

Abstract
Oxidative stress and subsequent lipid peroxidation are involved in the pathogenesis of numerous neurodegenerative conditions, including stroke. Cyclopentenone isoprostanes (IsoPs) are novel electrophilic lipid peroxidation products formed under conditions of oxidative stress via the isoprostane pathway. These cyclopentenone IsoPs are isomeric to highly bioactive cyclopentenone prostaglandins, yet it has not been determined if these products are biologically active or are formed in the brain. Here we demonstrate that the major cyclopentenone IsoP isomer 15-A2t-IsoP potently induces apoptosis in neuronal cultures at submicromolar concentrations. We present a model in which 15-A2t-IsoP induced neuronal apoptosis involves initial depletion of glutathione and enhanced production of reactive oxygen species, followed by 12-lipoxygenase activation and phosphorylation of extracellular signal-regulated kinase 1/2 and the redox sensitive adaptor protein p66shc, which results in caspase-3 cleavage. 15-A2t-IsoP application also dramatically potentiates oxidative glutamate toxicity at concentrations as low as 100 nM, demonstrating the functional importance of these molecules in neurodegeneration. Finally, we employ novel mass spectrometric methods to show that cyclopentenone IsoPs are formed abundantly in brain tissue under conditions of oxidative stress. Together these findings suggest that cyclopentenone IsoPs may contribute to neuronal death caused by oxidative insults, and that their activity should perhaps be addressed when designing neuroprotective therapies.
Keywords: apoptosis, isoprostane, lipid peroxidation, 12-lipoxygenase, oxidative stress, p66shc
Excessive production of reactive oxygen species (ROS) and subsequent lipid peroxidation have been implicated in the pathogenesis of many neurodegenerative conditions, including stroke and Parkinson’s disease (Beal 1995; Chan 2001; Jenner 2003). One of the most well known but least understood factors that contribute to ROS-induced cell death is lipid peroxidation. Significant evidence suggests that reactive lipid peroxidation products, such as 4-hydroxynonenal, contribute to the pathology of CNS oxidative injury (Kruman et al. 1997; Ou et al. 2002; Bruckner et al. 2003). However, the exact lipid species and molecular mechanisms by which lipid peroxidation contributes to free radical-mediated neuronal death are poorly defined.
Free radical-induced peroxidation of arachidonic acid, a ubiquitous polyunsaturated fatty acid found in membrane phospholipids, results in the formation of isoprostanes (IsoPs), a group of prostaglandin-like compounds (Morrow et al. 1990). Previously, the highly stable, chemically inert F-ring IsoPs (F2-IsoPs) have been quantified as markers of oxidative damage (Roberts and Morrow 2002), and have been shown to be elevated in brain tissue affected by numerous neurodegenerative conditions, including ischemia, Alzheimer’s, and Huntington’s diseases (Marin et al. 2000; Montine et al. 2001, 2002; Reich et al. 2001). Recently it was demonstrated that highly reactive A- and J-ring IsoPs, known as cyclopentenone IsoPs (Fig. 1a), are abundantly formed following oxidative injury via the same isoprostane pathway (Chen et al. 1999a). Esterified cyclopentenone IsoPs were detected in membrane phospholipids under basal conditions in rat liver, and their levels increased 22-fold following oxidant injury (Chen et al. 1999a). These cyclopentenone IsoPs, which are isomeric to the bioactive prostaglandins PGA2 and PGJ2, contain an electrophilic α,β-unsaturated carbonyl moiety in their cyclopentane ring that rapidly adducts cellular thiols, including those found in proteins and glutathione (GSH) via Michael addition (Chen et al. 1999a).
Fig. 1.
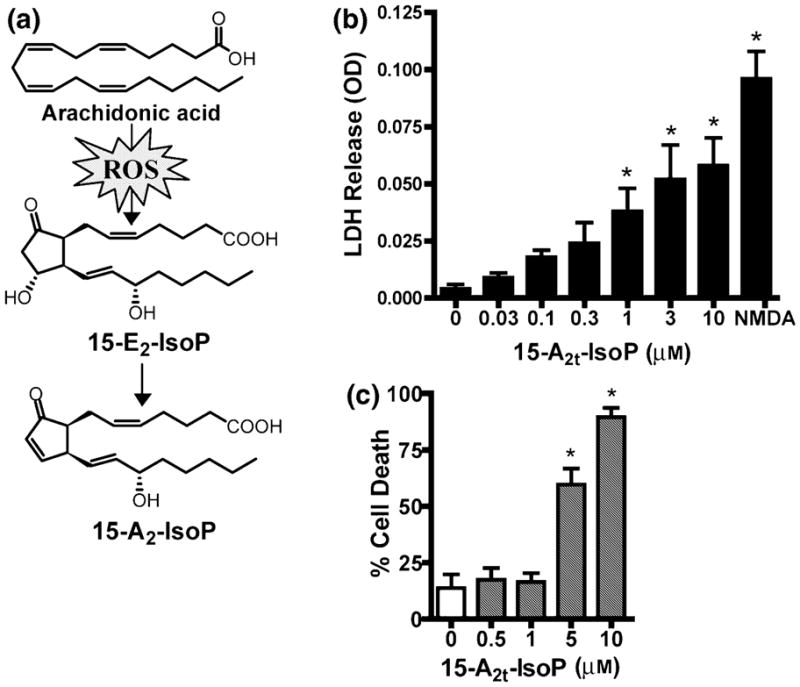
15-A2t-Isoprostane (15-A2t-IsoP) is toxic to neurons. (a) Diagram depicting the formation of 15-A2t-IsoP via the oxidation of arachidonic acid and subsequent spontaneous dehydration of 15-E2t-IsoP. (b) Mature primary cortical neurons were exposed to increasing concentrations of 15-A2t-IsoP for 48 h, at which time cell death was assessed by measuring release of lactate dehydrogenase (LDH) into the cell media from dying neurons. NMDA (100 μM) was used as a positive control, as it induces total cell death in these cultures. Note that 15-A2t-IsoP induces cell death in a dose-dependent manner with an LD50 of 950 nM. (c) The oxidative stress-sensitive HT22 hippocampal cell line was exposed to 15-A2t-IsoP for 18 h, after which cell viability was assessed by measuring cellular MTT reducing capacity. Like primary neurons, HT22 cells are killed by 15-A2t-IsoP with an LD50 of 3 μM. Data in (b) and (c) represent the mean ± SEM of at least three separate experiments, each performed at least in triplicate. *p < 0.05 vs. control by one-way ANOVA.
Due to their reactive cyclopentenone ring structure, cyclopentenone IsoPs are likely to exhibit potent biological actions (Levonen et al. 2004). However, the formation and biological activity of the reactive cyclopentenone IsoPs in the CNS remains unexplored. Cyclopentenone prostaglandins, which are produced enzymatically by cyclooxygenases and are isomeric to cyclopentenone IsoPs, have a wide variety of biological actions, including both neurotoxic (Rohn et al. 2001; Kondo et al. 2002) and neuroprotective (Qin et al. 2001; Wang et al. 2002) properties, suggesting that cyclopentenone IsoPs may also have potent bioactivity in neurons. In this work, we have for the first time determined that cyclopentenone IsoPs are indeed produced in abundance in the human brain and are elevated under oxidative and neurodegenerative conditions. Furthermore, we show that purified 15-A2t-IsoP, a major isomer formed in vivo (Fig. 1a) (Chen et al. 1999b), is a potent neuronal apoptogen. We have identified a discrete redox-sensitive cell death pathway activated by 15-A2t-IsoP which overlaps extensively with the cell death signal transduction pathways activated during ischemia and glutathione depletion. Indeed, application of 15-A2t-IsoP during mild oxidative or ischemic injury greatly exacerbates toxicity. Our results suggest that cyclopentenone IsoPs represent a novel class of neurotoxic lipid peroxidation products that contribute to ischemic and excitotoxic injury in the CNS, and reveal novel signaling pathways activated by these compounds which may represent pharmacological targets for protection from oxidative stress-induced neuro-degeneration.
Experimental procedures
Materials and reagents
15-A2t-Isoprostane was obtained by total synthesis (Zanoni et al. 2002). All cell culture media and supplements were purchased from Invitrogen (Gaithersburg, MD, USA) unless otherwise noted. 2′-Amino-3′-methoxyflavone (PD98059) and 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) and cyclohexamide (CHX) were from Calbiochem (La Jolla, CA, USA); N-tert-butyl-α-phenylnitrone (PBN), carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), and 4-hydroxy TEMPO (TEMPOL) were from Sigma (St. Louis, MO, USA), and baicalein and 2,2′-azobis(2-amidinopropane) hydrochloride (AAPH) were from Cayman (Ann Arbor, MI). Primary antibodies for activated caspase-3, extracellular signal-regulated kinase (ERK1/2), and phospho-ERK1/2 were from Cell Signaling Technologies (Beverly, MA, USA); Ku70 antibodies were purchased from BD Biosciences (San Jose, CA, USA), phospho-p66shc (serine 36) antibody was from Calbiochem and 12-lipoxygenase (12-LOX) antibody was from Cayman. Alexa 680-conjugated secondary antibodies were purchased from Molecular Probes (Eugene, OR, USA).
Cell culture
Neuron-enriched rat cortical cultures were prepared as described previously (McLaughlin et al. 1998). In brief, cortices from embryonic day 16 Sprague Dawley rat fetuses were isolated and incubated in trypsin (Sigma) for 30 min at room temperature. Cortices were dissociated in 10 mL of plating medium containing Dulbecco’s modified Eagle’s medium, 10% F12 nutrients, and 10% bovine calf serum (Hyclone, Logan, UT, USA). Cells were then plated on poly-L-ornithine coated coverslips and grown in plating medium. After 2 DIV, cells were treated for 24 h with 1–2 μM cytosine arabinoside to prevent glial proliferation, and then transferred to Neurobasal media containing 1 × B27 supplement, 100 U/mL penicillin, and 100 μg/mL streptomycin. These cultures were maintained for 21–28 days prior to use in experiments, allowing time for them to express a full complement of glutamate receptors (Sinor et al. 1997). HT22 cells, a mouse hippocampal cell line selected for its responsiveness to oxidative stress (Ratan et al. 1994a), were grown in 75-mm2 flasks in Dulbecco’s modified Eagle’s medium containing 10% bovine calf serum. HT22 cells were a generous gift of Dr Pamela Maher (Salk Institute).
15-A2t-isoprostane toxicity assays
Primary neuronal culture toxicity assays were performed by rinsing 28 DIV cells in 96-well plates with minimal essential medium (without phenol red), which was then partially replaced with 100 μL of minimal essential medium containing 25 mM HEPES, 0.001% bovine serum albumin, and 1 × N2 supplement, plus either 15-A2t-IsoP or vehicle. For experiments involving pharmacological inhibitors, 100 μL of media with inhibitor or vehicle was added for 30 min, and then replaced with media containing 15-A2t-IsoP and inhibitors. After 24–48 h, 40 μL of cell media was removed and used to assess cell viability using a lactate dehydrogenase (LDH)-based in vitro toxicity kit (Sigma). In order to account for variation in total LDH content, raw LDH values were normalized to the toxicity caused by 100 μM NMDA plus 10 μM glycine, which is known to cause 100% cell death in this system (McLaughlin et al. 2003). All experiments were performed using cells derived from at least two independent dissections.
Oxidative glutamate toxicity
Oxidative glutamate toxicity assays were performed as previously described (Ratan et al. 1994a). Briefly, immature dissociated fore-brain neurons (1–2 DIV) were rinsed twice with minimal essential medium, and then incubated in minimal essential medium containing 25 mM HEPES, 0.01% bovine serum albumin, 1 × N2 supplement, and 10 μM glycine, with or without 3 mM glutamate and 15-A2t-IsoP. LDH assays and microscopy were performed 18–24 h later.
For all toxicity experiments using HT22 cells, cells were plated at a density of 7500 cells/well in 96-well plates 24 h prior to experiments. Cells were rinsed with minimal essential medium, and then treated with minimal essential medium containing 0.01% fetal bovine serum, 2 mM glutamine, 10 μM glycine, and 5.5 g/L glucose, with or without 3 mM glutamate and 15-A2t-IsoP, for 18 h. Cell media was next replaced with minimal essential medium plus 10% fetal bovine serum. Cultures were then incubated with a 1 : 10 dilution of 5 mg/mL MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reagent (Sigma) dissolved in minimal essential medium. Cells were incubated for 3 h, allowing MTT reduction by living cells, then solubilized by 1 : 1 addition of acidified isopropanol (0.1 N HCl) containing 10% Triton X-100, and absorbance measured at 630 nm using a SPECRAfluor Plus fluorometric plate reader (Tecan, Durham, NC, USA). MTT measurements were normalized to toxicity caused by 1M sorbitol (100%). Serum-free media was used in all experiments. Slightly lower levels of cell death and more reproducible results were observed in serum-free media than if serum was included (3 mM glutamate induced ~85% cell death in 10% serum, but only ~50% death without serum). We believe this observation is a consequence of variability in manufacturer’s serum preparation as well as of residual glutamine in serum which would potentiate glutamate toxicity in this model.
Mass spectrometric analysis of cyclopentenone isoprostanes
Frozen brain samples (100–200 mg) were placed directly in 2 : 1 chloroform : methanol (10 mL) containing 0.005% butylated hydroxytolulene, then homogenized and extracted as previously described (Morrow and Roberts 1999). For experiments employing oxidized brain tissue, adult rat brain tissue was homogenized and oxidized via incubation in phosphate-buffered saline containing 1 mM 2,2′-azobis-(2-amidinopropane) (AAPH) at 37°C for 12 h, then extracted with 2 : 1 chloroform methanol. Lipid extracts were dried under N2, resuspended in diethyl ether, and esterified IsoPs were hydrolyzed via addition of Apis melifera phospholipase A2 (Sigma). Three nanograms of [2H4]PGA2 internal standard was then added to each of the samples, which were dried, resuspended in MeOH, then diluted in pH 3 water and subjected to solid-phase extraction on C18 SepPaks (Waters, Milford, MA, USA) as previously described, (Morrow and Roberts 1999) followed by resuspension in 2 : 1 methanol : water. Cyclopentenone IsoPs were then analyzed by liquid chromatography electrospray tandem mass spectrometry (LC/MS/MS). LC was carried out on a Mercury MS (Phenomenex, Torrance, CA, USA) C18 column (2.0 × 20 mm) utilizing a gradient mobile phase at 300 μL/min. Mobile phase Awas water, mobile phase B was acetonitrile/methanol (95/5); the gradient started with 80% A and 20% B and proceeded to 40% A and 60% B over 19 min. Cyclopentenone IsoPs were analyzed in the multiple-reaction monitoring (MRM) mode on a ThermoFinnigan TSQ Quantum 1.0 SR 1 mass spectrometer. The reaction monitored for both the samples and internal standard was decarboxylation and loss of water from the parent ion (loss of 62 from the parent m/z).
Human brain samples
Human postmortem samples of frontal cortex were kindly provided from the Human Brain and Spinal Fluid Resource Center by Dr Rashad Nagra of the VA West Los Angeles Healthcare Center. All included patients were diagnosed as neurpathologically normal. The average patient age was 72.8 + 13.9 years, and the average postmortem interval was 15.3 + 4.7 h. Samples were prepared and analyzed in the manner described above.
Measurement of total F2-isoprostanes
Total F2-IsoPs were measured in cell lysates and conditioned media from neuronal cultures by gas chromatography/mass spectrometry (GC/MS) as previously described (Morrow and Roberts 1999). Data was normalized to number of cells collected.
Immunofluorescence
Following exposure to 15-A2t-IsoPs or vehicle, cultures were fixed in 10% formaldehyde for 20 min, then rinsed with phosphate-buffered saline, permeabilized with 0.1% Triton X-100, and blocked for 1 h with 1% bovine serum albumin diluted in phosphate-buffered saline. Coverslips were then incubated overnight at 4°C in rabbit anti-activated caspase-3 (1 : 75) and mouse anti- microtubule-associated protein 2 (MAP2) (1 : 200), or rabbit polyclonal anti-12-LOX (1 : 100) primary antibodies. Cells were then washed in phosphate-buffered saline for a total of 20 min and incubated in cy-2 or cy-3-labeled secondary antibodies for 1 h. Cells were then washed and stained with 1.4 μM 4′-6-diamidino-2-phenylindole (DAPI) for 10 min, followed by further washes. Coverslips were mounted on microscope slides, and fluorescence was visualized with a Zeiss Axioplan microscope.
Glutathione assay
Cellular glutathione was measured using the ApoGSH glutathione detection kit (Biovision, Mountain View, CA, USA), following the manufacturers protocol. Briefly, cell lysates were incubated with a solution containing 2 μL of 25 mM monochlorobimane (MCB), a dye that has a high affinity for glutathione (GSH), and 2 μL of 50 U/mL glutathione-S-transferase. Free MCB is non-fluorescent, whereas the GSH–MCB adduct emits at 461 nm after excitation at 380 nm. Fluorescence measurements were performed using a SPECRAfluor Plus fluorometric plate reader (Tecan, Durham, NC, USA) and values were normalized to number of cells collected.
Western blots
Total protein extract was prepared as described previously (McLaughlin et al. 2001). Equal protein concentrations were separated using Criterion Tris-HCl gels (Bio-Rad, Hercules, CA, USA). Proteins were then transferred to polyvinylidene difluoride membranes (Amersham Biosciences, Piscataway, NJ, USA) and blocked for 1 h at room temperature with Odyssey blocking buffer (LiCor, Lincoln, NE, USA). Membranes were then incubated overnight in primary antibodies diluted in blocking solution. Membranes were washed in phosphate-buffered saline containing 0.1% Tween-20 and then incubated in Alexa 680-conjugated fluorescent secondary antibodies (Molecular Probes) for 1 h at room temperature. Protein bands were visualized using an Odyssey infrared imaging system and software (version 1.2, LiCor Biosciences).
Neuronal membrane preparation
Total cell membranes were isolated by the method of Nagamatsu et al. (1992) with slight modifications. Briefly, cells were homogenized in 1 mL of homogenization buffer (10 mM Tris-HCl, pH 7.4, 1 mM EDTA, 200 mM sucrose, 1 mM phenylmethylsulfonyl fluoride). The nuclei and cell debris were removed from the homogenate by centrifugation at 900 g for 10 min at 4°C. The resulting supernatant was centrifuged at 110 000 g for 75 min at 4°C (SW40 rotor, Beckman ultracentrifuge). The supernatant (cytosolic fraction) was stored at −70°C, whereas the membrane pellet was solubilized in buffer (10 mM Tris-HCl, pH 7.4, 1 mM EDTA, 0.5% Triton X-100, 1 mM phenylmethylsulfonyl fluoride) for a minimum of 1 h at 4°C and then stored at −70°C.
Results
Cyclopentenone isoprostanes are formed in the brain and are elevated following oxidative injury As cyclopentenone IsoPs exert neurotoxic effects, it is imperative to determine if they are formed in the brain. The formation of D2- and E2-IsoPs, the direct precursors of cyclopentenone IsoPs, are significantly increased in brain tissue following oxidant damage (Reich et al. 2001; Montine et al. 2003), suggesting that cyclopentenone IsoPs should be present. Thus, we analyzed cyclopentenone IsoP levels in rat brain tissue using a stable isotope dilution liquid chromatography tandem mass spectrometric method (LC/MS/MS) employing [2H4]PGA2 as an internal standard. Basal levels of cyclopentenone IsoPs in adult rat brain using this assay were 14.7 + 8.6 ng/g (n = 3), and were elevated to 179.0 + 84.2 ng/g (n = 3, p < 0.05) following 12-h exposure to the oxidant AAPH (1 mM), a 12-fold increase. In contrast, F2-IsoP levels in rat brain were 1.6 + 0.3 ng/g, nine-fold lower, and increased only two-fold following oxidation. These findings demonstrate that in brain tissue, cyclopentenone IsoPs may be the favored product of the IsoP pathway. We next examined the cyclopentenone IsoP levels in postmortem samples of frontal cortex tissue of neurologically normal human subjects. Cyclopentenone IsoPs were abundant in human brain tissue, present at levels of 33.5 + 3.5 ng/g wet brain tissue (n = 5), 6.8-fold higher than levels of F2-IsoPs in normal human cortex (4.9 + 0.3 ng/g) previously reported from our laboratory (Reich et al. 2001). Thus, cyclopentenone IsoPs are formed in abundance in the CNS and are increased by oxidant injury in parallel with F2-IsoPs.
The endogenous cyclopentenone isoprostane 15-A2t-isoprostane is neurotoxic to primary cortical neurons in culture
In order to evaluate the potential role of cyclopentenone IsoPs in neurodegeneration, we first examined the effects of 15-A2t-IsoP, one of the major cyclopentenone IsoPs formed in vivo (Chen et al. 1999b), on neuronal viability in primary neuronal cultures. 15-A2t-IsoP caused dose-dependent neuronal death in mature cortical cultures (LD50 = 950 nM, Fig. 1b), as determined by LDH release. Toxicity was seen at concentrations as low as 300 nM after 48-h exposure, though higher concentrations of 15-A2t-IsoP (30 μM) were required to induce neurotoxicity at 24 h (Fig. 2e). 15-A2t-IsoP was also highly toxic to the oxidative stress-sensitive HT22 hippocampal cell line, with an observed LD50 of 4.8 μM following 24 h exposure (Fig. 1c). Notably, 15-F2t-IsoP (also known as 8-iso-PGF2α), a non-reactive F2-IsoP which is structurally identical to 15-A2t-IsoP but lacks the cyclopentenone moiety, did not induce cell death under similar conditions (data not shown).
Fig. 2.
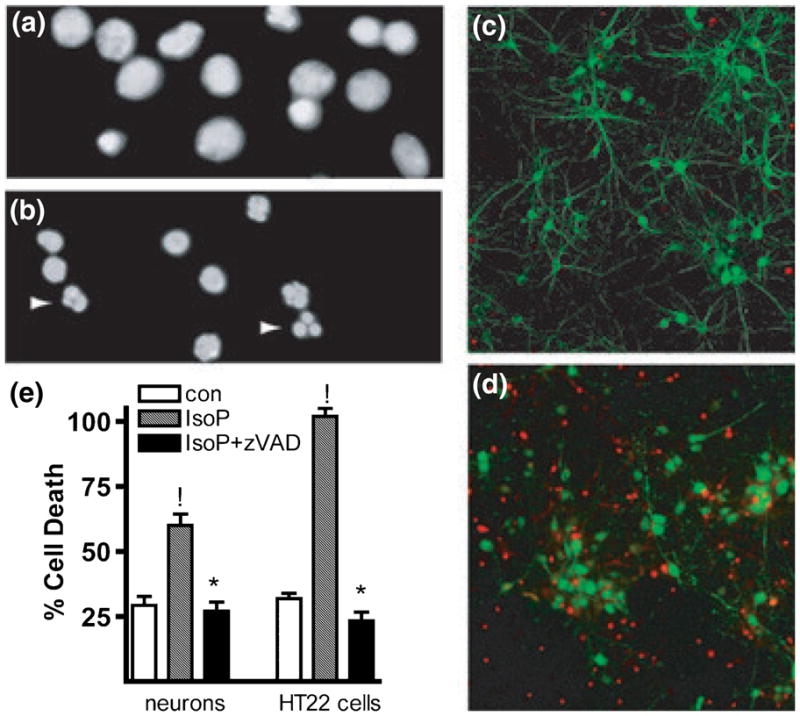
15-A2t-Isoprostane (15-A2t-IsoP) induces apoptotic neuronal death. (a, b) 4′-6-Diamidino-2-phenylindole (DAPI) staining of neurons treated with vehicle (a) or 30 μM 15-A2t-IsoP for 24 h (b) demonstrates an increase in asymmetric chromatin formations (arrows), an indication of apoptosis. (c, d) Neuronal culture treated for 48 h with vehicle (c) vs. 30 μM 15-A2t-IsoP (d). IsoP-treated cells (d) show a loss of microtubule-associated protein 2 (MAP-2) staining (green) and increased activated caspase-3 expression (red). Images in (a)–(d) are representative of three separate experiments. (e) Co-incubation of cells [primary cortical neurons (left) or HT22 cells (right)] with 20 μM zVAD-FMK, a broad-spectrum caspase inhibitor, caused a significant reduction in cell death following 24 h treatment with 15-A2t-IsoP as assessed by lactate dehydrogenase (LDH) release (neurons) or MTT assay (HT22s). Data represent the mean + SEM of four independent experiments, each performed in triplicate and analyzed by two-tailed paired t-test. !p < 0.05 vs. control, *p < 0.05 vs. IsoP + zVAD.
Neuronal death induced by 15-A2t-isoprostane is apoptotic
In order to more fully characterize 15-A2t-IsoP-induced cell death, nuclear morphology was assessed by DAPI nuclear staining in primary cultures treated with 30 μM 15-A2t-IsoP for 24 h. An abundance of asymmetric chromatin formations was observed in 15-A2t-IsoP-treated cells, consistent with apoptotic cell death (Figs 2a and b). Furthermore, analysis of activated caspase-3 staining in these cultures demonstrated greatly increased apoptotic protease immunoreactivity in 15-A2t-IsoP-treated neurons, as well as a reduction in the neuron specific marker microtubule-associated protein MAP2 staining (Figs 2c and d). These findings are consistent with the neurite retraction and beading that typify apoptotic cell death. Moreover, the increased caspase-3 cleavage in 15-A2t-IsoP-treated neurons was also confirmed after 24 h of treatment by western blot analysis (data not shown). In order to confirm that caspase activation was essential to the pathological cascade, neurons were co-incubated with 15-A2t-IsoPs in the presence of zVAD-FMK (20 μM). This broad-spectrum caspase inhibitor provided complete protection from 15-A2t-IsoP-induced cell death in both primary neuronal cultures (as assessed by LDH release) and HT22 cells (as assessed by MTT assay, Fig. 2e).
15-A2t-Isoprostane depletes cellular glutathione and induces oxidative stress
We have previously observed that 15-A2-IsoPs rapidly adduct glutathione (GSH) in non-neuronal cells (Milne et al. 2004). Because GSH depletion is fatal to neurons (Murphy et al. 1989), we next measured the GSH levels in immature neurons and HT22 cells exposed to 15-A2t-IsoP. Three-hour exposure to 15-A2t-IsoP resulted in a dose-dependent depletion of GSH in both neurons and HT22 cells (Fig. 3a). 15-A2t-IsoP depleted GSH levels as effectively as 5 mM glutamate, a well-characterized oxidative injury (Tan et al. 1998). Furthermore, we found that depletion of cellular GSH via pre-treatment with the compound buthionine sulfoximine (BSO) enhanced IsoP-induced HT22 cell death and that cell death could be blocked by pre-incubation with 100 μM N-acetyl cysteine (data not shown). Both of these observations are in keeping with those observed following glutamate exposure to HT22 cells (Ratan et al. 1994b). Thus, the rapid depletion of GSH induced by 15-A2t-IsoP likely contributes to the potent toxicity of this compound in both primary neuronal cultures and HT22 cells.
Fig. 3.
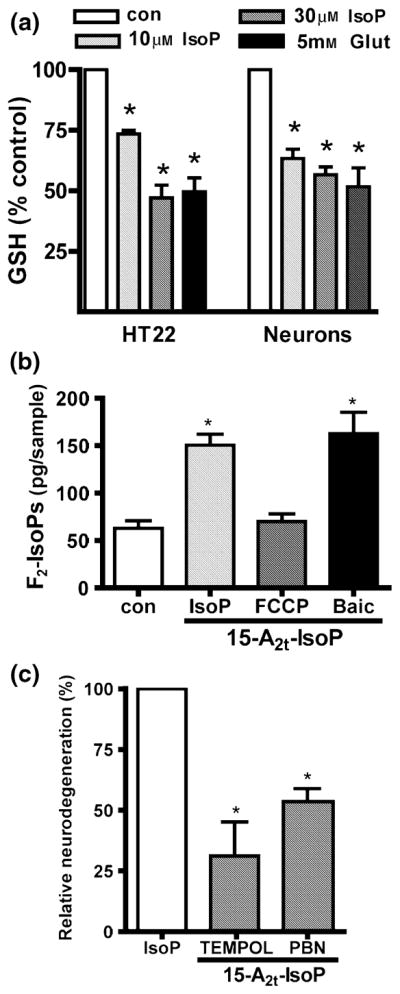
Glutathione depletion and oxidative stress contribute to 15-A2t-isoprostane (15-A2t-IsoP) toxicity. (a) Glutathione (GSH) levels were measured spectrophotometrically after immature neurons or HT22 cells were exposed for 3 h to vehicle (0.1% dimethylsulfoxide), 10 or 30 μM 15-A2t-IsoP, or 5 mM glutamate, which was used as a positive control. Data represent the mean + SEM of three separate experiments and was analyzed by two-tailed paired t-test. *p < 0.05 vs. control. (b) Neuronal cultures were exposed to vehicle or 15-A2t-IsoP (30 μM) + the mitochondrial uncoupling agent carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP: 10 μM) or baicalein (2 μM) for 3 h, then harvested and assayed for total F2-isoprostane content (F2-IsoPs) by gas chromatography/mass spectrometry (GC/MS). FCCP and baicalein were present 30 min before and during 15-A2t-IsoP exposure. Data represent the mean + SEM for three independent experiments, and were analyzed one-way ANOVA. *p < 0.05 vs. control. (c) Neurons were exposed to 15-A2t-IsoP (30 μM) + the antioxidants TEMPOL (1 mM) or N-tert-butyl-α-phenylnitrone (PBN, 0.5 mM). After 20–24 h, cell death was assessed by lactate dehydrogenase assay. Antioxidants were present for 30 min before and during IsoP exposure, and significantly decreased 15-A2t-IsoP-induced cell death. Data were normalized to the amount of cell death caused by 15-A2t-IsoP alone minus that caused by vehicle (100% relative neurodegeneration) in this and subsequent figures. Data represent the mean + SEM for three separate experiments, each performed in quadruplicate, and were analyzed by one-way ANOVA.*p < 0.05 vs. control (a, b) or IsoP alone (c).
As GSH depletion is associated with oxidant stress, we next examined oxidative damage in neurons following treatment with 15-A2t-IsoP. 15-A2t-IsoP exposure for 3 h induced a nearly three-fold increase in neuronal levels of F2-isoprostanes (F2-IsoPs), stable lipid peroxidation products measured as an index of oxidative stress (Fig. 3b) (Morrow and Roberts 1999). We found that 15-A2t-IsoP-induced lipid peroxidation production could be completely blocked by co-application of the mitochondrial uncoupling agent FCCP (10 μM), but not by the 12-lipoxygenase inhibitor baicalein (2 μM), suggesting that ROS production is dependent on functional mitochondria. We also observed progressive up-regulation of Ku70, a DNA repair protein involved in cell death, over the course of a 24 h exposure to 15-A2t-IsoP, indicative of oxidative DNA damage.
Our observation that 15-A2t-IsoP increases lipid peroxidation suggests that ROS contribute to the apoptotic process induced by this agent. Accordingly, the free radical spin traps 4-hydroxy-TEMPO (TEMPOL, 1 mM) and PBN (0.5 mM) provided significant protection from 15-A2t-IsoP (Fig. 3c, 68% and 54% protection, respectively). These observations suggest that 15-A2t-IsoP-induced ROS production could potentiate oxidative injury via a feed-forward mechanism.
12-Lipoxygenase activation is required for 15-A2t-Isoprostane-induced neurodegeneration
12-Lipoxygenase (12-LOX) oxidizes arachidonic acid to 12-hydroperoxides and plays a key role in neurotoxicity associated with a number of oxidative stress-inducing insults, including glutamate (Li et al. 1997; Khanna et al. 2003), thiol oxidants (Du et al. 2002), nitric oxide (Canals et al. 2003), β-amyloid (Lebeau et al. 2004), and peroxynitrite (Zhang et al. 2004). Furthermore, 12-LOX activation occurs secondary to GSH depletion in several of these models (Li et al. 1997; Canals et al. 2003).
Given that 15-A2t-IsoP both depletes GSH and induces oxidative stress, we examined the potential role of 12-LOX in 15-A2t-IsoP toxicity. Immunofluorescent analysis of 15-A2t-IsoP-treated neurons revealed striking translocation of 12-LOX from a nuclear/cytoplasmic localization toward the edges of the cell (Fig. 4a). As 12-LOX activation is dependent on its translocation to the membrane (Hagmann et al. 1993; Li et al. 1997; Du et al. 2002), we examined 12-LOX localization in subcellular fractions from immature neurons exposed to 15-A2t-IsoP for 3 h. As shown in Fig. 4b, nearly all cellular 12-LOX was present in the cytoplasm in control (vehicle-treated) cells. As a positive control, we used neurons exposed to 5 mM glutamate for 8 h, which has been previously shown to induce 12-LOX translocation and activation in these cells (Li et al. 1997). Exposure to 15-A2t-IsoP for 3 h also caused marked 12-LOX membrane translocation, which was blocked by co-application of the specific 12-LOX inhibitor baicalein (20 μM), which is known to prevent 12-LOX membrane localization (Du et al. 2002). Finally, baicalein at two concentrations (20 and 2 μM) also provided ~80% protection from 15-A2t-IsoP-induced neuronal death (Fig. 4d), demonstrating that 12-LOX activation is required for 15-A2t-IsoP-induced neuronal death.
Fig. 4.
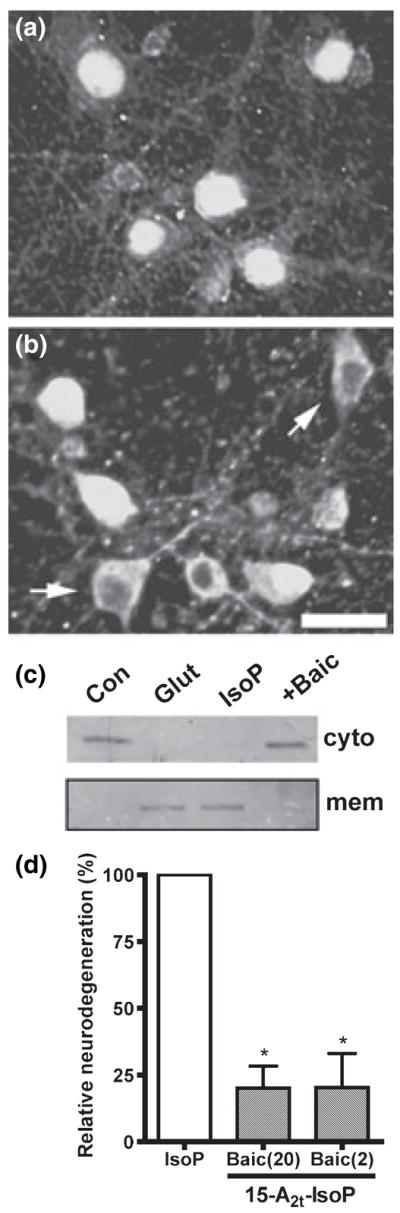
12-Lipoxygenase (12-LOX) activation in neurons following 15-A2t-isoprostane (15-A2t-IsoP) exposure causes cell death. Neurons were exposed to (a) vehicle or (b) 30 μM 15-A2t-IsoP for 3 h then fixed, stained with an anti-12-LOX antibody, and subjected to fluorescent microscopy. Translocation of 12-LOX to the membrane is associated with its activation (arrowheads). Photomicrographs are representative of results from three independent experiments. (c) Immature neurons were exposed to vehicle (Con), 30 μM 15-A2t-IsoP + baicalein (Baic: 20 μM) or 5 mM glutamate (Glut, used as a positive control) for 3 h, then membrane and cytoplasmic fractions were prepared and analyzed for 12-LOX by western blot. Blots are representative of two independent experiments. (d) Neurons were exposed to 15-A2t-IsoP (30 μM) + the specific 12-LOX inhibitor baicalein (Baic: 20 or 2 μM). After 20–24 h, cell death was assessed by lactate dehydrogenase (LDH) assay. Baicalein was present for 30 min before and during IsoP exposure. Data represents the mean + SEM for at least three independent experiments, and was analyzed by one-way ANOVA. *p < 0.05 vs. IsoP alone.
Although baicalein has been reported to possess antioxid-ant properties, the 2 μM concentration of this inhibitor is well below its antioxidant EC50 (Gao et al. 1999). Furthermore, this low concentration of baicalein failed to inhibit 15-A2t- IsoP-induced lipid peroxidation (Fig. 3b), but provided significantly better protection than a 500-fold greater concentration of the potent antioxidant TEMPOL (Fig. 3c). Thus, the early activation of 12-LOX is a critical component of 15-A2t-IsoP-induced cell death.
Extracellular signal-regulated kinase and p66shc phosphorylation participate in 15-A2t-Isoprostane neurotoxicity
We next sought to determine if known redox-sensitive kinases are involved in 15-A2t-IsoP-induced cell death. Phosphorylation of the stress-responsive kinases ERK1/2 plays a role in several neurodegenerative diseases (Alessandrini et al. 1999; Perry et al. 1999; Zhu et al. 2002). Moreover, blockade of ERK attenuates neuronal death in response to oxidative stimuli. (Stanciu et al. 2000; Du et al. 2002; Lee et al. 2003). We examined ERK1/2 phosphorylation in 15-A2t-IsoP-treated neurons and observed robust ERK1/2 phosphorylation within 30 min of 15-A2t-IsoP exposure. ERK phosphorylation returned to baseline within 60 min post-treatment, and then showed a more subtle and sustained increase at 8 and 24 h (Fig. 5a). Consistent with a role for ERK1/2 phosphorylation in 15-A2t-IsoP neurotoxicity, application of the MEK inhibitor PD98059 (20 μM) completely inhibited ERK phosphorylation, and provided 55% neuroprotection from 15-A2t-IsoP exposure (Figs 5b and d). Co-application of either FCCP (1 μM) or TEMPOL (1 mM) blocked early ERK activation (Fig. 5b), suggesting that 15-A2t-IsoP-induced mitochondrial ROS production is upstream of ERK phosphorylation. Although ERK stimulation is capable of inducing a host of transcriptional responses, we found that the protein synthesis inhibitor cyclohexamide (CHX, 3.5 μM) did not protect neurons from 15-A2t-IsoP (Fig. 5d).
Fig. 5.
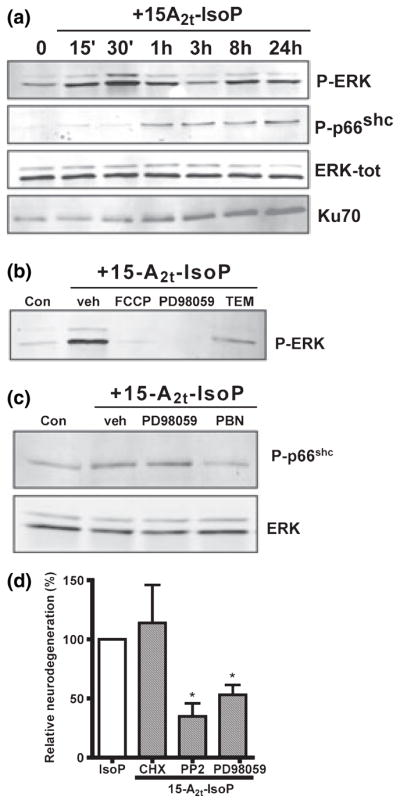
15-A2t-Isoprostane (15-A2t-IsoP) neurotoxicity involves phosphorylation of extracellular signal-regulated kinase (ERK) and p66shc and up-regulation of Ku70. (a) Neuronal cultures were exposed to 30 μM 15-A2t-IsoP or vehicle for various amounts of time, then whole- cell extracts were harvested and subjected to western blot analysis. ERK is displayed as a loading control. (b) Neurons were exposed to 30 μM 15-A2t-IsoP + the MEK inhibitor PD98059 (20 μM), TEMPOL (1 mM), or carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP: 1 μM), then harvested at 20 h and subjected to western blot using a phospho-ERK specific antibody. (c) Neurons were exposed to 30 μM 15-A2t-IsoP + PD98059 (20 μM), N-tert-butyl-α-phenylnitrone (PBN: 0.5 mM), or baicalein (20 μM), then harvested at 1 h and subjected to western blot analysis. (d) Neurons were exposed to 15-A2t-IsoP (30 μM) + cyclohexamide (3.5 μM), the Src kinase inhibitor PP2 (50 nM), or the MEK inhibitor PD98059 (20 μM). Cell death was assessed after 20–24 h by lactate dehydrogenase (LDH) assay. Data represent the mean ± SEM of three independent experiments; each performed in triplicate, and were analyzed by one-way ANOVA. Blots shown in (a), (b) and (c) are representative of at least three independent experiments. Inhibitors in (b), (c) and (d) were present for 30 min before and during IsoP exposure. PD98059, 2′-amino-3′-methoxyflavone; PP2, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo-[3,4-d]pyrimidine.
We next sought to evaluate the activation of the adaptor protein p66shc, which has been shown to be serine phosphorylated in response to oxidative stress and may contribute to oxidant and β-amyloid induced cell death (Migliaccio et al. 1999; Trinei et al. 2002; Smith et al. 2005). Western blot analysis demonstrated that the redox sensitive serine 36 residue of p66shc is strongly and transiently phosphorylated 1 h following 15-A2t-IsoP treatment, and returns to base-line within 3 h (Fig. 5a), though more persistent phosphorylation was seen in some experiments (data not shown). Co-application of the antioxidant PBN (0.5 mM) prevented p66shc phosphorylation, suggesting that p66shc activation is downstream of ROS generation (Fig. 5c). However, co-application with PD98059 did not prevent IsoP-induced p66shc phosphorylation, demonstrating that ERK signals independently of p66shc (Fig. 5c). These results illustrate independent roles for ERK and p66shc phosphorylation in the 15-A2t-IsoP neuronal death pathway, and suggest that ROS production precedes both events.
p66shc belongs to a family of Src kinases which are redox-responsive and have been implicated in ROS-induced cell death (Devary et al. 1992; Balamurugan et al. 2002). The Src kinase inhibitor PP2 (50 nM), which also blocks p66shc activity, provided significant protection from 15-A2t-IsoP toxicity (65% neuroprotection, Fig. 5d), suggesting a contributory role for p66shc or other Src kinases in the 15-A2t-IsoP toxicity pathway.
15-A2t-Isoprostane potentiates oxidative glutamate toxicity in immature neuronal cultures and HT22 cells
Given that 15-A2-IsoPs are formed under conditions of oxidative stress, we next examined the effects of 15-A2t-IsoP on cell death in an in vitro model of oxidative neurodegeneration. Exposure of immature primary neurons or HT22 cells, neither of which express functional NMDA receptors, to millimolar concentrations of extracellular glutamate causes inhibition of cellular cystine import and subsequent depletion of GSH and oxidative stress-dependent cell death (Murphy et al. 1989; Ratan et al. 1994b; Li et al. 1997). Neither 1.5 mM glutamate nor 5 μM 15-A2t-IsoP alone were toxic to immature neurons following 18 h of continuous exposure (Fig. 6a). However, application of both agents together greatly potentiated cell death (Fig. 6a). Similarly, exposure of HT22 cells to 3 mM glutamate for 18 h led to ~50% cell death (Fig. 6b), whereas exposure to 1 μM 15-A2t-IsoP alone did not effect cell viability (Fig. 1c). However, application of these concentrations of glutamate and 15-A2t-IsoP together resulted in 95% cell death (Fig. 6b). Indeed, 15-A2t-IsoP concentrations as low as 100 nM significantly potentiated glutamate (3 mM) toxicity in this model, demonstrating that low concentrations of these oxidation products can synergize with other oxidative insults to augment cell death.
Fig. 6.
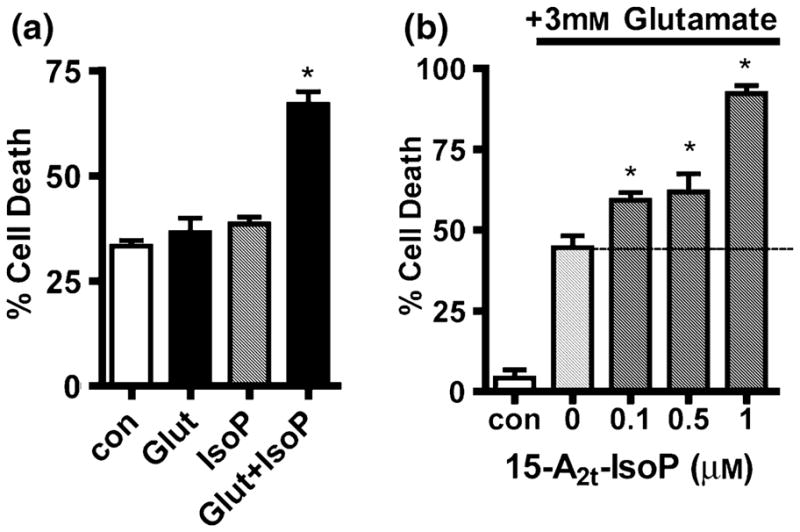
15-A2t-Isoprostane (15-A2t-IsoP) potentiates oxidative glutamate toxicity in neurons and HT22 cells. (a) Immature (1–2 DIV) primary cortical neurons were exposed to 1.5 mM glutamate or 5 μM 15-A2t-IsoP alone or in combination. Cell death was assessed after 18 h by lactate dehydrogenase (LDH) assay. (b) HT22 cells were exposed to glutamate plus increasing concentrations of 15-A2t-IsoP. Cell viability was assessed by MTT assay after 20–24 h. Note that in both (a) and (b), 15-A2t-IsoP and glutamate in combination resulted in significantly increased cell death. Data in (a) and (b) represent the mean ± SEM of three independent experiments, each performed in triplicate, and were analyzed by one-way ANOVA. *p < 0.05 vs. control.
Discussion
In this work we report the novel observations that reactive cyclopentenone IsoPs are formed abundantly in human cerebral cortex, and that their levels are elevated following oxidation. Moreover, a major cyclopentenone IsoP isomer, 15-A2t-IsoP, is highly bioactive and is a potent neuronal apoptogen. The mechanism of 15-A2t-IsoP toxicity occurs via depletion of cellular GSH, induction of ROS production, and activation of redox-sensitive signaling molecules, including 12-LOX, p66shc, and ERK1/2, which culminate in caspase-3-dependent neuronal apoptosis (Fig. 7). Finally, we have demonstrated that 15-A2t-IsoP can potentiate neuronal death in response to oxidative glutamate toxicity at biologically relevant concentrations. Thus, we propose that cyclopentenone IsoPs represent novel bioactive compounds that contribute to oxidative injury-induced neurodegeneration.
Fig. 7.
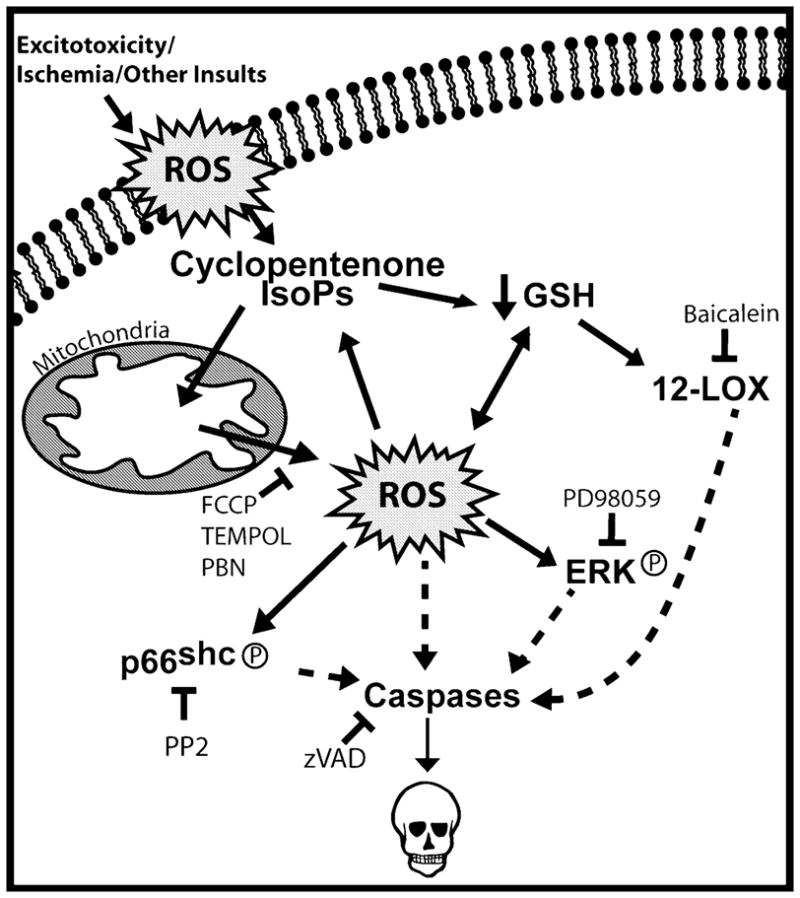
Model of signaling events in 15-A2t-isoprostane (15-A2t-IsoP)- induced neurodegeneration. Based on our findings in this work, we suggest that 15-A2t-IsoPs are formed in membranes (plasma membrane is depicted, but other membranes are included) following an initial oxidant injury. 15-A2t-IsoPs could then induce mitochondrial dysfunction and glutathione (GSH) depletion, causing marked increases in intracellular reactive oxygen species (ROS) production and further lipid peroxidation. This results in phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2) and serine 36 of p66shc, both of which contribute to cell death. The loss of GSH leads to the translocation and activation of 12-lipoxygenase (12-LOX), which contributes to cell death. As 15-A2t-IsoP is a product and inducer of oxidative stress, the production of 15-A2t-IsoPs in response to an initial oxidant injury sets in motion a feed-forward cycle that leads to a loss of intracellular redox homeostasis and cell death. Interruption of this cycle with baicalein, 4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo[3,4-d]pyrimidine (PP2), or antioxidants can prevent cell death. At the earliest stage, inhibition of mitochondrial function with carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) blocks 15-A2t-IsoP-induced oxidative injury and ERK phosphorylation.
IsoPs are formed when free or membrane-bound arachidonic acid is exposed to ROS (Morrow et al. 1992). Non-reactive F2-IsoPs have long been quantified as markers of lipid peroxidation (Musiek and Morrow 2005), and elevated cerebral F2-IsoPs levels have been detected in human Alzheimer’s and Huntington’s diseases (Montine et al. 1999; Reich et al. 2001), as well as in animal models of stroke, encephalitis, and seizure (Marin et al. 2000; Patel et al. 2001; Milatovic et al. 2002).
Our data demonstrate that cyclopentenone IsoPs are not only formed much more abundantly than F2-IsoPs in peroxided brain tissue, but are 6.8-fold more plentiful in human brain. Cumulative oxidative stress is higher in long lived species, such as humans, than it is in mice. Our data are not necessarily incongruent with this finding – they may represent two ends of the spectrum with mice that are 4 months old and humans that averaged 73 years old. Moreover, the brains of the mice in this work were flash frozen whereas those of the humans had an average of a 15 h postmortem delay.
Interestingly, we have detected esterified cyclopentenone IsoPs in oxidized rat brain tissue at levels of 179 ng/g, which roughly converts to ~550 nM. This concentration is within the biologically relevant range, as we noted increased cell death and exacerbation of glutamate toxicity at concentrations as low as 100 nM. It is difficult to directly compare the concentrations of IsoPs measured in vivo with those required to exert toxicity in our cell culture system for several reasons. First, we are currently only able to measure the esterified pool of cyclopentenone IsoPs, which fails to account for the free acid pool of IsoPs, and does not supply information about the rate of formation, adduction, and metabolism of these compounds. However, recent studies have identified the major urinary metabolite of 15-A2t-IsoP in rats exposed to oxidant injury, demonstrating that cyclopentenone IsoPs, which are largely formed esterified to membrane phospholipids, are then hydrolyzed to free compounds, adducted to proteins and GSH, and excreted in vivo (Milne et al. 2005). Further, the exogenous application of reactive IsoPs to cells in culture certainly requires much higher concentrations to exert effects than would the endogenous, intracellular generation of these compounds, as exogenous IsoPs readily react with albumin and other proteins in the cell media and may not cross the cell membrane with perfect efficiency. Taking these factors into account, we would predict from our current data that cyclopentenone IsoPs are also formed in neurological diseases in which F2-IsoPs have been detected, and could reach biologically relevant concentrations under pathophysiological conditions. Further studies are required to more thoroughly examine the formation of cyclopentenone IsoPs in specific neurodegenerative diseases.
15-A2t-IsoPs are metabolized via adduction to GSH (Milne et al. 2004), and rapidly deplete cellular GSH levels in neurons. 15-A2t-IsoP also promotes oxidative stress, as evidenced by an increase in membrane-bound F2-IsoPs following exposure. 15-A2t-IsoP-induced lipid peroxidation can be abrogated by uncoupling mitochondrial respiration with FCCP, which inhibits mitochondrial ROS production in neurons (Tan et al. 1998; Liu et al. 2002) and is in keeping with previous studies of cyclopentenone PGs, which are capable of inducing oxidant stress (Kondo et al. 2001; Li et al. 2001). This observation suggests that an initial oxidative insult can set in motion a feed-forward cycle of increasing oxidative stress in cells, as cyclopentenone IsoPs are formed as products of oxidative stress, then induce further GSH depletion, ROS production, and oxidant injury (Fig. 7).
Data from several groups now suggest that overexpression of GST A4-4, the specific GST isoform which metabolizes several electrophilic lipids including cyclopentenone IsoPs, protects non-neuronal cells from oxidative insults (Zimniak et al. 1997; Cheng et al. 1999; Hubatsch et al. 2002). There is mounting evidence that GSH depletion in neurons is intimately linked to 12-LOX activation (Li et al. 1997; Arai et al. 2001; Du et al. 2002; Canals et al. 2003; Khanna et al. 2003; Pratico et al. 2004). 12-LOX is the primary lipoxygenase expressed in neurons and oxidizes arachidonic acid at the 12 position to hydroperoxides (12-HPETEs) and alcohols (12-HETEs), both of which have been implicated in neuronal signaling (Nishiyama et al. 1993; Watanabe et al. 1993). Inhibition of 12-LOX activity protects neurons from numerous oxidative stress-associated insults, including glutamate, thiol oxidants, peroxynitrite, and β-amyloid (Li et al. 1997; Du et al. 2002; Lebeau et al. 2004; Zhang et al. 2004). We have shown that 12-LOX activation occurs in response to 15-A2t-IsoP exposure and is toxic to neurons. Although 12-LOX has been implicated as a source of ROS following GSH depletion (Li et al. 1997; Tan et al. 1998), we found that early 15-A2t-IsoP-induced ROS production cannot be blocked with the 12-LOX inhibitor baicalein, suggesting that 12-LOX activation is downstream of 15-A2t-IsoP-induced GSH depletion.
Once thought to be random destructive agents, it has become increasingly clear that ROS production is carefully regulated and that ROS are capable of activating discrete signaling pathways (Martindale and Holbrook 2002; Finkel 2003). The contribution of the bioactivity of 15-A2t-IsoP to enhancing ROS production has numerous downstream signaling consequences, including the activation of stress-responsive kinases. Indeed, the Shc adaptor protein isoform p66shc is phosphorylated at serine 36 in response to ROS-inducing stimuli, and cells and animals lacking p66shc are highly resistant to oxidative injury (Migliaccio et al. 1999). p66shc is also phosphorylated in skeletal muscle following ischemia/reperfusion injury and contributes to cell death in this setting (Zaccagnini et al. 2004), and is activated in neurons in response to OGD (R. Breeding, E. Musiek and B.A. McLaughlin, unpublished data). p66shc translocation to the mitochondria has recently been shown to induce opening of the permeability transition pore (Orsini et al. 2004) and to enhance cellular ROS production (Trinei et al. 2002). Our data demonstrate that p66shc is ser36 phosphorylated following 15-A2t-IsoP treatment and ensuing ROS production. 15-A2t-IsoP is the first lipid peroxidation product shown to activate this important cell death pathway and provides an important molecular link in neurons between ROS stress and p66shc activation.
15-A2t-IsoP-induced ROS production also induces a biphasic phosphorylation of ERK1/2. Considerable evidence now suggests that ERK can mediate cell death in response to oxidative injury (Colucci-D’Amato et al. 2003; Lee et al. 2003; Chu et al. 2004). Intriguingly, the subcellular localization of ERK may dictate its effects, with extended nuclear or mitochondrial retention of phosphorylated ERK leading to cell death (Stanciu and DeFranco 2002; Chu et al. 2004). Indeed, inhibition of ERK1/2 phosphorylation provides partial protection from 15-A2t-IsoP toxicity and supports an emerging role of ERK activation in cell death mediated by oxidative stress.
To further understand the actions of cyclopentenone IsoPs in neurodegeneration, we sought to examine a potential role for these molecules in the neuronal death caused by oxidative insults. Ratan et al. (1996) have developed methods to mimic pathophysiological oxidative injury induced by glutamate exposure. We have used two of these model systems: immature primary cortical cultures as well as HT22 cells (which lack functional NMDA receptors) that have been exposed to millimolar concentrations of glutamate resulting in inhibition of the glutamate-cystine antiporter and rapid GSH depletion followed by oxidative stress-dependent cell death (Murphy et al. 1989). Like 15-A2t-IsoP-induced cell death, oxidative glutamate toxicity proceeds via a pathway requiring mitochondrial ROS production, 12-LOX activation, and ERK phosphorylation (Li et al. 1997; Tan et al. 1998; Stanciu et al. 2000). Our observations that cyclopen- tenone IsoP levels increase eight-fold during glutamate treatment and that concentrations of 15-A2t-IsoPs as low as 100 nM can markedly potentiate oxidative glutamate toxicity demonstrate the potent toxicity of these molecules under pathophysiological conditions. Furthermore, GSH depletion would be expected to impair the detoxification of ROS and cyclopentenone isoprostanes (Milne et al. 2004), exacerbating the effects of these molecules. These findings are relevant to acute neurological insults such as stroke, in which GSH depletion is an essential feature (Schubert and Piasecki 2001; Lerouet et al. 2002), as well as Parkinson’s disease, where the depletion of GSH in the substantia nigra is an early hallmark of the disease (Perry and Yong 1986).
In conclusion, the cyclopentenone IsoPs are formed in neural tissue during oxidative stress, and application of these compounds recapitulates many of the pathogenic signaling events observed during neurodegeneration caused by oxidative insults. Given the abundance of cyclopentenone IsoPs formed following oxidative stress, as well as the potent ability of these compounds to induce neuronal apoptosis and exacerbate oxidant injury, cyclopentenone IsoPs may represent important mediators of oxidative neuropathology. As such, reactive cyclopentenone IsoPs and the pathogenic kinase pathways activated by these molecules merit further evaluation as targets for neuroprotective therapies.
Acknowledgments
The authors would like to thank Dr Gregg Stanwood for microscopy assistance, Ms. Kylie Beck for her outstanding graphic artwork, Alessio Porta for work in synthesizing 15-A2t-IsoP, Samantha Jefferies for technical assistance, and Dr Rashed Nagra for providing tissue samples from the Human Brain and Spinal Fluid Resource Center, VA West Los Angeles Healthcare Center, which is sponsored by the NINDS/NIMH, National MS Society, and Department of Veterans Affairs. This work was supported in part by NICHD Grant P30HD15052, NINDS grant NS050396, and by NIH grants GM15431 and DK48831. Support for ESM was provided by a grant from the PhRMA foundation.
Abbreviations used
- AAPH, 2
2′-azobis(2-amidinopropane) hydrochloride
- BSO
buthionine sulfoximine
- DAPI
4′-6-diamidino-2-phenylindole
- ERK1/2
extracellular signal-regulated kinase 1/2
- FCCP
carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- GC
gas chromatography
- GSH
glutathione
- GST
glutathione transferase
- IsoP
isoprostane
- LDH
lactate dehydrogenase
- LC
liquid chromatography
- 12-LOX
12-lipoxygenase
- MCB
monochlorobimane
- MS
mass spectrometry
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PBN
N-tert-butyl-α-phenylnitrone
- PG
prostaglandin
- ROS
reactive oxygen species
References
- Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci USA. 1999;96:12 866–12 869. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai K, Nishiyama N, Matsuki N, Ikegaya Y. Neuro-protective effects of lipoxygenase inhibitors against ischemic injury in rat hippocampal slice cultures. Brain Res. 2001;904:167–172. doi: 10.1016/s0006-8993(01)02491-x. [DOI] [PubMed] [Google Scholar]
- Balamurugan K, Rajaram R, Ramasami T, Narayanan S. Chromium (III) – induced apoptosis of lymphocytes: death decision by ROS and Src-family tyrosine kinases. Free Radic Biol Med. 2002;33:1622–1640. doi: 10.1016/s0891-5849(02)01115-2. [DOI] [PubMed] [Google Scholar]
- Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- Bruckner SR, Perry G, Estus S. 4-hydroxynonenal contributes to NGF withdrawal-induced neuronal apoptosis. J Neurochem. 2003;85:999–1005. doi: 10.1046/j.1471-4159.2003.01749.x. [DOI] [PubMed] [Google Scholar]
- Canals S, Casarejos MJ, de Bernardo S, Rodriguez-Martin E, Mena MA. Nitric oxide triggers the toxicity due to glutathione depletion in midbrain cultures through 12-lipoxygenase. J Biol Chem. 2003;278:21 542–21 549. doi: 10.1074/jbc.M213174200. [DOI] [PubMed] [Google Scholar]
- Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- Chen Y, Morrow JD, Roberts LJ., 2nd Formation of reactive cyclopentenone compounds in vivo as products of the isoprostane pathway. J Biol Chem. 1999a;274:10863–10868. doi: 10.1074/jbc.274.16.10863. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zackert WE, Roberts LJ, 2nd, Morrow JD. Evidence for the formation of a novel cyclopentenone isoprostane, 15-A2t-isoprostane (8-isoprostaglandin A2) in vivo. Biochim Biophys Acta. 1999b;1436:550–556. doi: 10.1016/s0005-2760(98)00168-4. [DOI] [PubMed] [Google Scholar]
- Cheng JZ, Singhal SS, Saini M, Singhal J, Piper JT, Van Kuijk FJ, Zimniak P, Awasthi YC, Awasthi S. Effects of mGST A4 transfection on 4-hydroxynonenal-mediated apoptosis and differentiation of K562 human erythroleukemia cells. Arch Biochem Biophys. 1999;372:29–36. doi: 10.1006/abbi.1999.1479. [DOI] [PubMed] [Google Scholar]
- Chu CT, Levinthal DJ, Kulich SM, Chalovich EM, DeFranco DB. Oxidative neuronal injury. The dark side of ERK1/2. Eur J Biochem. 2004;271:2060–2066. doi: 10.1111/j.1432-1033.2004.04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colucci-D’Amato L, Perrone-Capano C, di Porzio U. Chronic activation of ERK and neurodegenerative diseases. Bio-essays. 2003;25:1085–1095. doi: 10.1002/bies.10355. [DOI] [PubMed] [Google Scholar]
- Devary Y, Gottlieb RA, Smeal T, Karin M. The mammalian ultraviolet response is triggered by activation of Src tyrosine kinases. Cell. 1992;71:1081–1091. doi: 10.1016/s0092-8674(05)80058-3. [DOI] [PubMed] [Google Scholar]
- Du S, McLaughlin B, Pal S, Aizenman E. In vitro neurotoxicity of methylisothiazolinone, a commonly used industrial and household biocide, proceeds via a zinc and extracellular signal-regulated kinase mitogen-activated protein kinase-dependent pathway. J Neurosci. 2002;22:7408–7416. doi: 10.1523/JNEUROSCI.22-17-07408.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- Gao Z, Huang K, Yang X, Xu H. Free radical scavenging and antioxidant activities of flavonoids extracted from the radix of Scutellaria Georgi. Biochim Biophys Acta. 1999;1472:643–650. doi: 10.1016/s0304-4165(99)00152-x. [DOI] [PubMed] [Google Scholar]
- Hagmann W, Kagawa D, Renaud C, Honn KV. Activity and protein distribution of 12-lipoxygenase in HEL cells: induction of membrane association by phorbol ester TPA, modulation of activity by glutathione and 13-HPODE, and Ca(2+)-dependent translocation to membranes. Prostaglandins. 1993;46:471–477. doi: 10.1016/0090-6980(93)90066-g. [DOI] [PubMed] [Google Scholar]
- Hubatsch I, Mannervik B, Gao L, Roberts LJ, Chen Y, Morrow JD. The cyclopentenone product of lipid peroxidation, 15-A(2t)-isoprostane (8-isoprostaglandin A(2)), is efficiently conjugated with glutathione by human and rat glutathione transferase A4–4. Chem Res Toxicol. 2002;15:1114–1118. doi: 10.1021/tx020027r. [DOI] [PubMed] [Google Scholar]
- Jenner P. Oxidative stress in Parkinson’s disease. Ann Neurol. 2003;53(Suppl 3):S26–S36. doi: 10.1002/ana.10483. discussion S36–S38. [DOI] [PubMed] [Google Scholar]
- Khanna S, Roy S, Ryu H, Bahadduri P, Swaan PW, Ratan RR, Sen CK. Molecular basis of vitamin E action: tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J Biol Chem. 2003;278:43 508–43 515. doi: 10.1074/jbc.M307075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo M, Oya-Ito T, Kumagai T, Osawa T, Uchida K. Cyclopentenone prostaglandins as potential inducers of intracellular oxidative stress. J Biol Chem. 2001;276:12 076–12 083. doi: 10.1074/jbc.M009630200. [DOI] [PubMed] [Google Scholar]
- Kondo M, Shibata T, Kumagai T, Osawa T, Shibata N, Kobayashi M, Sasaki S, Iwata M, Noguchi N, Uchida K. 15-Deoxy-Delta(12,14)-prostaglandin J(2): the endogenous electrophile that induces neuronal apoptosis. Proc Natl Acad Sci. 2002;99:7367–7372. doi: 10.1073/pnas.112212599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci. 1997;17:5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeau A, Terro F, Rostene W, Pelaprat D. Blockade of 12-lipoxygenase expression protects cortical neurons from apoptosis induced by beta amyloid peptide. Cell Death Differ. 2004;11:875–884. doi: 10.1038/sj.cdd.4401395. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Cho HN, Soh JW, Jhon GJ, Cho CK, Chung HY, Bae S, Lee SJ, Lee YS. Oxidative stress-induced apoptosis is mediated by ERK1/2 phosphorylation. Exp Cell Res. 2003;291:251–266. doi: 10.1016/s0014-4827(03)00391-4. [DOI] [PubMed] [Google Scholar]
- Lerouet D, Beray-Berthat V, Palmier B, Plotkine M, Margaill I. Changes in oxidative stress, iNOS activity and neutrophil infiltration in severe transient focal cerebral ischemia in rats. Brain Res. 2002;958:166–175. doi: 10.1016/s0006-8993(02)03685-5. [DOI] [PubMed] [Google Scholar]
- Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, Zanoni G, Morrow JD, Darley-Usmar VM. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defenses in response to electrophilic lipid oxidation products. Biochem J. 2004;378:373–382. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- Li L, Tao J, Davaille J, Feral C, Mallat A, Rieusset J, Vidal H, Lotersztajn S. 15-deoxy-Delta 12,14-prostaglandin J2 induces apoptosis of human hepatic myofibroblasts. A pathway involving oxidative stress independently of peroxisomeproliferator- activated receptors. J Biol Chem. 2001;276:38 152–38 158. doi: 10.1074/jbc.M101980200. [DOI] [PubMed] [Google Scholar]
- Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- Marin JG, Cornet S, Spinnewyn B, Demerle-Pallardy C, Auguet M, Chabrier PE. BN 80933 inhibits F2-isoprostane elevation in focal cerebral ischaemia and hypoxic neuronal cultures. Neuroreport. 2000;11:1357–1360. doi: 10.1097/00001756-200004270-00041. [DOI] [PubMed] [Google Scholar]
- Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- McLaughlin BA, Nelson D, Silver IA, Erecinska M, Chesselet MF. Methylmalonate toxicity in primary neuronal cultures. Neuroscience. 1998;86:279–290. doi: 10.1016/s0306-4522(97)00594-0. [DOI] [PubMed] [Google Scholar]
- McLaughlin B, Pal S, Tran MP, Parsons AA, Barone FC, Erhardt JA, Aizenman E. p38 activation is required upstream of potassium current enhancement and caspase cleavage in thiol oxidant-induced neuronal apoptosis. J Neurosci. 2001;21:3303–3311. doi: 10.1523/JNEUROSCI.21-10-03303.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin B, Hartnett KA, Erhardt JA, Legos JJ, White RF, Barone FC, Aizenman E. Caspase 3 activation is essential for neuroprotection in preconditioning. Proc Natl Acad Sci. 2003;100:715–720. doi: 10.1073/pnas.0232966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- Milatovic D, Zhang Y, Olson SJ, Montine KS, Roberts LJ, 2nd, Morrow JD, Montine TJ, Dermody TS, Valyi-Nagy T. Herpes simplex virus type 1 encephalitis is associated with elevated levels of F2-isoprostanes and F4-neuroprostanes. J Neurovirol. 2002;8:295–305. doi: 10.1080/13550280290100743. [DOI] [PubMed] [Google Scholar]
- Milne GL, Zanoni G, Porta A, Sasi S, Vidari G, Musiek ES, Freeman ML, Morrow JD. The cyclopentenone product of lipid peroxidation, 15-A2t isoprostane, is efficiently metabolized by HepG2 cells via conjugation with glutathione. Chem Res Toxicol. 2004;17:17–25. doi: 10.1021/tx034213o. [DOI] [PubMed] [Google Scholar]
- Milne GL, Gao L, Zanoni G, Vidari G, Morrow JD. Identification of the major urinary metabolite of the highly reactive cyclopentenone isoprostane 15-a2t isoprostane in vivo. J Biol Chem. 2005;280:25 178–25 184. doi: 10.1074/jbc.M502891200. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Beal MF, Robertson D, Cudkowicz ME, Biaggioni I, O’Donnell H, Zackert WE, Roberts LJ, Morrow JD. Cerebrospinal fluid F2-isoprostanes are elevated in Huntington’s disease. Neurology. 1999;52:1104–1105. doi: 10.1212/wnl.52.5.1104. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Kaye JA, Montine KS, McFarland L, Morrow JD, Quinn JF. Cerebrospinal fluid abeta42, tau, and f2-isoprostane concentrations in patients with Alzheimer disease, other dementias, and in age-matched controls. Arch Pathol Lab Med. 2001;125:510–512. doi: 10.5858/2001-125-0510-CFATAF. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Neely MD, Quinn JF, Beal MF, Markesbery WR, Roberts LJ, Morrow JD. Lipid peroxidation in aging brain and Alzheimer’s disease. Free Radic Biol Med. 2002;33:620–626. doi: 10.1016/s0891-5849(02)00807-9. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Montine KS, Reich EE, Terry ES, Porter NA, Morrow JD. Antioxidants significantly affect the formation of different classes of isoprostanes and neuroprostanes in rat cerebral synaptosomes. Biochem Pharmacol. 2003;65:611–617. doi: 10.1016/s0006-2952(02)01607-6. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Roberts LJ., 2nd Mass spectrometric quantification of F2-isoprostanes in biological fluids and tissues as measure of oxidant stress. Meth Enzymol. 1999;300:3–12. doi: 10.1016/s0076-6879(99)00106-8. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ., 2nd A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci USA. 1990;87:9383–9387. doi: 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow JD, Awad JA, Boss HJ, Blair IA, Roberts LJ., 2nd Noncyclooxygenase- derived prostanoids (F2-isoprostanes) are formed in situ on phospholipids. Proc Natl Acad Sci USA. 1992;89:10 721–10 725. doi: 10.1073/pnas.89.22.10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- Musiek ES, Morrow JD. F2-Isoprostanes as Markers of Oxidant Stress: An Overview. In: Costa LG, Hodgson E, Lawrence D, Reed DJ, editors. Current Protocols in Toxicology. Suppl 24. John Wiley and Sons; Edison, NJ: 2005. pp. 17.15–17.16. [DOI] [PubMed] [Google Scholar]
- Nagamatsu S, Kornhauser JM, Burant CF, Seino S, Mayo KE, Bell GI. Glucose transporter expression in brain. cDNA sequence of mouse GLUT3, the brain facilitative glucose transporter isoform, and identification of sites of expression by in situ hybridization. J Biol Chem. 1992;267:467–472. [PubMed] [Google Scholar]
- Nishiyama M, Watanabe T, Ueda N, Tsukamoto H, Watanabe K. Arachidonate 12-lipoxygenase is localized in neurons, glial cells, and endothelial cells of the canine brain. J Histochem Cytochem. 1993;41:111–117. doi: 10.1177/41.1.8417106. [DOI] [PubMed] [Google Scholar]
- Orsini F, Migliaccio E, Moroni M, et al. The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J Biol Chem. 2004;279:25 689–25 695. doi: 10.1074/jbc.M401844200. [DOI] [PubMed] [Google Scholar]
- Ou JJ, Zhang Y, Montine TJ. In vivo assessment of lipid peroxidation products associated with age-related neurodegenerative diseases. Exp Neurol. 2002;175:363–369. doi: 10.1006/exnr.2002.7923. [DOI] [PubMed] [Google Scholar]
- Patel M, Liang LP, Roberts LJ., 2nd Enhanced hippocampal F2- isoprostane formation following kainate-induced seizures. J Neurochem. 2001;79:1065–1069. doi: 10.1046/j.1471-4159.2001.00659.x. [DOI] [PubMed] [Google Scholar]
- Perry TL, Yong VW. Idiopathic Parkinson’s disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci Lett. 1986;67:269–274. doi: 10.1016/0304-3940(86)90320-4. [DOI] [PubMed] [Google Scholar]
- Perry G, Roder H, Nunomura A, et al. Activation of neuronal extracellular receptor kinase (ERK) in Alzheimer disease links oxidative stress to abnormal phosphorylation. Neuroreport. 1999;10:2411–2415. doi: 10.1097/00001756-199908020-00035. [DOI] [PubMed] [Google Scholar]
- Pratico D, Zhukareva V, Yao Y, Uryu K, Funk CD, Lawson JA, Trojanowski JQ, Lee VM. 12/15-lipoxygenase is increased in Alzheimer’s disease: possible involvement in brain oxidative stress. Am J Pathol. 2004;164:1655–1662. doi: 10.1016/S0002-9440(10)63724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin ZH, Wang Y, Chen RW, Wang X, Ren M, Chuang DM, Chase TN. Prostaglandin A (1) protects striatal neurons against excitotoxic injury in rat striatum. J Pharmacol Exp Ther. 2001;297:78–87. [PubMed] [Google Scholar]
- Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994a;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 1994b;62:376–379. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- Ratan RR, Lee PJ, Baraban JM. Serum deprivation inhibits glutathione depletion-induced death in embryonic cortical neurons: evidence against oxidative stress as a final common mediator of neuronal apoptosis. Neurochem Int. 1996;29:153–157. doi: 10.1016/0197-0186(95)00115-8. [DOI] [PubMed] [Google Scholar]
- Reich EE, Markesbery WR, Roberts LJ, 2nd, Swift LL, Morrow JD, Montine TJ. Brain regional quantification of Fring and D-/E-ring isoprostanes and neuroprostanes in Alzheimer’s disease. Am J Pathol. 2001;158:293–297. doi: 10.1016/S0002-9440(10)63968-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts LJ, 2nd, Morrow JD. Products of the isoprostane pathway: unique bioactive compounds and markers of lipid peroxidation. Cell Mol Life Sci. 2002;59:808–820. doi: 10.1007/s00018-002-8469-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn TT, Wong SM, Cotman CW, Cribbs DH. 15-deoxy-delta 12,14-prostaglandin J2, a specific ligand for peroxisome proliferator-activated receptor gamma, induces neuronal apoptosis. Neuroreport. 2001;12:839–843. doi: 10.1097/00001756-200103260-00043. [DOI] [PubMed] [Google Scholar]
- Schubert D, Piasecki D. Oxidative glutamate toxicity can be a component of the excitotoxicity cascade. J Neurosci. 2001;21:7455–7462. doi: 10.1523/JNEUROSCI.21-19-07455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinor JD, Boeckman FA, Aizenman E. Intrinsic redox properties of N-methyl-D-aspartate receptor can determine the developmental expression of excitotoxicity in rat cortical neurons in vitro. Brain Res. 1997;747:297–303. doi: 10.1016/s0006-8993(96)01237-1. [DOI] [PubMed] [Google Scholar]
- Smith WW, Norton DD, Gorospe M, Jiang H, Nemoto S, Holbrook NJ, Finkel T, Kusiak JW. Phosphorylation of p66Shc and forkhead proteins mediates Abeta toxicity. J Cell Biol. 2005;169:331–339. doi: 10.1083/jcb.200410041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanciu M, DeFranco DB. Prolonged nuclear retention of activated extracellular signal-regulated protein kinase promotes cell death generated by oxidative toxicity or proteasome inhibition in a neuronal cell line. J Biol Chem. 2002;277:4010–4017. doi: 10.1074/jbc.M104479200. [DOI] [PubMed] [Google Scholar]
- Stanciu M, Wang Y, Kentor R, et al. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem. 2000;275:12 200–12 206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- Tan S, Sagara Y, Liu Y, Maher P, Schubert D. The regulation of reactive oxygen species production during programmed cell death. J Cell Biol. 1998;141:1423–1432. doi: 10.1083/jcb.141.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinei M, Giorgio M, Cicalese A, et al. A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene. 2002;21:3872–3878. doi: 10.1038/sj.onc.1205513. [DOI] [PubMed] [Google Scholar]
- Wang X, Qin ZH, Leng Y, Wang Y, Jin X, Chase TN, Bennett MC. Prostaglandin A1 inhibits rotenone-induced apoptosis in SH-SY5Y cells. J Neurochem. 2002;83:1094–1102. doi: 10.1046/j.1471-4159.2002.01224.x. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Medina JF, Haeggstrom JZ, Radmark O, Samuelsson B. Molecular cloning of a 12-lipoxygenase cDNA from rat brain. Eur J Biochem. 1993;212:605–612. doi: 10.1111/j.1432-1033.1993.tb17699.x. [DOI] [PubMed] [Google Scholar]
- Zaccagnini G, Martelli F, Fasanaro P, et al. p66ShcA modulates tissue response to hindlimb ischemia. Circulation. 2004;109:2917–2923. doi: 10.1161/01.CIR.0000129309.58874.0F. [DOI] [PubMed] [Google Scholar]
- Zanoni G, Porta A, Vidari G. First total synthesis of A (2) isoprostane. J Org Chem. 2002;67:4346–4351. doi: 10.1021/jo025652f. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang H, Li J, Jimenez DA, Levitan ES, Aizenman E, Rosenberg PA. Peroxynitrite-induced neuronal apoptosis is mediated by intracellular zinc release and 12-lipoxygenase activation. J Neurosci. 2004;24:10 616–10 627. doi: 10.1523/JNEUROSCI.2469-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JH, Kulich SM, Oury TD, Chu CT. Cytoplasmic aggregates of phosphorylated extracellular signal-regulated protein kinases in Lewy body diseases. Am J Pathol. 2002;161:2087–2098. doi: 10.1016/S0002-9440(10)64487-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimniak L, Awasthi S, Srivastava SK, Zimniak P. Increased resistance to oxidative stress in transfected cultured cells overexpressing glutathione S-transferase mGSTA4–4. Toxicol Appl Pharmacol. 1997;143:221–229. doi: 10.1006/taap.1996.8070. [DOI] [PubMed] [Google Scholar]