthine phosphoribosyl-transferase, we used 5′-TGAAGAGCTACTGTAATGATCAGTCAA and 5′-AGCAAGCTTGCAACCTTAACCA.
ELISPOT assays
These assays were performed according to published techniques (25).
Results
Expression of a single-donor MHCII gene prevents rejection of fully allogeneic heart transplants
As depicted in Fig. 1, CBA (H-2k) mice were reconstituted with syngeneic BMCs that were previously transduced with a vector containing the allogenic MHCII Ab gene (MHCII IAb.RV) or a control GFP gene (GFP.RV) (Fig. 2A). Hereafter, these mice are referred to as IAb-CBA and GFP-CBA mice, respectively. These animals were monitored for repopulation of blood cells and showed normal levels by 8 wk after bone marrow (BM) infusion compared with naive CBA animals (data not shown). At that time, IAb-CBA mice received one injection of anti-CD8 mAb and were transplanted with fully allogeneic, but IAb-matched, B6 (H-2b) or third-party BALB/c (H-2d) hearts. Control animals included GFP-CBA mice injected with anti-CD8 and transplanted with B6 hearts as well as untreated CBA recipients of B6 or BALB/c hearts. As expected, untreated CBA recipients rejected B6 (mean survival time [MST], 25 ± 4 d; n = 4) and BALB/c allogeneic hearts (MST, 18 ± 7 d; n = 15; data not shown). Likewise, GFP-CBA recipients acutely rejected B6 heart transplants (MST, 29 ± 6 d; Fig. 2B). In contrast, the vast majority of B6 cardiac allografts, placed in IAb-CBA recipients, survived indefinitely (>150 d). A second series of IAb-CBA mice transplanted with a third-party BALB/c (H-2d) heart acutely rejected their grafts within 19 d post-transplantation (Fig. 2B). Thus, the transfer of a single donor MHCII IAb gene in recipient BMCs was sufficient to prevent the acute rejection of IAb+ grafts that were fully allogeneic with regard to MHC class I and minor Ags to the host genotype. T cell tolerance induced by MHCII gene therapy seemed to be donor-specific, because third-party allografts were promptly rejected.
FIGURE 1.
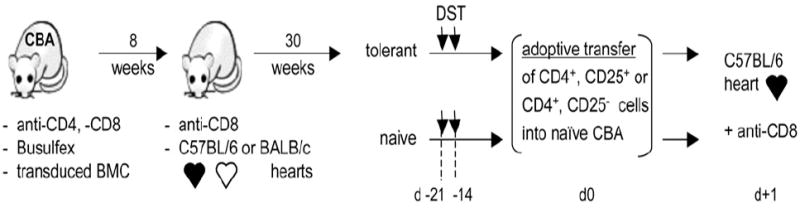
Conditioning protocol for IAb gene therapy. CBA (IAk, IEk) recipients were conditioned with anti-CD4, anti-CD8 mAbs (days −3 and −1) and Busulfex (days −4, −3, −2, and −1) prior to transplantation of syngeneic BMCs that were transduced with the IAb.RV or GFP.RV vector (Fig. 2A). Eight weeks after BM transplantation, animals received B6 hearts (IAb, IE0) with anti-CD8. For adoptive transfers, tolerant (POD 170) and naive CBA mice received DST at days −21 and −14 relative to the adoptive transfer. Purified CD4+CD25+ or CD4+CD25− T cells from tolerant or naive CBA mice were infused at day 0 into naive CBA recipients, together with 1 mg of anti-CD8. Animals were transplanted with B6 cardiac allografts the following day.
FIGURE 2.
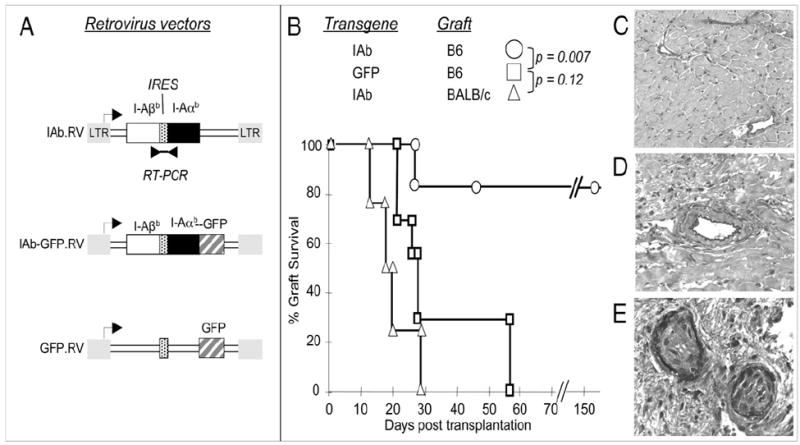
Survival of IAb+ cardiac transplants in IAb-CBA and GFP-CBA recipient mice. A, The IAb-RV vector contained cDNA for the IAαb- and IAβb-chains, spaced by an IRES. Transcription was under long terminal repeat promoter control (arrow). Intact vector transcripts were detected by RT-PCR (double arrowheads). The IAb-GFP.RV vector was constructed on the same backbone with the addition of the GFP sequence that was fused, in frame, to that of I-Aαb. The GFP.RV vector included the GFP sequence. B, IAb-transduced CBA mice (n = 15) received B6 (n = 11) or third-party BALB/c hearts (n = 4) that were monitored for survival. Controls were CBA mice (n = 7) engineered with the GFP.RV vector and transplanted with B6 hearts. Representative histology of two long-term accepted B6 hearts from IAb-CBA mice (C, D) and a B6 transplant rejected by a control GFP-CBA mouse (E) (Verhoeff elastic stain, original magnifications ×200 (C), ×320 (D), and ×640 (E) at POD 177, 175, and 23, respectively). CAV lesions were quantified as detailed in Materials and Methods.
MHCII gene transfer markedly reduces chronic rejection characterized by CAV
Chronic rejection characterized by CAV is the main cause of heart graft failure after the first year of transplantation in patients. In this study, we investigated whether long-term surviving B6 hearts from IAb-CBA recipients exhibited CAV lesions. The onset and severity of CAV were analyzed using our standard histomorphometric test to evaluate intimal thickening and subsequent luminal occlusion on numerous graft vessels (23). The B6 heart grafts being rejected by control GFP-CBA mice (Fig. 2B) displayed severe CAV associated with fibrosis and intimal occlusion that was detectable as early as 14–28 d posttransplantation (Fig. 2E). In contrast, the B6 hearts collected from IAb-CBA mice had no signs of chronic rejection and displayed normal vessel architecture for as long as 177 d after transplantation (Fig. 2C, 2D). Therefore, in addition to preventing acute allograft rejection, expression of donor-type MHCII gene in hematopoietic cells of graft recipients also thwarted chronic rejection of heart allografts.
Expression of donor MHCII in graft recipients modulates antidonor alloresponses
Acute rejection of allotransplants is dependent upon the activation/expansion of proinflammatory T cells secreting type 1 cytokines (IL-2 and IFN-γ) (26). Conversely, T cells secreting type 2 cytokines (IL-4 and -10) have been implicated in the prolongation of allograft survival (27), presumably by suppressing inflammatory T cell immunity (28). These observations prompted us to examine whether the IAb gene transfer in CBA mice had altered their ability to mount a T cell-mediated alloresponse to B6 allogeneic cells (Fig. 3). The frequency and specificity of activated T cells producing type 1 (IL-2 and IFN-γ) or type 2 (IL-4) cytokines were measured in control CBA and IAb-CBA mice. To test this, T cells from control CBA and IAb-CBA mice were collected prior to and 10 d after placement of a B6 cardiac allotransplant. These cells were then cultured for 24–48 h in the presence of donor B6 or third-party BALB/c irradiated stimulators and the numbers of types 1 and 2 cytokine-producing T cells were assessed using an ELISPOT assay. A similar frequency of IL-2 producers (~120 spots/million T cells) was observed for CBA and IAb-CBA T cells responding to third-party BALB/c stimulators, irrespective of the presence of B6 allografts (Fig. 3A, 3D). A comparable trend developed in anti-BALB/c, IFN-γ responses of CBA and IAb-CBA mice (Fig. 3B, 3E, top). Thus, expression of IAb in IAb-CBA mice did not affect the anti-third party (BALB/c H-2d) T cell response that, in an MLR, is mainly directed to allogeneic MHCII Ags (Ad and Ed) (29). We next measured the inflammatory (type 1) alloresponse of CBA and IAb-CBA mice to B6 stimulators. As seen in Fig. 3A and 3B, these mice developed similar Th1 responses (~280 spots/million T cells) prior to transplantation, suggesting that the primary anti-MHCII IAb response was intact in IAb-CBA mice. In contrast, the type 1 inflammatory alloresponse was abrogated in the IAb-CBA mice transplanted with a B6 heart compared with the control allotransplanted CBA recipients (Fig. 3D, 3E). In turn, high frequencies of activated donor-specific T cells secreting type 2 cytokine T cells were found in allotransplanted IAb-CBA mice (Fig. 3F). Therefore, the acceptance of IAb+ B6 transplants in IAb-CBA mice is associated with a shifting from a type 1 proinflammatory antidonor T cell response toward an alloresponse dominated by T cells secreting type 2 cytokines.
FIGURE 3.
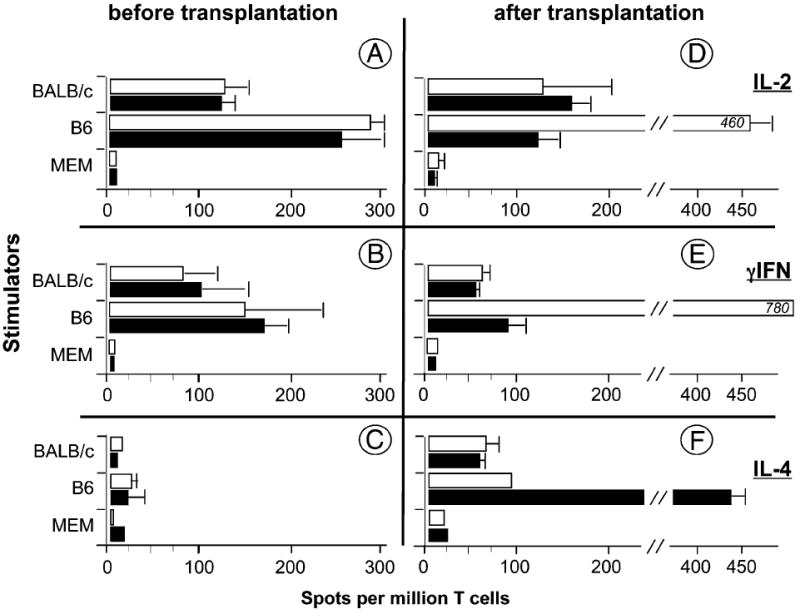
T cell alloresponses in IAb-CBA mice. IL-2 (A, D), IFN-γ (B, E), and IL-4 (C, F) ELISPOT assays were performed on splenocytes from naive CBA (white bars) and IAb-CBA (black bars) mice. Assays were done 8 wk after BM transplantation, prior to heart transplantation (n = 3) and 10 d after transplantation of B6 heart grafts (n = 3). Stimulator cells were irradiated splenocytes from BALB/c or B6 mice. MEM: control medium. Results are mean ± SD.
Transferred IAb is expressed in lymphoid tissues and in BM-derived DCs
Eight weeks after transfusion of transduced BMCs, the IAb-CBA mice received a B6 heart under anti-CD8 therapy (Fig. 1). The presence of IAb transcripts was detected in PBMCs from 100% of animals 2 wk after BMC injection, whereas 77% (14/18) were positive at the time of heart transplantation (Fig. 4B). The IAb signal faded out over time in all IAb-treated animals to become undetectable in PBMCs by 18 wk posttransduction (Fig. 4B). A similar pattern of GFP expression was observed when the control GFP.RV vector was used for transduction. In addition, PBLs from control GFP-CBA mice were RT-PCR negative for IAb transcripts (Fig. 4A). Expression of IAb was further analyzed in lymphoid tissues from CBA-IAb mice that were collected at the time of allograft rejection (2/11 mice) or at postoperative days (PODs) 150–177 in six long-term tolerant animals (Fig. 4B).
FIGURE 4.

Time course analysis of IAb transcription in vivo. A, RT-PCR analysis of IAb and control CD2 gene transcription in tissues from long-term tolerant CBA #13 (POD 176). Proviral IAb sequences were detected by annealing to a [32P]-IRES–specific oligonucleotide. B, IAb transgene transcription in PBMCs, lymph nodes (LN), spleen, and thymus. Two rejector IAb-CBA (Rej; tested 11 wk after BMC infusion) and six long-term tolerant IAb-CBA (Tol; 30–32 wk) mice were monitored by RT-PCR for proviral IAb transcription. *Number of animals positive for IAb/number tested. Rej, rejector IAb-CBA mouse; Tol, long-term tolerant IAb-CBA mouse.
To investigate IAb surface expression in transduced BMCs, we examined cells collected 48 h after ex vivo transduction. Surprisingly, IAb.RV-transduced BMCs were negative for surface IAb (Fig. 5A), whereas 10–15% of cells stained with specific Abs for endogenous IAk (data not shown). To ascertain whether the absence of surface IAb was due to poor transduction or to cell type-specific constraints, IAb expression was further studied in DCs derived from transduced BM, because DCs are key players in thymocyte negative selection (2). Bulk cultures of DCs derived from control GFP transduction were fluorescent, as were immature DCs (iDCs) and mDCs that differentiated from those cultures (Fig. 5B). Unexpectedly, iDCs and mDCs obtained from superinfected IAb-GFP BMCs were negative for IAb-GFP and positive for endogenous IAk (Fig. 5C, panels 1–3). This suggests a defective IAb gene transcription in DCs and/or an intracellular cleavage of the ectopic IAb α- and β-chains. The former hypothesis was tested by comparing GFP transcript and protein levels in IAb-GFP–transduced DCs and control fibroblast cells (L cells). Results presented in Fig. 5C, panels 4–6, indicated that transcription of IAb-GFP in DCs was not compromised. Finally, monensin was used to block protein transport in A20 monocytic, MHCII+ cells transduced with IAb-GFP.RV. Results suggested that cytosolic IAb-GFP fusion protein was short-lived (Fig. 5C, panels 7 and 8).
FIGURE 5.
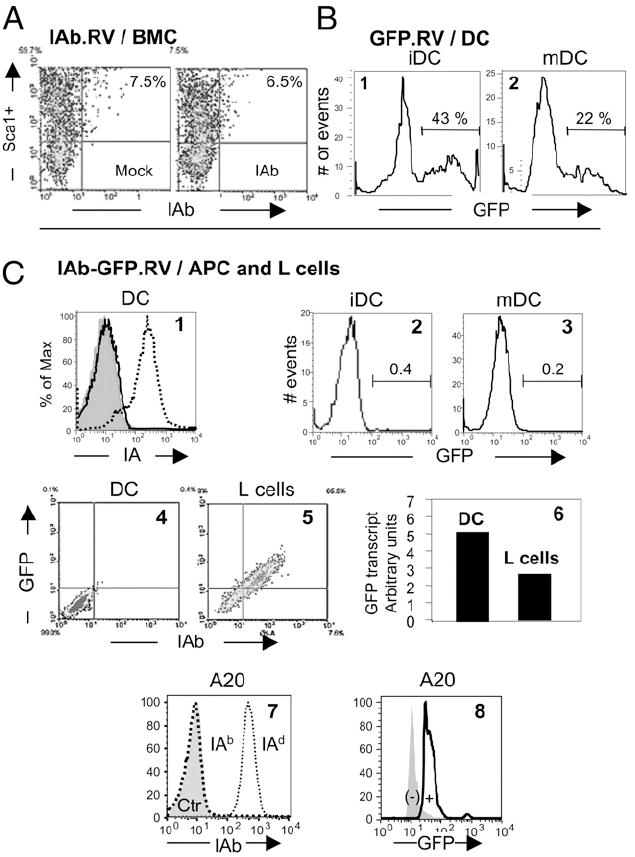
Patterns of IAb transgene expression in APCs. A, BM Sca1+ progenitors from CBA mice transduced with the empty (mock) or IAb.RV (IAb) vectors were analyzed 48 h after transduction for IAb expression. B, Subsets of DCs, derived from BMCs from CBA mice, were sorted as iDCs (CD11c+IAkloCD86lo) or mDCs (CD11c+IAkhiCD86hi). Each subset was transduced with the GFP.RV vector and analyzed 5 d later for GFP expression. Data presented are from one of two experiments. C, 1: Unsorted DC cultures from CBA mice were transduced with the IAb-GFP.RV vector and tested for surface expression of recipient (IEk, dotted line) and vector-derived MHCII (IAb, black line) 7 d posttransduction. Shaded curve represents Ig isotype control. 2, 3: GFP expression in the iDC and mDC subsets that were derived from the same bulk DC culture transduced with the IAb-GFP.RV vector. Representative results from one of three experiments. 4, 5: DCs and L cells transduced with IAb-GFP.RV were analyzed for expression of the IAb-GFP fusion protein 1 wk after transduction. Similar data were obtained in four additional experiments using DC or other APC lines. 6: Real-time RT-PCR analysis of GFP transcripts in transduced DC and L cells from panels 4 and 5. 7: The IAd+ IEd+ lymphoblastic cell line A20 was transduced with the IAb-GFP.RV and tested 4 d later for endogenous (IAd, fine dotted line) and ectopic (IAb, bold dotted line) expression. Shaded curve represents staining with isotype control (Ctr). 8: A20 cells from panel C7 were also tested for GFP expression in the absence (−) or presence (+) of monensin.
Tregs from tolerant IAb gene recipients transfer transplantation tolerance to naive animals
Our data on indefinite survival of fully allogeneic B6 heart grafts implanted in IAb-CBA recipients (Fig. 2B) lend support to the notion of complete inactivation of type 1 inflammatory alloresponses to B6 graft major and minor Ags, a phenomenon tentatively attributed to Tregs (30). Therefore, we examined whether acceptance of B6 allogeneic hearts could be achieved in naive CBA mice, recipients of purified Tregs from long-term tolerant IAb-CBA mice. The in vitro suppressive function of IAb-CBA Tregs was first assessed on two tolerant mice at 177 and 178 d post-heart transplantation by a standard Treg suppression assay (31). Results indicated that CD4+CD25+ Tregs from IAb-CBA and naive CBA mice were poorly and equally suppressive of CD4+ CD25− effector T cell proliferation (data not shown). To activate recipient Tregs in vivo, tolerant IAb-CBA mice (>160 d post-transplantation) were infused with two consecutive donor-specific blood transfusions (32). Two weeks later, the CD4+CD25+Foxp3+ Treg and CD4+CD25−Foxp3− effectors subsets were isolated (purity >90%) from DST-treated IAb-CBA recipients as well as from naive CBA mice injected or not with DST (Fig. 1). These cell populations were injected separately into naive CBA mice together with one dose of anti-CD8. Treated animals were challenged the following day with B6 allografts, according to the protocol detailed in Fig. 1. Transfer of CD25− or CD25+ T cell subsets, isolated from untreated CBA mice, did not result in statistically significant improvements in graft survival over that of non-transferred controls (Fig. 6, left panel; 33 ± 3 d; p = 0.2). CBA mice that received CD25+ or CD25− T cells collected from mice treated with DST only experienced accelerated rejection of B6 hearts (Fig. 6, middle panel; 16 ± 2 d; p = 0.01). More importantly, four of five naive CBA mice that were injected with purified CD25+ Tregs from tolerant IAb-CBA mice showed significant prolongation of graft survival (Fig. 6, right panel; p = 0.03). In contrast, three of four recipients of CD25− cells from tolerant IAb-CBA mice rejected their transplants in the same time course as untreated CBA controls. These data underscored the potent role of tolerant Tregs at transferring tolerance, because a single dose of Tregs fostered significant prolongation of graft survival in immunocompetent recipients. Fig. 7 tentatively presents a model of Treg control of alloresponses, which will be discussed in detail below.
FIGURE 6.
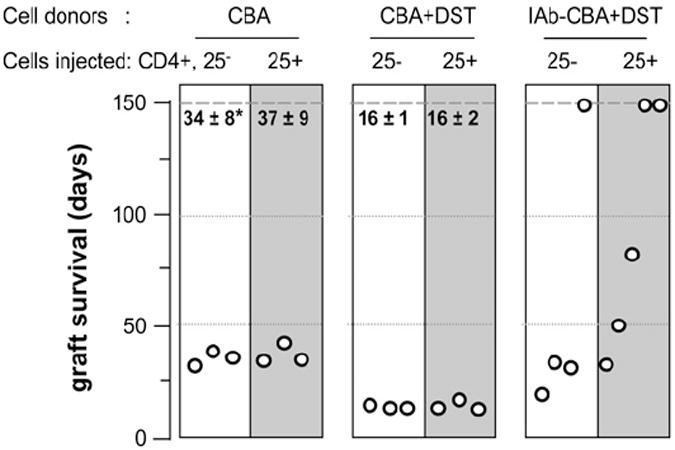
Tregs from tolerant recipients transfer IAb-induced tolerance to B6 heart grafts. CD4+CD25− and CD4+CD25+ T cells were purified from naive CBA mice, CBA mice infused with a donor-specific transfusion (CBA+DST), or long-term tolerant IAb-CBA mice treated with DST (IAb-CBA+DST). Each T cell subset was injected separately into naive CBA mice that also received a dose of anti-CD8 mAb and B6 heart grafts on the following day. The effects of adoptive transfer of CD4+CD25− (white columns) and CD4+CD25+ (gray columns) T cells on graft survival were monitored. Each point represents survival data from a single mouse. *Survival time (days; mean ± SD).
FIGURE 7.
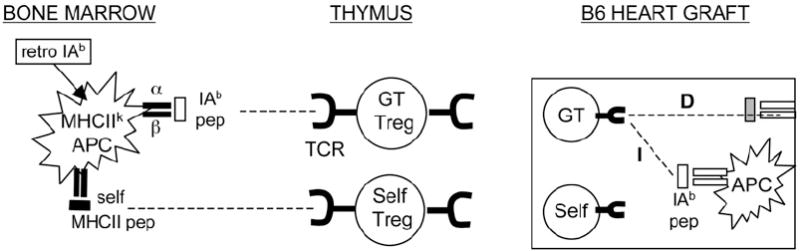
MHCII gene therapy to induce Treg-mediated tolerance to vascularized transplants. Transduction of BM-derived APCs from recipient mice (H-2k) with the IAb.RV (retro IAb) vector leads to IAb peptides (pep) presented on MHCIIk heterodimers. These MHCII/peptide complexes participate in the thymic differentiation of IAb-specific Tregs (GT Tregs). Self-Tregs would differentiate on self-MHCII peptides/MHCII complexes. In these animals, transplanted hearts from B6, but not from third-party BALB/c donors, provide IAb signals for the in situ activation of GT Tregs via the direct (D) or indirect (I) presentation pathways. In turn, locally activated GT Tregs repress the entire Th1 antigraft alloresponse (spreading tolerance) and prevent rejection.
Discussion
The present study demonstrated that mice expressing a single donor-type MHCII molecule at the time of transplantation of fully allogeneic heart grafts become tolerant to the whole donor/recipient antigenic disparity, a phenomenon referred to as linked or infectious tolerance (33). The tolerogenic effects of MHCII gene therapy is not unique to the murine heart transplantation model; we previously reported that MHCII gene transfer induced tolerance to MHCII transgene-matched renal allografts in large animals (16). Transfer of fibroblasts expressing donor MHC class I and MHCII prolonged cardiac allograft survival in mice (34), suggesting an equal role for MHCI and MHCII genes in regulating immune responses. However, in contrast to our gene-therapy approach, these experiments did not induce transplantation tolerance. In addition, retroviral transfer of MHCI Kb gene in BMCs also failed to induce spreading tolerance to Kb+ grafts coexpressing third-party Ags (35). Thus, our findings strongly suggest that induction of spreading tolerance is a property of MHCII molecules. Although transduction rates were not as high as those for CBA BMCs, extended prolongation of graft survival was also observed in BALB/c mice receiving IAb genes and B6 transplants (75 ± 6 d; n = 10). This approach also established that linked tolerance was preferentially carried out by Tregs (Fig. 6). These points imply that the immune regulatory function of MHCII genes, which has been the cornerstone of their discovery (36), resides, in part, in their unique ability to generate Tregs.
The long-term surviving heart transplants (>170 d) presented no sign of chronic rejection, the major cause of late graft loss in clinical transplantation (Fig. 2) (37). Prevention of CAV development has been reported in other models using heavy conditioning with costimulatory blockade, cytolytic drugs, and donor BM infusions (38). Remarkably, our study showed that transduction of recipient BMCs with a single donor MHCII gene can achieve tolerance to fully allogeneic heart transplants, a result showing that activation of donor-specific Tregs can be sufficient to prevent CAV.
B6 heart transplants survived indefinitely in IAb-treated CBA mice, whereas third-party BALB/c cardiac allografts were acutely rejected. This supports a model of MHCII allele-specific tolerance involving the recognition of ectopic IAb heterodimers (direct recognition) and/or IAb-derived peptides presented by recipient MHC H-2k molecules (indirect recognition). Testing for IAb surface expression on transduced BMCs revealed no detectable IAb signal on whole BM or Sca1+ckit + hematopoietic precursors, although IAb transcripts were consistently detected by RT-PCR. Likewise, blood cells from IAb-CBA mice exhibited no IAb on their surface but expressed IAb transcripts (Fig. 4). Analysis of IAb expression in DCs derived from IAb-transduced BMCs confirmed the absence of surface IAb molecules, despite levels of IAb transcripts that were compatible with surface expression (Fig. 5). Results from preliminary studies suggest that the α/β-chain pairing, rather than a limiting pool of the invariant chain, is a critical checkpoint controlling ectopic IAb chain cleavage. Because DCs are involved in the establishment of CD4+ T cell tolerance to self-Ags (39, 40), the absence of surface IAb molecules on IAb-transduced DCs suggests that the presentation of IAb intracellular chains or IAb -derived peptides by recipient MHC molecules underlies tolerance induction in this model. Additional data from our group support the MHCII peptide hypothesis. We observed that human B cell lines, transduced with a pig MHCII DR gene, did not express surface pig DR but stimulated autologous human T cell proliferation, which was only blocked by anti-human MHCII mAbs (41).
Immunocompetent CBA recipients were used to demonstrate acceptance of B6 allografts following the injection of Tregs from tolerant IAb-CBA mice (Fig. 6). Although other successful Treg/ tolerance transfer protocols have been described using immunoincompetent Rag−/− recipients (42), to our knowledge, this is the first report on efficient transfer of graft acceptance in immunocompetent recipients via a single treatment with isolated Tregs. The initial in vivo priming to donor blood cells, and likely to cognate IAb, was potentially the cause of this favorable outcome, because injection of MHCII− cells abolished the DST effect (43).
Treg TCR specificity was shown to be different from that of Th cells that they ultimately suppress (44). Our results suggest that MHCII allele-specific tolerance may be driven by the recognition of IAb peptides displayed on BM-derived APCs. This suggests that naturally occurring Tregs might differentiate in the thymus following recognition of MHCII/MHCII peptide complexes. We hypothesize that the transfer of the IAb gene in CBA mice promoted the development of IAb peptide-specific Tregs (Fig. 7; gene therapy [GT] Tregs). IAb-treated animals would only accept IAb+ transplants because the latter provide IAb determinants via the direct or indirect pathways for secondary stimulation of GT Tregs and suppression of the antigraft responses. Tolerance to autografts tissues would proceed along the same mechanism involving, this time, native CBA Tregs. Data from autoimmune models have stressed the pivotal role played by MHCII-derived peptides in the reversal of defective Treg functions. For instance, the injection of NOD mice with self-IEb peptide led to Treg activation (45), whereas binding of IE-derived peptides on IA heterodimers prevented collagen-induced arthritis (46). The specificity of naturally occurring Tregs for self-MHCII determinants is consistent with results from studies that showed Treg thymic precursors differentiating in the medulla (3), a compartment that is known to selectively display MHCII peptide/MHCII complexes (47). Such bias of Treg specificity for MHCII determinants may explain how MHCII genes control the effector as well as the regulatory phases of T lymphocyte responses.
Acknowledgments
The authors are grateful to Kaela Golstein for technical assistance and to Dr. Christene Huang for critical review of the manuscript.
This work was supported by National Institutes of Health Grants R01EY13310 (to G.B.) and R01AI063408 (to C.L.). Y.A. was the recipient of a Sangstat fellowship from the American Society of Transplantation.
Abbreviations used in this paper
- BM
bone marrow
- BMC
bone marrow cell
- CAV
cardiac allograft vasculopathy
- Ctr
control
- D
direct
- DC
dendritic cell
- DST
donor-specific transfusion
- eGFP
enhanced GFP
- I
indirect
- GT
gene therapy
- iDC
immature dendritic cell
- IRES
internal ribosome entry site
- LN
lymph node
- mDC
mature dendritic cell
- MHCII
MHC class II
- MST
mean survival time
- pep
peptides
- pMHCII
MHC class II molecules plus self-peptide
- POD
postoperative day
- qRT-PCR
quantitative RT-PCR
- Rej
rejector IAb-CBA mouse
- Tol
long-term tolerant IAb-CBA mouse
- Treg
regulatory T cell
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol. 2003;21:139–176. doi: 10.1146/annurev.immunol.21.120601.141107. [DOI] [PubMed] [Google Scholar]
- 2.Sprent J, Kishimoto H. The thymus and negative selection. Immunol Rev. 2002;185:126–135. doi: 10.1034/j.1600-065x.2002.18512.x. [DOI] [PubMed] [Google Scholar]
- 3.Watanabe N, Wang YH, Lee HK, Ito T, Wang YH, Cao W, Liu YJ. Hassall’s corpuscles instruct dendritic cells to induce CD4+CD25+ regulatory T cells in human thymus. Nature. 2005;436:1181–1185. doi: 10.1038/nature03886. [DOI] [PubMed] [Google Scholar]
- 4.Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–352. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 5.McDevitt HO, Deak BD, Shreffler DC, Klein J, Stimpfling JH, Snell GD. Genetic control of the immune response. Mapping of the Ir-1 locus. J Exp Med. 1972;135:1259–1278. doi: 10.1084/jem.135.6.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shevach EM, Rosenthal AS. Function of macrophages in antigen recognition by guinea pig T lymphocytes. II. Role of the macrophage in the regulation of genetic control of the immune response. J Exp Med. 1973;138:1213–1229. doi: 10.1084/jem.138.5.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hunt DF, Michel H, Dickinson TA, Shabanowitz J, Cox AL, Sakaguchi K, Appella E, Grey HM, Sette A. Peptides presented to the immune system by the murine class II major histocompatibility complex molecule I-Ad. Science. 1992;256:1817–1820. doi: 10.1126/science.1319610. [DOI] [PubMed] [Google Scholar]
- 8.Ashton-Rickardt PG, Bandeira A, Delaney JR, Van Kaer L, Pircher HP, Zinkernagel RM, Tonegawa S. Evidence for a differential avidity model of T cell selection in the thymus. Cell. 1994;76:651–663. doi: 10.1016/0092-8674(94)90505-3. [DOI] [PubMed] [Google Scholar]
- 9.Fukui Y, Ishimoto T, Utsuyama M, Gyotoku T, Koga T, Nakao K, Hirokawa K, Katsuki M, Sasazuki T. Positive and negative CD4+ thymocyte selection by a single MHC class II/peptide ligand affected by its expression level in the thymus. Immunity. 1997;6:401–410. doi: 10.1016/s1074-7613(00)80283-6. [DOI] [PubMed] [Google Scholar]
- 10.Schaeffer EB, Sette A, Johnson DL, Bekoff MC, Smith JA, Grey HM, Buus S. Relative contribution of “determinant selection” and “holes in the T-cell repertoire” to T-cell responses. Proc Natl Acad Sci USA. 1989;86:4649–4653. doi: 10.1073/pnas.86.12.4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gelder C, Davenport M, Barnardo M, Bourne T, Lamb J, Askonas B, Hill A, Welsh K. Six unrelated HLA-DR-matched adults recognize identical CD4+ T cell epitopes from influenza A haemagglutinin that are not simply peptides with high HLA-DR binding affinities. Int Immunol. 1998;10:211–222. doi: 10.1093/intimm/10.2.211. [DOI] [PubMed] [Google Scholar]
- 12.Erlich HA, Opelz G, Hansen J. HLA DNA typing and transplantation. Immunity. 2001;14:347–356. doi: 10.1016/s1074-7613(01)00115-7. [DOI] [PubMed] [Google Scholar]
- 13.Rosengard BR, Ojikutu CA, Guzzetta PC, Smith CV, Sundt TM, 3rd, Nakajima K, Boorstein SM, Hill GS, Fishbein JM, Sachs DH. Induction of specific tolerance to class I-disparate renal allografts in miniature swine with cyclosporine. Transplantation. 1992;54:490–497. doi: 10.1097/00007890-199209000-00020. [DOI] [PubMed] [Google Scholar]
- 14.LeGuern C, Shimada H, Emery DW, Germana S, Shafer GE, Sachs DH. Retrovirus-mediated transfer of MHC class II cDNA into swine bone marrow cells. J Mol Med. 1995;73:269–278. doi: 10.1007/BF00231613. [DOI] [PubMed] [Google Scholar]
- 15.Emery DW, Sablinski T, Shimada H, Germana S, Gianello P, Foley A, Shulman S, Arn S, Fishman J, Lorf T, et al. Expression of an allogeneic MHC DRB transgene, through retroviral transduction of bone marrow, induces specific reduction of alloreactivity. Transplantation. 1997;64:1414–1423. doi: 10.1097/00007890-199711270-00007. [DOI] [PubMed] [Google Scholar]
- 16.Sonntag KC, Emery DW, Yasumoto A, Haller GW, Germana S, Sablinski T, Shimizu A, Yamada K, Shimada H, Arn S, et al. Tolerance to solid organ transplants through transfer of MHC class II genes. J Clin Invest. 2001;107:65–71. doi: 10.1172/JCI11015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orbaek Andersen H. Heart allograft vascular disease: an obliterative vascular disease in transplanted hearts. Atherosclerosis. 1999;142:243–263. doi: 10.1016/s0021-9150(98)00291-3. [DOI] [PubMed] [Google Scholar]
- 18.Miller AD, Rosman GJ. Improved retroviral vectors for gene transfer and expression. Biotechniques. 1989;7:980–982. 984–986, 989–990. [PMC free article] [PubMed] [Google Scholar]
- 19.Banerjee PT, Kanor GC, Muthukumar S, Denaro M, Shimada H, Zhu S, Rosa MD, Sachs DH, LeGuern C. A polycistronic retrovirus vector for expression of swine MHC class II DR α/β heterodimers. Xenotransplantation. 1997;4:161–173. [Google Scholar]
- 20.Andersson G, Denaro M, Johnson K, Morgan P, Sullivan A, Houser S, Patience C, White-Scharf ME, Down JD. Engraftment of retroviral EGFP-transduced bone marrow in mice prevents rejection of EGFP-transgenic skin grafts. Mol Ther. 2003;8:385–391. doi: 10.1016/s1525-0016(03)00210-7. [DOI] [PubMed] [Google Scholar]
- 21.Andersson G, Illigens BM, Johnson KW, Calderhead D, LeGuern C, Benichou G, White-Scharf ME, Down JD. Nonmyeloablative conditioning is sufficient to allow engraftment of EGFP-expressing bone marrow and subsequent acceptance of EGFP-transgenic skin grafts in mice. Blood. 2003;101:4305–4312. doi: 10.1182/blood-2002-06-1649. [DOI] [PubMed] [Google Scholar]
- 22.Corry RJ, Winn HJ, Russell PS. Primarily vascularized allografts of hearts in mice. The role of H-2D, H-2K, and non-H-2 antigens in rejection. Transplantation. 1973;16:343–350. doi: 10.1097/00007890-197310000-00010. [DOI] [PubMed] [Google Scholar]
- 23.Houser SL, McMorrow IM, LeGuern C, Schwarze ML, Fuchimoto DH, Sachs DH, Madsen JC. Histomorphometric comparison of cardiac allograft vasculopathy in miniature swine. J Heart Lung Transplant. 2004;23:50–60. doi: 10.1016/s1053-2498(03)00065-2. [DOI] [PubMed] [Google Scholar]
- 24.Ozato K, Sachs DH. Monoclonal antibodies to mouse MHC antigens. III Hybridoma antibodies reacting to antigens of the H-2b haplotype reveal genetic control of isotype expression. J Immunol. 1981;126:317–321. [PubMed] [Google Scholar]
- 25.Benichou G, Valujskikh A, Heeger PS. Contributions of direct and indirect T cell alloreactivity during allograft rejection in mice. J Immunol. 1999;162:352–358. [PubMed] [Google Scholar]
- 26.Csencsits KL, Bishop DK. Contrasting alloreactive CD4+ and CD8+ T cells: there’s more to it than MHC restriction. Am J Transplant. 2003;3:107–115. doi: 10.1034/j.1600-6143.2003.00036.x. [DOI] [PubMed] [Google Scholar]
- 27.Strom TB, Roy-Chaudhury P, Manfro R, Zheng XX, Nickerson PW, Wood K, Bushell A. The Th1/Th2 paradigm and the allograft response. Curr Opin Immunol. 1996;8:688–693. doi: 10.1016/s0952-7915(96)80087-2. [DOI] [PubMed] [Google Scholar]
- 28.Oldenhove G, de Heusch M, Urbain-Vansanten G, Urbain J, Maliszewski C, Leo O, Moser M. CD4+ CD25+ regulatory T cells control T helper cell type 1 responses to foreign antigens induced by mature dendritic cells in vivo. J Exp Med. 2003;198:259–266. doi: 10.1084/jem.20030654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Bont ES, Reilly CR, Lo D, Glimcher LH, Laufer TM. A minimal level of MHC class II expression is sufficient to abrogate autoreactivity. Int Immunol. 1999;11:1295–1306. doi: 10.1093/intimm/11.8.1295. [DOI] [PubMed] [Google Scholar]
- 30.Cobbold SP, Waldmann H. Infectious tolerance. Curr Opin Immunol. 1998;10:518–524. doi: 10.1016/s0952-7915(98)80217-3. [DOI] [PubMed] [Google Scholar]
- 31.Suri-Payer E, Amar AZ, Thornton AM, Shevach EM. CD4+CD25 + T cells inhibit both the induction and effector function of autoreactive T cells and represent a unique lineage of immunoregulatory cells. J Immunol. 1998;160:1212–1218. [PubMed] [Google Scholar]
- 32.Roelen DL, van Rood JJ, Brand A, Claas FH. Immunomodulation by blood transfusions. Vox Sang. 2000;78(Suppl. 2):273–275. [PubMed] [Google Scholar]
- 33.Qin S, Cobbold SP, Pope H, Elliott J, Kioussis D, Davies J, Waldmann H. “Infectious” transplantation tolerance. Science. 1993;259:974–977. doi: 10.1126/science.8094901. [DOI] [PubMed] [Google Scholar]
- 34.Madsen JC, Superina RA, Wood KJ, Morris PJ. Immunological unresponsiveness induced by recipient cells transfected with donor MHC genes. Nature. 1988;332:161–164. doi: 10.1038/332161a0. [DOI] [PubMed] [Google Scholar]
- 35.Fraser CC, Sykes M, Lee RS, Sachs DH, LeGuern C. Specific unresponsiveness to a retrovirally-transferred class I antigen is controlled through the helper pathway. J Immunol. 1995;154:1587–1595. [PubMed] [Google Scholar]
- 36.McDevitt HO. Discovering the role of the major histocompatibility complex in the immune response. Annu Rev Immunol. 2000;18:1–17. doi: 10.1146/annurev.immunol.18.1.1. [DOI] [PubMed] [Google Scholar]
- 37.Libby P, Pober JS. Chronic rejection. Immunity. 2001;14:387–397. doi: 10.1016/s1074-7613(01)00119-4. [DOI] [PubMed] [Google Scholar]
- 38.Shirasugi N, Adams AB, Durham MM, Lukacher AE, Xu H, Rees P, Cowan SR, Williams MA, Pearson TC, Larsen CP. Prevention of chronic rejection in murine cardiac allografts: a comparison of chimerism-and nonchimerism-inducing costimulation blockade-based tolerance induction regimens. J Immunol. 2002;169:2677–2684. doi: 10.4049/jimmunol.169.5.2677. [DOI] [PubMed] [Google Scholar]
- 39.Adler AJ, Marsh DW, Yochum GS, Guzzo JL, Nigam A, Nelson WG, Pardoll DM. CD4+ T cell tolerance to parenchymal self-antigens requires presentation by bone marrow-derived antigen-presenting cells. J Exp Med. 1998;187:1555–1564. doi: 10.1084/jem.187.10.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Umemura A, Monaco AP, Maki T. Donor MHC class II antigen is essential for induction of transplantation tolerance by bone marrow cells. J Immunol. 2000;164:4452–4457. doi: 10.4049/jimmunol.164.9.4452. [DOI] [PubMed] [Google Scholar]
- 41.Denaro M, Kolber-Simonds D, Schad V, Muthukumar S, Germana S, White-Scharf ME, Banerjee PT, LeGuern C, Andersson G. Expression of xenogeneic MHC class II molecules in HLA-DR+ and -DR(−) cells: influence of retrovirus vector design and cellular context. Xenotransplantation. 2002;9:115–124. doi: 10.1034/j.1399-3089.2002.1o038.x. [DOI] [PubMed] [Google Scholar]
- 42.Kingsley CI, Karim M, Bushell AR, Wood KJ. CD25+CD4+ regulatory T cells prevent graft rejection: CTLA-4- and IL-10-dependent immunoregulation of alloresponses. J Immunol. 2002;168:1080–1086. doi: 10.4049/jimmunol.168.3.1080. [DOI] [PubMed] [Google Scholar]
- 43.van Rood JJ, Claas FH. The influence of allogeneic cells on the human T and B cell repertoire. Science. 1990;248:1388–1393. doi: 10.1126/science.1972596. [DOI] [PubMed] [Google Scholar]
- 44.Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 2000;164:183–190. doi: 10.4049/jimmunol.164.1.183. [DOI] [PubMed] [Google Scholar]
- 45.Chaturvedi P, Agrawal B, Zechel M, Lee-Chan E, Singh B. A self MHC class II β-chain peptide prevents diabetes in nonobese diabetic mice. J Immunol. 2000;164:6610–6620. doi: 10.4049/jimmunol.164.12.6610. [DOI] [PubMed] [Google Scholar]
- 46.Gonzalez-Gay MA, Nabozny GH, Bull MJ, Zanelli E, Douhan J, 3rd, Griffiths MM, Glimcher LH, Luthra HS, David CS. Protective role of major histocompatibility complex class II Ebd transgene on collagen-induced arthritis. J Exp Med. 1994;180:1559–1564. doi: 10.1084/jem.180.4.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murphy DB, Rath S, Pizzo E, Rudensky AY, George A, Larson JK, Janeway CA., Jr Monoclonal antibody detection of a major self peptide. MHC class II complex. J Immunol. 1992;148:3483–3491. [PubMed] [Google Scholar]